
CHAPTER 44
Identification of Differentially Expressed Genes (DEGs)
GVPPSR Kumar, A Kumar and AP Sahoo
Animal Biotechnology Division, IVRI, UP, India
This chapter is discussed in three sections:
- Section I – Quality filtering of data using PRINSEQ
- Section II – Identification of Differentially expressed genes – I (Using Cufflinks)
- Section III – Identification of Differentially expressed genes – II (Using RSEM–DE packages – EBSeq, DESeq2, and edgeR)
44.1 SECTION I. QUALITY FILTERING OF DATA USING PRINSEQ
44.1.1 Introduction
The data generated from most of the platforms are in FASTQ format (i.e., base call data). The data files for this chapter are designated as control.fastq and infected.fastq. Both the fastq files have paired end reads. These data need to be initially checked and quality trimmed for further use. The most commonly used program for quality filtering/trimming is prinseq‐lite.pl. There are several options in Prinseq‐lite for data trimming and/or filtering. First, trimming is done, followed by execution of the filtering commands. Trimming is commonly done to remove the adapter sequences present in the raw data generated. It is also used to remove the poly A tail at the end of the read.
44.1.2 Quality check analyses using PRINSEQ
From a data set, summary statistics, filtered, reformatted and trimmed quality data can be generated using PRINSEQ. This can be used for all types of sequence data. PRINSEQ can be accessed through a web interface or can be used, standalone.
The command for quality filtering is given below (Figure 44.1):
perl prinseq‐lite.pl ‐fastq control.fastq ‐out_format 5 ‐min_len 50 ‐min_qual_mean 25

FIGURE 44.1 The basic command for running PRINSEQ‐lite.
For any further help please type:‐ perl prinseq‐lite.pl ‐h on the command line.
Prinseqlite is to be run on both the data files as given below:
For the control sample:
perl prinseq‐lite.pl –fastq control_R1.fastq –out_format 5 –min_len 50 –min_qual_mean 25
For the infected sample:
perl prinseq‐lite.pl –fastq infected_R1.fastq –out_format 5 –min_len 50 –min_qual_mean 25
These quality‐filtered data are further analyzed through different pipelines. Here, we initially discuss Cufflinks, and then the RSEM–DE package. The summary statistics, good and bad files generated from control_R1.fastq, are given below.
44.1.3 Summary statistics
A summary of total input sequences, the number of good sequences as per the details provided in the command, the number of bad sequences, and so on, is obtained for each dataset (Figure 44.2).
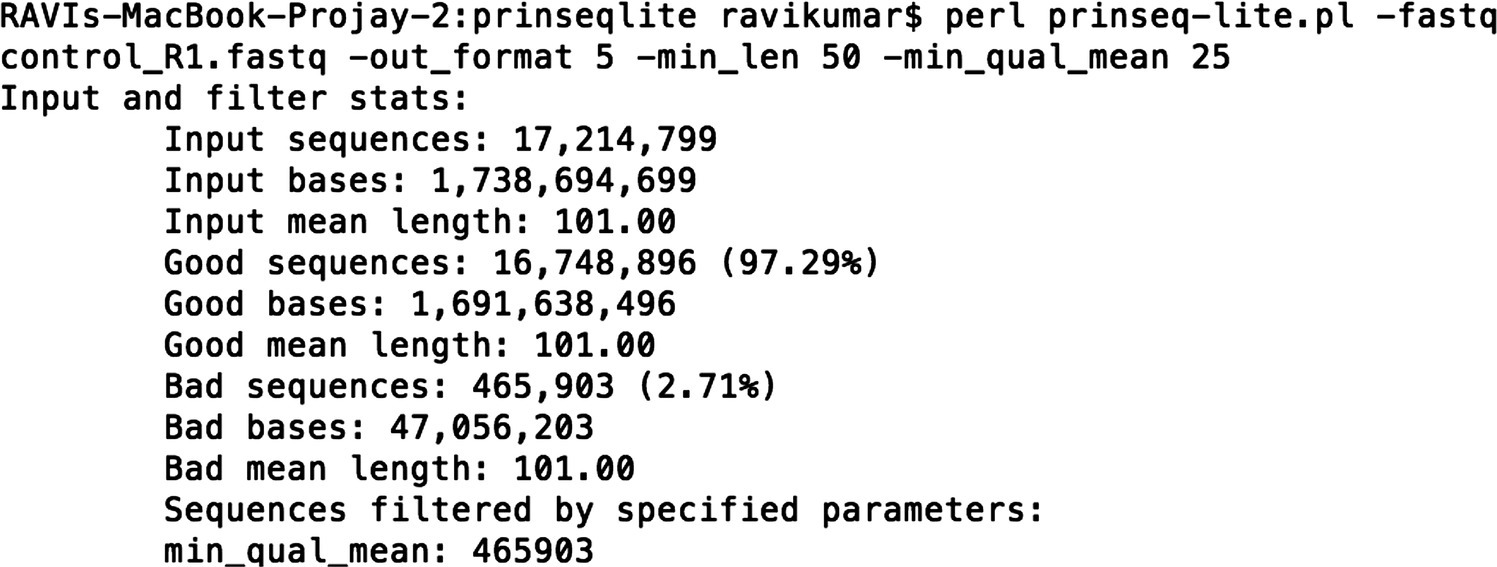
FIGURE 44.2 Summary statistics after running prinseq‐lite.pl.
44.1.4 Good and bad files generated after running Prinseq
With the output format 5, six files (three “good” and three “bad”) are generated in the folder from which the command is run (Figure 44.3). The next steps of the analysis pipeline will use the “Good” fastq file.

FIGURE 44.3 Six files generated after running prinseq‐lite.pl.
44.2 SECTION II. IDENTIFICATION OF DIFFERENTIALLY EXPRESSED GENES – I (USING CUFFLINKS)
44.2.1 Introduction to Cufflinks
“Cufflinks” stands for the suite of software tools as well as the program, which assembles and estimates abundances of transcripts and evaluates differential expression in samples. It accepts mapped reads and assembles them into a parsimonious set of transcripts. “Cuffdiff” then estimates FPKM or RPKM by normalizing for both the library size and gene length (Trapnell et al., 2012).
The prinseq‐lite output (the good files) of all the data files can be analyzed either by mapping to the reference genome or by de novo assembling the transcriptome. Here, we will be illustrating data analysis using a reference‐based approach, by mapping the reads using GMAP‐GSNAP.
Before proceeding with GMAP‐GSNAP, we need to initially download the GTF and the FASTA file sequence of the reference genome. Note that the reference FASTA file and the GTF file should be downloaded from the same genome browser. The most commonly used genome browsers are NCBI, UCSC, and Ensembl. Here, we download the GTF and FASTA files from the UCSC genome browser.

44.2.2 Downloading the FASTA file from the UCSC genome browser
Go to the UCSC genome browser, and click on downloads (Figure 44.5) and then on genomics data, to select the species of your interest (Figure 44.6). Here we select the cow to open the cow genome files.

FIGURE 44.5 UCSC genome browser.


FIGURE 44.6 Click on downloads, genomics data and then select “cow”.
When you click on the bosTau8.fa.gz, you will be able to download a file of 866.1 MB which, on gunzipping, would give a file of 2.72 GB (Figure 44.7).

FIGURE 44.7 Zip file and FASTA file of the cow genome.
44.2.3 Downloading the GTF file
The GTF file can be downloaded from UCSC by clicking on the table browser, and then selecting the options shown below (Figure 44.8).

FIGURE 44.8 Downloading the GTF file.
44.2.4 Genome mapping and alignment using GMAP‐GSNAP
GMAP‐GSNAP is a standalone program for mapping and aligning reads to a genome. This program does a fast batch processing of large sequence sets by aligning sequences with minimal startup time and memory requirements. The program generates accurate gene structures without using probabilistic splice site models (Wu and Watanabe, 2005); even plenty of polymorphisms and sequence errors are present in the data. The genome sequence that is downloaded is initially indexed, and this index is further used for mapping the filtered reads to generate the Sequence Alignment/Map (SAM) file(s).
44.2.5 Identifying the differentially expressed genes
Identifying the differentially expressed genes, starting from indexing the genome (Step 1, Figures 44.9 and 44.10), mapping the reads to the indexed genome (Step 2) to generate SAM files, and converting the SAM files to BAM files using Samtools (Step 3), to differential expression using cufflinks suite (Steps 3, 4, 5 and 6, Figures 44.11 – 44.13), is explained in six steps below:
Step 1: Command for indexing the genome: gmap_build ‐d btau8 bosTau8.fa.
Here, the FASTA reference genome (bosTau8.fa) is indexed as btau8.

FIGURE 44.9 Indexing the genome using GMAP.
The index files created are as below in the folder btau8.

FIGURE 44.10 Indexing files generated after indexing.
Step 2: Mapping the reads to the genome.
The good fastq files from the prinseq‐lite.pl output for control and infected samples are renamed Control_R1.fastq and infected_R1.fastq, respectively.
Note: R1 and R2 paired end reads of the same sample are treated as replicates for further analysis.
Command for mapping:
gsnap –d <genome> –t <nthreads> <fastq_file> > <output_file.sam>
Example:
For the control sample:
gsnap –d btau8 –t 4 control_R1.fastq> control_R1.sam
For the infected sample:
gsnap –d btau8 –t 4 infected_R1.fastq> infected_R1.sam
The end product of the GMAP‐GSNAP aligner is a SAM file, which needs to be converted into a BAM file for further analysis in cufflinks. Repeat the same for the other replicates. A total of four SAM files are generated separately for two replicates of each sample.
Step 3: Converting SAM to BAM using Samtools.
Samtools is useful for manipulating alignments in the SAM and BAM formats. It imports from and exports to the SAM format, and does sorting, merging and indexing (Li et al., 2009a, 2009b).
Command for SAM to BAM conversion: ./samtools view –bsh aln.sam >aln.bam
–b: Output in the BAM format. –s: Input in the SAM format. –h: Include header in the output
Example:
For the control sample:
./samtools view –bsh control_R1.sam >control_R1.bam
For the infected sample:
./samtools view –bsh infected_R1.sam >infected_R1.bam
Step 4: Sorting BAM using samtools
Command for sorting: ./samtools sort aln.bam aln.sorted
Example:
For the control sample:
./samtools sort control_R1.bam control_R1_sorted
For the infected sample:
./samtools sort infected_R1.bam infected_R1_sorted
The BAM files generated can be analyzed in two ways:
- The BAM files can be used to generate a merged assembly of transcripts via cufflinks and cuffmerge. This merged assembly (i.e. merged.gtf) is used in Cuffdiff to generate differentially expressed genes.
- Cuffdiff can be used directly to generate differentially expressed genes using the BAM files generated.
Step 5 (Option 1): Differential expression using cufflinks, cuffmerge, and cuffdiff.
Command for running Cufflinks on a BAM file (Figures 44.11 and 44.12):
For the control sample:
cufflinks ‐G btau8refflat.gtf ‐g btau8refflat.gtf ‐b bosTau8.fa ‐u ‐L CN control_R1_sorted.bam

FIGURE 44.11 Files generated after running cufflinks on control BAM file.
For the infected sample:
cufflinks ‐G btau8refflat.gtf ‐g btau8refflat.gtf ‐b bosTau8.fa ‐u ‐L CN infected_R1_sorted.bam

FIGURE 44.12 Files generated after running cufflinks on infected BAM file.
The transcript.gtf files (Figures 44.10 and 44.11) for each replicate are renamed as per the sample and replicate, and are further used in cuffmerge to generate a merged assembly. This merged assembly is then used in Cuffdiff to generate differentially expressed genes.
Command for running Cuffmerge:
cuffmerge ‐g btau8refflat.gtf ‐s bosTau8.fa ‐p 8 assemblies.txt
assemblies.txt is the file with the list of all the GTFs (transcripts.gtf) for all the replicates of all the samples. The file assemblies.txt is a text file, which looks like the file below (Figure 44.13).

FIGURE 44.13 The assemblies.txt file.
The Cuffmerge command generates a merged.gtf in the merged_asm folder. This file is used in the next Cuffdiff command.
Command for running cuffdiff:
CuffDiff computes differentially expressed genes. The design of experiment should consider at least two contrasting groups of experimental subjects (e.g., healthy vs. diseased) for identifying the differentially expressed genes. CuffDiff should always be run on replicates (i.e., infected vs. control).
cuffdiff merged.gtf control_R1_sorted.bam control_R2_sorted.bam infected_R1_sorted.bam infected_R2_sorted.bam
This command generates many files, out of which, gene_exp.diff is the file to look for the differentially expressed genes.
Step 5 (Option 2): Differential expression using CuffDiff directly from the sorted bam file.
Command:
Cuffdiff –p –N transcripts.gtf
–p: num‐threads <int>. –N
44.2.6 Running Cuffdiff for our BAM files
cuffdiff –p 3 –N bostau8refflat.gtf
control_R1_sorted.bam,control_R2_sorted.bam infected_R1_sorted.bam,infected_R2_sorted.bam –o cuffdiff_out
The gene_exp.diff is the file in which to look for the differentially expressed genes. The file contains the fields as marked below (Figure 44.14).

FIGURE 44.14 gene_exp.diff file giving the fold change of the genes, along with significance.
Calculation of Log2fold change for A1BG gene (row 3 in Figure 44.14 above):
Log2fold change = Log2(FPKM infected/FPKM of control)
= Log2(0.576748/3.92513) = –2.76673
44.3 SECTION III. IDENTIFICATION OF DIFFERENTIALLY EXPRESSED GENES – II (USING RSEM‐DE PACKAGES EBSEQ, DESEQ2 AND EDGER)
44.3.1 Introduction
RSEM is a cutting‐edge RNASeq analysis package that is an end‐to‐end solution for differential expression, and simplifies the whole process (Li and Dewey, 2011). It also introduces a new more robust unit of RNASeq measurement called TPM. Calculating expression counts using RSEM should be initially taken up. These counts for all the samples and their replicates are further used in differential expression (DE) packages for identifying differential expressed genes (DEGs).

FIGURE 44.15 Workflow for identifying DEGs using RSEM and DE packages.
Calculating expression counts using RSEM is explained in nine steps below:
Step 1: Downloading RSEM and installing.
By using the wget command, RSEM can be downloaded using the link below. After unzipping the folder, run “make” to install RSEM.
wget http://deweylab.biostat.wisc.edu/rsem/src/rsem‐1.2.19.tar.gz
tar –xvzf rsem‐1.2.19.tar.gz
cd rsem‐1.2.19/make
Step 2: Prerequisites required for running RSEM.
Perl, R, and Bowtie need to be installed. Perl and R are normally present on most computers. Bowtie 2 needs to be added to your path (explained in steps 3 and 4 below).
Step 3: Downloading Bowtie and installing
Download Bowtie from http://sourceforge.net/projects/bowtie‐bio/files/bowtie/1.1.1/
Step 4: Copy bowtie in your path or add bowtie path in bash profile.
Copying bowtie in your path:
sudo cp –R/Users/appleserver/Desktop/bowtie2/usr/local/bin
Add bowtie path in bash profile (preferred). Open the.bash_profile (Figure 44.16), add the path below to the file and run the source from the ~/.bash_profile:
export PATH="/Users/ravikumar/Desktop/bowtie2:$PATH"
run source ~/.bash_profile
echo $PATH – to check whether the path has been added

FIGURE 44.16 .bash_profile with the path added.
To check whether the path has been added to the.bash_profile, type ‐ echo $PATH (Figure 44.17).

FIGURE 44.17 Echo $PATH indicating that the path is added.
Step 5: Downloading the reference, gunzipping and concatenating
Download Bos taurus genome from Ensembl genome browser. An easier alternative is to use the wget command for a direct download on HPC (Figure 44.18):
wget –m ftp://ftp.ensembl.org/pub/release‐81/fasta/bos_taurus/dna/&or f in $(find.–name "*.gz")

FIGURE 44.18 wget command downloading the genome from the ensemble genome browser.
The folder that is created after the download is ftp.ensembl.org(Figure 44.19). This folder contains FASTA files of all chromosomes (Figure 44.20). These FASTA files are further concatenated into a single file (combined.fa), having all chromosomes.

FIGURE 44.19 Folder ftp.ensembl.org created after the download.

FIGURE 44.20 The chromosome gunzip files in the folder ftp.ensembl.org.
A direct download of each chromosome from the ftp site can also be done as given below (Figure 44.21). However, this is time‐consuming. The first option, downloading using the wget command, is faster.


FIGURE 44.21 Direct download from the.ftp site.
The files downloaded are gunzipped using:
gunzip Bos_taurus.UMD3.1.dna.chromosome.*.fa.gz
Concatenating/combining all the fasta files into a combined fasta file (reference):
cat Bos_taurus.UMD3.1.dna.chromosome.*.fa > combined.fa
Step 6: Download annotation file in gtf format.
Command for downloading the gtf: wget –m
ftp://ftp.ensembl.org/pub/release‐81/gtf/bos_taurus
The gtf file downloaded needs to be modified for RSEM to extract only the exon annotations. This is done by using an “awk” command to create a filtered.gtf file.
awk command to extract the exon annotations from gtf:
awk ‘$3 == “exon”’ Bos_taurus.UMD3.1.8.1.gtf> filtered.gtf
Step 7: Prepare reference using RSEM
To prepare the reference sequence, run the “rsem‐prepare‐reference” program.
Command for preparing the reference is simply indexing the reference sequence. This creates 12 files as index files (Figure 44.22) with the name of BT and extension bt2.

FIGURE 44.22 Index files created after indexing using bowtie 2.0.
Step 8: Calculating expression values in counts, TPM and FPKM:
To calculate expression values, the “rsem‐calculate‐expression” program is used.
The command for running rsem‐calculate‐expression should be run for each of the replicates (_R1 and _R2) of both the samples. This will generate six files, as shown in Figure 44.23, of which genes.results is the most important file among the six for identifying the differentially expressed genes.
For the control sample:
./rsem‐calculate‐expression –‐bowtie2 control_R1.fastq BT ControlR1

FIGURE 44.23 Six files generated after running the calculate expression command.
There will be six files generated as shown above, and genes.results is the most important file among the six for identifying the differentially expressed genes.
For the infected sample:
./rsem‐calculate‐expression –‐bowtie2 infected_R1.fastq BT infectedR1
The output ControlR1.genes.results gives the expected counts, TPM and FPKM for each of the ensemblIDs (Figure 44.24).

FIGURE 44.24 Expected counts, TPM and FPKM of each of the ensemblIDs.
Step 9: Combining RSEM genes.results of all the files. The expected counts of all the ensemblIDs for all four files (two replicates each of control and infected) are combined (Figure 44.25).
Command for combining the RSEM genes.results of all the files:
./rsem‐generate‐data‐matrix *.genes.results > genes.results

FIGURE 44.25 Combining the counts of all the files and rounding them to the nearest integer.
After rounding these expected counts values to the nearest integer (Figure 44.25), they can be used in programs such as EBSeq, DESeq, or edgeR to identify differentially expressed genes.
44.4 USE OF DE PACKAGES FOR IDENTIFYING THE DIFFERENTIALLY EXPRESSED GENES
(using EBSeq, DESeq2 and edgeR)
44.4.1 Differentially expression usingEBSeq (Leng et al., 2013)
EBSeq is an R package for identifying differentially expressed genes (DEGs) across biological conditions. EBSeq uses RSEM counts as input to identify differentially expressed genes. RSEM counts as input to identify differentially expressed genes. Identifying the DEGs using EBSeq is explained in six steps below.
Step 1: Installing EBSeq.
To install, type the following commands in R:
source("https://bioconductor.org/biocLite.R")
biocLite("EBSeq")
Step 2: Command for loading the package EBSeq (Figure 44.26).
>library(EBSeq)
Step 3: Command for getting the working directory.
>getwd()

FIGURE 44.26 Loading the EBSeq package in R.
Step 4: Command for setting the working library (Figure 44.27).

FIGURE 44.27 Input file for EBSeq.
> setwd()
Set the working directory to RSEM.
Step 5: Input requirement for Gene level DE analysis:
The input file formats supported by EBSeq are.csv,.xls, or.xlsx,.txt (tab delimited). In the input file, rows should be the genes, and columns should be the samples. An example of the data set in .txt format (genesresults.txt) is given in Figure 44.27.
Step 6: Commands to Run EBSeq (the details of each of the commands are given in explaining the commands (https://www.bioconductor.org/packages/3.3/bioc/vignettes/EBSeq/inst/doc/EBSeq_Vignette.pdf)):
> x=data.matrix(read.table("genesresults.txt"))> dim(x)[1] 24596 4> str(x)num [1:24596, 1:4] 615 3 0 473 1 286 832 362 103 17 …– attr(*, "dimnames")=List of 2..$ : chr [1:24596] "ENSBTAG00000000005" "ENSBTAG00000000008" "ENSBTAG00000000009" "ENSBTAG00000000010" …..$ : chr [1:4] "infectedR1.genes.results" "infectedR2.genes.results" "ControlR1.genes.results" "ControlR2.genes.results"> Sizes=MedianNorm(x)> EBOut=EBTest(Data=x,+ Conditions=as.factor(rep(c("C1","C2"),each=2)),sizeFactors=Sizes,+ maxround=5)Removing transcripts with 75th quantile < = 1012071 transcripts will be testediteration 1 donetime 0.12iteration 2 donetime 0.13iteration 3 donetime 0.08iteration 4 done> PP=GetPPMat(EBOut)> str(PP)num [1:12071, 1:2] 1 1 0 0 1 …– attr(*, "dimnames")=List of 2..$ : chr [1:12071] "ENSBTAG00000000005" "ENSBTAG00000000010" "ENSBTAG00000000012" "ENSBTAG00000000013" …..$ : chr [1:2] "PPEE" "PPDE"> DEfound=rownames(PP)[which(PP[,"PPDE"]>=.95)]> str(DEfound)chr [1:6528] "ENSBTAG00000000012" "ENSBTAG00000000013" "ENSBTAG00000000015" "ENSBTAG00000000019" "ENSBTAG00000000021" "ENSBTAG00000000025" "ENSBTAG00000000026" "ENSBTAG00000000032" …> write.table(DEfound,"DE.txt",sep = " ",quote = F,col.names=F)> GeneFC=PostFC(EBOut)> write.table(GeneFC,"FC.txt",sep = " ",quote = F,col.names=F)
Running of EBSeq in R
The output file – FC.txt
The other output file – DE.txt
Explaining the commands: (https://www.bioconductor.org/packages/3.3/bioc/vignettes/EBSeq/inst/doc/EBSeq_Vignette.pdf)
44.4.1.1 Calling of the input file into EBSeq
The object data should be a G × S matrix containing the expression values for each gene and each sample;
where: “G”: number of genes
“S”: number of samples.
These values should exhibit raw counts, without normalizing over the samples. The dim(X) command gives us the dimensions of the matrix; str(x) command gives the structure of the data; num(x) gives the details of the values of the samples; attr(x) gives the details of row names and column names.
> x=data.matrix(read.table("genesresults.txt"))> dim(x)[1] 24596 4> str(x)num [1:24596, 1:4] 615 3 0 473 1 286 832 362 103 17 …– attr(*, "dimnames")=List of 2..$ : chr [1:24596] "ENSBTAG00000000005" "ENSBTAG00000000008" "ENSBTAG00000000009" "ENSBTAG00000000010" …..$ : chr [1:4] "infectedR1.genes.results" "infectedR2.genes.results" "ControlR1.genes.results" "ControlR2.genes.results"In our analysis, object “x” is a simulated data matrix containing 24 596 rows of genes and four columns of samples. The genes are named “ENSBTAG0000000000 5”, “ENSBTAG00000000008”… (Figure 44.28).

FIGURE 44.28 Running iterations of EBSeq.
44.4.1.2 Obtaining the library size factor
EBSeq requires the library size factors for each of the samples. This is achieved by the function MedianNorm, which uses the median normalization approach.
> Sizes=MedianNorm(x)
44.4.1.3 Identifying DE genes – running EBSeq to get gene expression estimates
The function EBTest is used to detect DE genes. We define the conditions and size factors.
Explaining the conditions to EBseq:
The object conditions should be a vector of length S that indicates to which condition each sample belongs. For example, if there are two conditions and sample‐pair in each, then S = 4 and conditions may be given by as.factor(c("C1","C1","C2","C2")). This means that we have simulated the first two samples to be in condition 1 and the other two in condition 2, and thus defined conditions as:
Conditions=as.factor(rep(c("C1","C2"),each=2))
Normalization using sizeFactors:
Similarly, sizeFactors in the EBTest command is used to define the library size factor of each sample. It could be obtained by summing up the total number of reads per sample. We can opt for median normalization, scaling normalization, upper‐quantile normalization or some other such approach. Here, we are doing a median normalization and running the EM algorithm by setting the number of iterations to five via maxround=5, which can be seen in the output of step 6 above (Figure 44.28).
> EBOut=EBTest(Data=x,+ Conditions=as.factor(rep(c("C1","C2"),each=2)),sizeFactors=Sizes,+ maxround=5)Calculating the probabilities for the DE genes:The list of DE genes and the posterior probabilities of being DE are obtained as follows (Figure 44.29):> PP=GetPPMat(EBOut)> str(PP)num [1:12071, 1:2] 1 1 0 0 1 …– attr(*, "dimnames")=List of 2..$ : chr [1:12071] "ENSBTAG00000000005" "ENSBTAG00000000010" "ENSBTAG00 000000012" "ENSBTAG00000000013" …..$ : chr [1:2] "PPEE" "PPDE"
PPEE gives the posterior probability of equally expressed and PPDE gives the posterior probability of differentially expressed. This indicates that 12 071 genes are differentially expressed (Figure 44.29).

FIGURE 44.29 Identifying DEGs in EBSeq.
Differentially expressed genes at the 5% level of significance:
To get the DE genes with a probability at the level of significance 5%, we run the DEfound command. DEfound is a list of genes identified with PPDE >=0.95 or FDR < 0.05. EBSeq found 6528 genes significantly (P < 0.05) differentially expressed (Figure 44.29).
> DEfound=rownames(PP)[which(PP[,"PPDE"]>=.95)]> str(DEfound)chr [1:6528] "ENSBTAG00000000012" "ENSBTAG00000000013" "ENSBTAG0000000001 5" "ENSBTAG00000000019" "ENSBTAG00000000021" "ENSBTAG00000000025" "ENSBTAG00000000026" "ENSBTAG00000000032" …
Calculating the fold change:
“PostFC” calculates the posterior fold change for each transcript across conditions (Figure 44.29).
> GeneFC=PostFC(EBOut)
Writing the files:
write.table is used to write the fold changes and the differentially expressed genes into a file. Here the fold changes are saved as fc.txt and DEGs as DE.txt files (Figures 44.30 and 44.31).

FIGURE 44.30 Fold change of all the ensemblIDs.
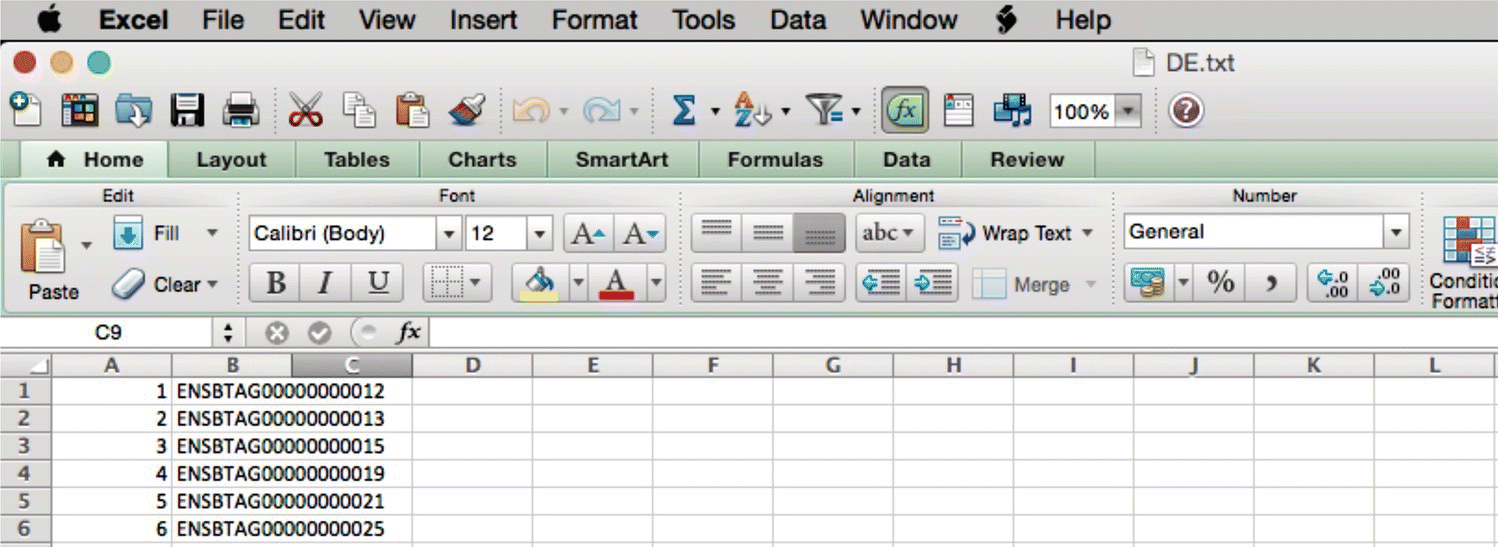
FIGURE 44.31 Significant DE genes.
44.4.2 Differentially expression usingDESeq2 (Love et al., 2014)
This is a differential expression analysis based on the negative binomial distribution. DESeq2 uses RSEM counts as input to identify differentially expressed genes.
44.4.2.1 Installing DESeq2
To install, type the following commands in R:
>source("https://bioconductor.org/biocLite.R")>biocLite("DESeq2")
44.4.2.2 Command to load the library (Figure 44.32)
>library(DESeq2)
Running DESeq2 in R:

FIGURE 44.32 Loading DESeq2 package.
Note: commands for getting the working directory and setting the working directory are the same as for step 3 and step 4 of EBSeq.
Step 5: Input requirement for Gene level DE analysis:
The input file formats supported by DESeq are .csv, .xls, or .xlsx, .txt (tab delimited). In the input file, rows should be the genes and the columns should be the samples.
Example of the data set in .txt format (roundedfn.txt) that is used here (Figure 44.33):

FIGURE 44.33 Example input data set.
Step 6: Commands to Run DESeq2:
> counts <– read.table(file = "roundedfn.txt", header = TRUE,row.names=1)> class(counts)[1] "data.frame"> countdata=data.matrix(counts)> class(countdata)[1] "matrix"> Design = data.frame(+ row.names = colnames(counts),+ condition = c("Control", "Control", "infected", "infected"),+ libType = c("single‐end", "single‐end", "single‐end", "single‐end"))> Designcondition libTypeC_R1 Control single‐endC_R2 Control single‐endT_R1 infected single‐endT_R2 infected single‐end> dds <– DESeqDataSetFromMatrix(countData = countdata,+ colData = Design,+ design = ~ condition)> ddsclass: DESeqDataSetdim: 24596 4exptData(0):assays(1): countsrownames(24596): ENSBTAG00000000005 ENSBTAG00000000008 … ENSBTAG00000048316 ENSBTAG00000048317rowData metadata column names(0):colnames(4): C_R1 C_R2 T_R1 T_R2colData names(2): condition libType> dds <– DESeq(dds)estimating size factorsestimating dispersionsgene‐wise dispersion estimatesmean‐dispersion relationshipfinal dispersion estimatesfitting model and testing> res <– results(dds)> resOrdered <– res[order(res$padj),]> head(resOrdered)log2 fold change (MAP): condition infected vs ControlWald test p‐value: condition infected vs ControlDataFrame with 6 rows and 6 columns

> write.table(resOrdered,"DEDEseq2.txt",sep = " ",quote = F,col.names=F)
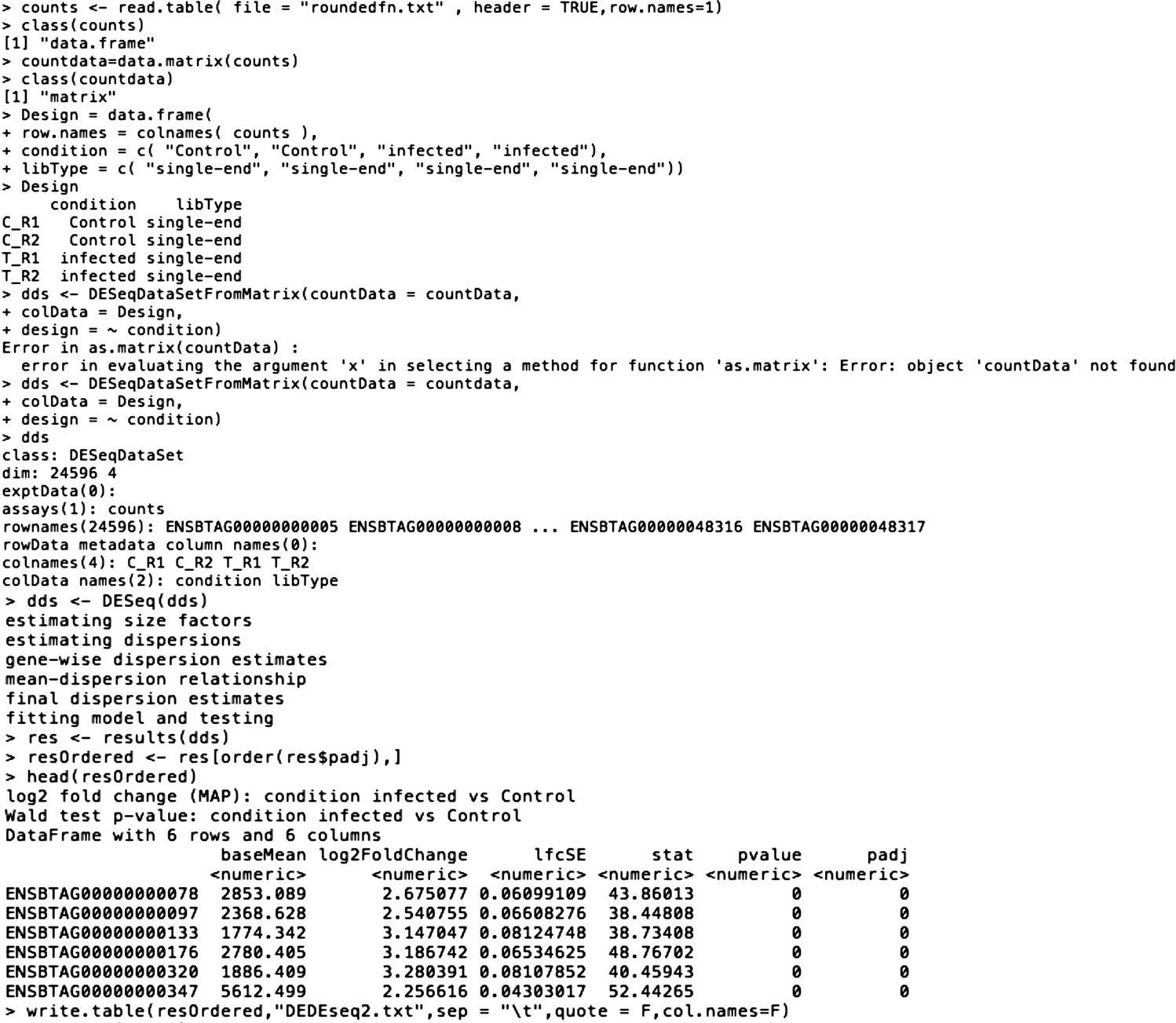
The output file is DEDESeq2.txt.

FIGURE 44.36 Fold change and significance of ensemble IDs in the file DEDESeq2.txt.
44.4.2.3 Differentially expressed genes at 5% level of significance
To get the DE genes with probability at the 5% level of significance, we select genes with a p value of < 0.05. DESeq found 8249 genes with a p‐value or padj < 0.05 (Figure 44.37).

FIGURE 44.37 Significant DEGs in DEDEseq2.txt.
Explaining the commands: https://www.bioconductor.org/packages/3.3/bioc/vignettes/DESeq2/inst/doc/DESeq2.pdf
- Calling the input file into DESeq2 and defining the dataset: The data are first read with read.table to initially create a counts object, which is then read as a data matrix into Countdata. Countdata should be a matrix of read counts, where columns correspond to different samples. Design is an object where we explain to the software the details of the input file, by giving what the columns are and what they represent – namely, control/infected. We also explain the library type of the data that are being called into DESeq2.
> counts <– read.table(file = "roundedfn.txt", header = TRUE,row.names=1> countdata=data.matrix(counts)> Design = data.frame(+ row.names = colnames(counts),+ condition = c("Control", "Control", "infected", "infected"),+ libType = c("single‐end", "single‐end", "single‐end", "single‐end"))With the countData and sample information in Design, we can construct a DESeqDataSet, which is the actual dataset used to identify differentially expressed genes:
> dds <– DESeqDataSetFromMatrix(countData = countdata,+ colData = Design,+ design = ~ condition)>ddsclass: DESeqDataSetdim: 24596 4exptData(0):assays(1): countsrownames(24596): ENSBTAG00000000005 ENSBTAG00000000008 … ENSBTAG00000048316 ENSBTAG00000048317rowData metadata column names(0):colnames(4): C_R1 C_R2 T_R1 T_R2colData names(2): condition libType - Identifying DE genes – Running DESeq2 to get differential gene expression: The DESeq function takes care of normalization, and identifies differentially expressed genes. This will print out a message for the various steps it performs. The estimation of size factors – controlling for differences in the sequencing depth of the samples, the estimation of dispersion values for each gene, and fitting a generalized linear model. The result tables are generated using the function results(), which extracts a results table with log2 fold changes, p values and adjusted p values. We can order our results table by the smallest adjust P value by running the resOrdered function.
> dds <– DESeq(dds)estimating size factorsestimating dispersionsgene‐wise dispersion estimatesmean‐dispersion relationshipfinal dispersion estimatesfitting model and testing> res <‐ results(dds)> resOrdered <‐ res[order(res$padj),]> head(resOrdered)log2 fold change (MAP): condition infected vs ControlWald test p‐value: condition infected vs ControlDataFrame with 6 rows and 6 columnsNote:
- basemean: the average of the normalized count values, dividing by size factors, taken over all samples in the DESeqDataSet.
- log2FoldChange: the effect size estimate. This tells us how much the gene’s expression would be changed in infected samples in comparison with control samples.
- lfcSE: the standard error estimate for the log2 fold change estimate.
- p‐value: the probability of a fold change as strong as the observed one, or even stronger.

FIGURE 44.38 reOrdered command ouput and the various column IDs generated.
44.4.3 Differential gene expression using edgeR (Robinson et al., 2010)
Step 1: Installing edgeR.
To install, type the following commands in R:
source("https://bioconductor.org/biocLite.R")
biocLite("edgeR")
Step 2: Command to load the library.
>library(edgeR)

Step 3: Input requirement for Gene level DE analysis.
The input file formats supported by edgeR are .csv,.xls, or.xlsx,.txt (tab delimited). In the input file, the rows should be the genes, and the columns should be the samples.
Example of the data set in.txt format (roundedfnedge.txt) that is used here:

(Note: commands for getting the working directory and setting the working directory are the same as step 3 and step 4 of EBSeq)
Step 5: Commands to run edgeR:
> raw.data <– read.table(file = "roundedfnedge.txt", header = TRUE)> counts <– raw.data[, ‐c(1,ncol(raw.data))]> head(counts)C_R1 C_R2 T_R1 T_R21 884 855 615 5882 5 5 3 23 0 0 0 04 658 647 473 4665 0 0 1 16 121 116 286 275> rownames(counts) <‐ raw.data[, 1]> head(counts)C_R1 C_R2 T_R1 T_R2ENSBTAG00000000005 884 855 615 588ENSBTAG00000000008 5 5 3 2ENSBTAG00000000009 0 0 0 0ENSBTAG00000000010 658 647 473 466ENSBTAG00000000011 0 0 1 1ENSBTAG00000000012 121 116 286 275> colnames(counts) <‐ paste(c(rep("C_R",2), rep("T_R",2)), c(1:2,1:2), sep="")> dim(counts)[1] 24596 4> colSums(counts)C_R1 C_R2 T_R1 T_R29348648 9150009 10517019 10334348> colSums(counts)/1e06C_R1 C_R2 T_R1 T_R29.348648 9.150009 10.517019 10.334348> table(rowSums(counts))[1:30]> group <– c(rep("C", 2), rep("T", 2))> cds <– DGEList(counts, group = group)> names(cds)[1] "counts" "samples"> cds <– cds[rowSums(1e+06 * cds$counts/expandAsMatrix(cds$samples$lib.size, dim(cds)) > 1) >= 3,]> cds <– calcNormFactors(cds)> cds$samplesgroup lib.size norm.factorsgroup lib.size norm.factorsC_R1 C 9348648 1.2798014C_R2 C 9150009 1.2807311T_R1 T 10517019 0.7805419T_R2 T 10334348 0.7816336> cds <– estimateCommonDisp(cds)> names(cds)[1] "counts" "samples" "common.dispersion" "pseudo.counts" "pseudo.lib.size" "AveLogCPM"> cds <– estimateTagwiseDisp(cds)> names(cds)[1] "counts" "samples" "common.dispersion" "pseudo.counts" "pseudo.lib.size" "AveLogCPM"[7] "prior.n" "tagwise.dispersion"> summary(cds$tagwise.dispersion)Min. 1st Qu. Median Mean 3rd Qu. Max.1.571e‐06 1.571e‐06 1.571e‐06 1.972e‐05 1.571e‐06 6.434e‐03> de.tgw <– exactTest(cds,dispersion = "common", pair = c("C", "T")) or> de.tgw <– exactTest(cds,dispersion = "tagwise", pair = c("C", "T"))> de.tgwAn object of class "DGEExact"$tablelogFC logCPM PValueENSBTAG00000000005 0.008546278 6.205706 8.892866e‐01ENSBTAG00000000010 0.065301786 5.816468 3.023431e‐01ENSBTAG00000000012 1.782004256 4.336904 6.581677e‐63ENSBTAG00000000013 2.043132781 5.820494 1.076701e‐219ENSBTAG00000000014 0.021064053 5.484187 7.743713e‐0110813 more rows …$comparison[1] "C" "T"$genesNULL> options(digits = 3)> topTags(de.tgw, n = 20, sort.by = "p.value")Comparison of groups: T‐ClogFC logCPM PValue FDRENSBTAG00000009012 8.89 11.55 0 0ENSBTAG00000033748 7.81 8.25 0 0ENSBTAG00000007883 7.81 7.41 0 0ENSBTAG00000008951 7.55 9.53 0 0ENSBTAG00000014762 7.52 8.86 0 0ENSBTAG00000037608 7.39 5.91 0 0ENSBTAG00000009206 7.26 6.92 0 0ENSBTAG00000007881 7.25 11.49 0 0ENSBTAG00000014707 7.02 11.29 0 0> Z <– topTags(de.tgw, sort.by = "p.value")> options(digits = 5)> Z <– topTags(de.tgw, sort.by = "p.value")>write.table(Z,"DEEdgeR.txt",sep = " ",quote = F,col.names=F)> Z <– topTags(de.tgw, n = 10000, sort.by = "p.value")>write.table(Z,"DEEdgeR.txt",sep = " ",quote = F,col.names=F)
Running of edgeR in R


The output file is DEEdgeR.txt.
Differentially expressed genes at the 5% level of significance:
To get the DE genes at the 5% level of significance, we select genes with p value < 0.05. edgeR found 9113 genes significantly (P < 0.05) differentially expressed.
Explaining the commands:
https://www.bioconductor.org/packages/3.3/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf
- Calling the input file into edgeR and defining the dataset. The dataset accepted in edgeR should contain counts with the row names as the gene ids and the column names as the sample ids. The given commands take care of reading the data and defining the data set. The counts object is created by reading the input file roundedfnedge.txt, and the row names and column names are defined using rownames(counts) and colnames (counts) function. The group command will group the columns into control and treated.
> raw.data <– read.table(file = "roundedfnedge.txt", header = TRUE)> counts <– raw.data[, ‐c(1,ncol(raw.data))]> head(counts)> rownames(counts) <– raw.data[, 1]> head(counts)>colnames(counts) <– paste(c(rep("C_R",2),rep("T_R",2)),c(1:2,1:2),sep="")> dim(counts)> colSums(counts)> colSums(counts)/1e06> table(rowSums(counts))[1:30] - Identifying DE genes – running to get differential gene expression. The function DGEList()coverts the count matrix into an edgeR object. In addition to the count matrix, we define a group variable that tells edgeR about the sample groups, which is supplied to DGEList. The elements that the object contains can be seen by using the names() function. Normalization factors can also be estimated.
> group <– c(rep("C", 2), rep("T", 2))> cds <– DGEList(counts, group = group)> names(cds)The low count reads need to be filtered out to detect differential expression. In edgeR, only those genes that have at least one read per million in at least three samples are kept for further analysis. After filtering, normalization factors, which correct for the different compositions of the samples, are calculated. The product of the actual library sizes, and these factors, give effective library sizes.
> cds <– cds[rowSums(1e+06 * cds$counts/expandAsMatrix(cds$samples$lib.size, dim(cds)) > 1) >= 3,]> cds <– calcNormFactors(cds)> cds$samplesThe following commands are used to estimate common dispersion and tagwise (i.e., genewise) dispersion:
> cds <– estimateCommonDisp(cds)> names(cds)> cds <– estimateTagwiseDisp(cds)> names(cds)> summary(cds$tagwise.dispersion)The pair‐wise test for differential expression between two groups is performed by the function exactTest(). One of the lists of elements generated in the output of exactTest() is a table of results. However, the table from exactTest() does not contain p‐values adjusted for multiple testing. These can be obtained by using the function topTags(). This takes the account from ExacTest() and adjusts the raw p‐values (by False Discovery rate (FDR) correction) to return the top differentially expressed genes. But for a column of adjusted p‐values sorted in increasing order, the output is similar to that of exactTest(). The sort.by argument sorts the table by p‐value or fold‐change. The topTags() function generates the top differentially expressed genes. If the n parameter is set to the total number of genes, the entire topTags() results table can be saved. Write.table writes the output to a txt file (Figure 44.42)
> de.tgw <– exactTest(cds, pair = c("C", "T"))> de.tgw> options(digits = 3)> topTags(de.tgw, n = 20, sort.by = "p.value")> Z <– topTags(de.tgw, sort.by = "p.value")> options(digits = 5)> Z <– topTags(de.tgw, n = 10000, sort.by = "p.value")> write.table(Z,"DEEdgeR.txt",sep = " ", quote = F, col.names=F)

FIGURE 44.42 Fold change and significance of ensemblIDs in DEEdgeR.txt.
44.5 QUESTIONS
- 1. Why is there a need to filter and trim RNA‐Seq data?
- 2. The gtf file downloaded from Ensembl cannot be used directly in RSEM. Why?
- 3. If cufflinks : RPKM; then RSEM : ______ ; and edgeR : _________.