
Late-Onset Caloric Restriction Alters Skeletal Muscle Metabolism
Mechanisms from Animal and Human Studies
Chiao-nan Chen, Chang Gung University, Taoyuan, Taiwan
Abstract
Skeletal muscle is an organ critical for generating movement and fueling metabolism. Age-related muscle dysfunction, including loss of muscle mass and strength, not only impairs elder persons’ physical function but also decreases their peripheral insulin sensitivity. The key contributor to the age-related muscle dysfunction is impaired mitochondrial function. Mitochondrial dysfunction results in the alteration of cellular metabolic patterns away from mitochondrial respiration and toward glycolysis, which decreases cells’ energy-generation capacity. In addition, impaired mitochondria release more free radicals, which increases cellular oxidative stress and protein oxidative modification and degradation. Caloric restriction (CR), with the characteristic of boosting cellular antioxidant capacity, was found beneficial for aging skeletal muscles. CR has been shown to retard age-related muscle loss, increase muscle antioxidant capacity, decrease muscle oxidative stress, improve muscle mitochondrial function, improve muscle insulin sensitivity and glucose uptake, and reprogram skeletal muscle metabolism from glycolysis to mitochondrial oxidative phosphorylation. In summary, CR provides beneficial effects on aging skeletal muscles partly by decreasing oxidative stress and modulating cellular metabolism.
Keywords
Diet; aging; mitochondria; glycolysis; oxidative stress
Skeletal muscle is an organ that enables individuals to move, protecting and supporting the skeleton and regulating the whole body’s glucose homeostasis. Energy-production systems in skeletal muscles are important in terms of skeletal muscle function because the generation of force and movement requires energy. In this chapter, we review skeletal muscle metabolism and its regulators. We discuss the changes in skeletal muscle metabolism with aging. The impacts and the mechanisms of the age-related metabolic changes in skeletal muscles are also discussed. Lastly, we show evidence from animal and human studies about the effects of caloric restriction on age-related changes in skeletal muscle metabolism. The impacts and mechanisms of changes related to calorie restriction (CR) in skeletal muscles are also discussed.
Skeletal Muscle Metabolism
Skeletal muscle is the largest organ in human. The key functions of skeletal muscle are force generation and movement, both of which require energy. Thus, energy-production systems in muscles not only are important for normal cellular maintenance but also critical for an individual’s capacity for movement. Fuel metabolism and energy production in muscles are mainly regulated by anaerobic and aerobic metabolism. In anaerobic metabolism where oxygen is not required, glucose that is transported from the bloodstream into muscle cells, is broken down into a net of two units of adenosine triphosphate (ATP) and two lactates (Benard et al., 2010). In aerobic metabolism where oxygen is required, glucose is broken down into two pyruvates in cytoplasm. Pyruvates are then transported into the matrix of the mitochondria by mitochondrial pyruvate carrier, where it is transformed into acetyl-CoA. Acetyl-CoA is also generated in the mitochondrial matrix by β oxidation where fatty acids are broken down. Acetyl-CoA is the primary substrate that enters the tricarboxylic acid (TCA) cycle (also known as the citric acid cycle or Krebs cycle), where a series of chemical reactions happen and reducing equivalents (nicotinamide adenine dinucleotide and FADH2) are produced. The reducing equivalents produced from the TCA cycle are oxidized by complexes in the electron-transport chain (ETC) on the mitochondrial inner membrane. Complexes I to IV transfer electrons and pump protons from the mitochondrial matrix into the intermembrane space, creating a proton gradient (an electrochemical gradient) across the mitochondrial inner membrane. As the protons flow back across the inner membrane through the ATP synthase (also called complex V), it drives ATP synthesis. The process happening on the ETC is called oxidative phosphorylation (OXPHOS), because proton gradient created by oxidizing reduced cofactors is used to phosphorylate adenosine diphosphate (ADP) to ATP. The net energy production in aerobic metabolism is 36 ATP. Collectively, energy production in skeletal muscles is mainly through anaerobic metabolism and aerobic metabolism. Anaerobic metabolism contains fewer steps of reactions and is faster in generating ATP compared to aerobic metabolism. Thus, anaerobic metabolism is effective for high-intensity and short-duration activities. Aerobic metabolism generates greater amounts of ATP than anaerobic metabolism, but it takes longer. Energy provided from aerobic metabolism is critical for sustained activities.
The key player in the cellular aerobic metabolism is the mitochondrion. The number and efficiency of mitochondria determine the capacity of cellular aerobic metabolism. The number increases by mitochondrial biogenesis, which is upregulated in conditions of energy deprivation (Lopez-Lluch et al., 2008; Peterson et al., 2012). During energy deprivation, the ratio of adenosine monophosphate (AMP) to ATP increases. This increased ratio activates AMP-activated protein kinase (AMPK) (Canto and Auwerx, 2011). The activated AMPK activates another nutrient-sensing protein silent mating type information regulation 2 homolog 1 (SIRT-1). AMPK and SIRT-1 activate the key regulator of mitochondrial biogenesis peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) by phosphorylation and deacetylation, respectively (Canto and Auwerx, 2011). Activated PGC-1α translocates to the nucleus and activates several nuclear transcription factors involving in mitochondrial biogenesis, including nuclear respiratory factor-1 (NRF-1), NRF-2, and mitochondrial transcription factor A (TFAM) (Anderson et al., 2008) (Fig. 28.1).

Mitochondrial efficiency is another factor that influences the energy-production capacity of cells. It is affected by the coupling status between proton flux from the intermembrane space to the matrix and the phosphorylation of ADP. When there is uncoupling (e.g., protons move back to matrix without going through ATP synthases), the efficiency of ETC decreases (Bellanti et al., 2013). Uncoupling proteins are shown to contribute to the mitochondrial uncoupling in muscle tissues (Bellanti et al., 2013). In addition to the uncoupling between the proton flux and the phosphorylation, any impairment of complexes in ETC results in decreased enzymatic functions of complexes and thus decreases the efficiency of mitochondria.
Skeletal Muscle Metabolism Changes with Aging
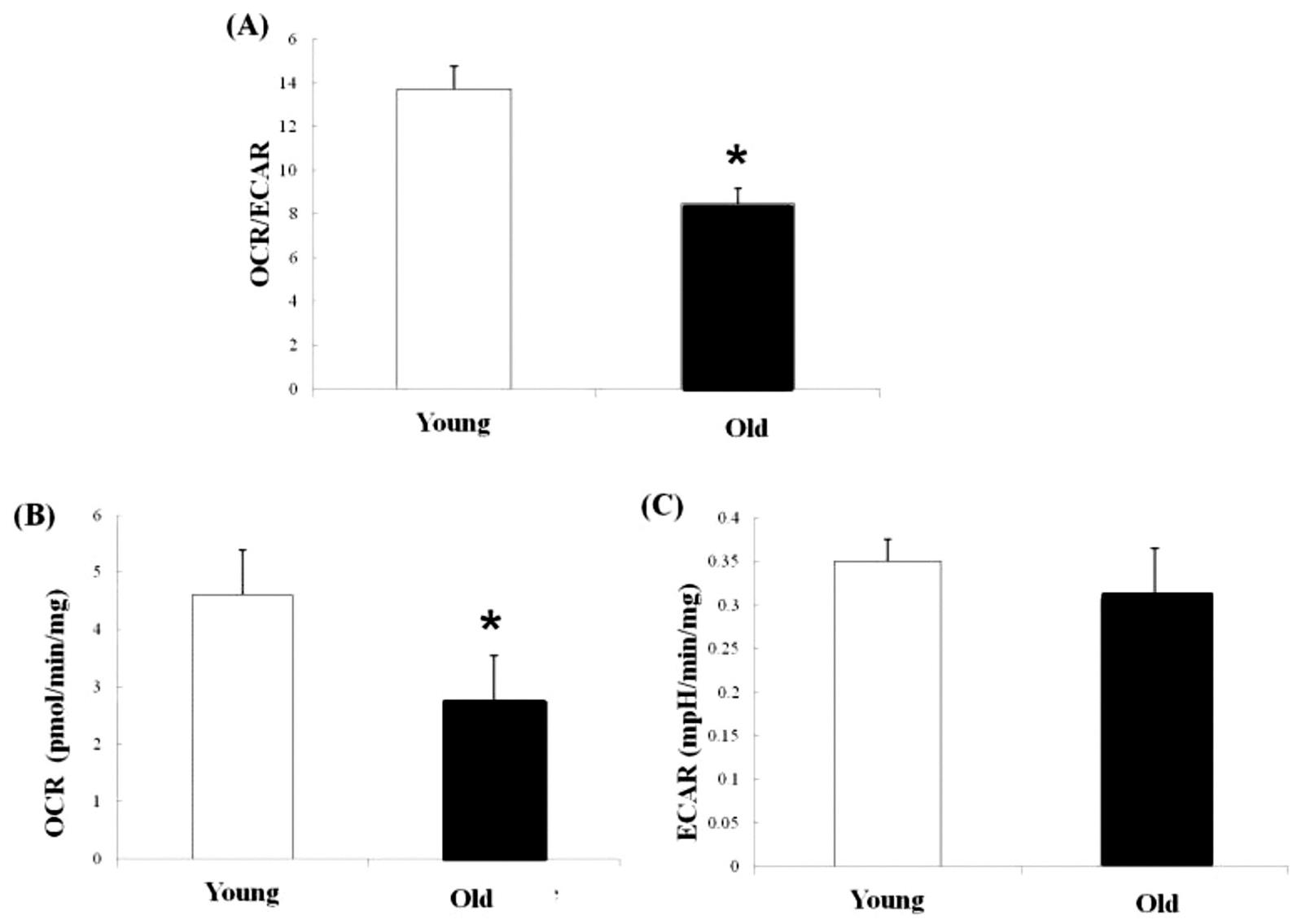
Metabolism in skeletal muscles changes significantly with aging. The pattern of cellular metabolism is defined as the ratio of mitochondrial OXPHOS to glycolysis, which decreases with aging (Chen et al., 2015) (Fig. 28.2A). This change suggests aging muscles shift energy metabolism away from mitochondrial respiration toward glycolysis. The metabolic shift was also reported in aging heart and liver cells, and in skeletal muscles with disuse atrophy (Bellanti et al., 2013; Stein and Wade, 2005). By assessing mitochondrial OXPHOS to glycolysis simultaneously, Chen et al. found that mitochondrial OXPHOS in muscles decreases significantly with aging but the glycolysis does not show significant changes with aging (Chen et al., 2015) (Fig. 28.2B, C). These findings suggest that the decreased mitochondrial OXPHOS is the main contributor to the age-related changes of the metabolic pattern in skeletal muscles.
Many studies have demonstrated that muscle mitochondria change with aging. Morphologically, aging muscles have a greater percentage of muscle fibers with large deletion mutations in mitochondrial DNA and abnormal mitochondrial ETC enzymes (McKiernan et al., 2011). Mitochondria in aging muscles show lower variation in their shape, and the cristae are less visible and defined (Figueiredo et al., 2008). Mitochondrial content in skeletal muscles also decreases with aging (Gouspillou et al., 2014; Lanza et al., 2012; Miller et al., 2012). Genomic profiling reveals that genes that associate with glucose metabolism, TCA cycles, and oxidative phosphorylation are downregulated with aging. Proteomic analysis also indicated the expression of several mitochondrial proteins decreases with aging, including complexes in ETC. The activities of citrate syntheses and complexes in ETC were found to be decreased in aging muscles when normalized to muscle weight (Ibebunjo et al., 2013; Mansouri et al., 2006). The age-related changes of mitochondria in skeletal muscles observed in animals are also seen in humans. Using principle component analysis and parametric analysis of gene-set enrichment, Mercken et al. found that pathways associated with mitochondrial electron transport and oxidative phosphorylation are downregulated in middle-aged individuals with a Western diet compared to young individuals with a Western diet (Mercken et al., 2013). Taken together, muscles of old animals contain greater amounts of mitochondria with DNA large deletion mutations. Mitochondrial content and structure change consistently with aging. Mitochondrial content in muscles decreases with aging. The expression of mitochondrial proteins and enzyme activities in muscles also decreases with aging.
In addition to the changes in structure and composition, mitochondrial function in muscles decreases with aging. The research of Mansouri et al. reported that ATP production per milligram of protein in the muscles of 28-month-old rats is only 70% of that in muscles of seven-month-old rats (Mansouri et al., 2006). The mitochondrial oxygen consumption rate (OCR) was found to be decreased with aging in both slow-twitch and fast-twitch muscles in many experimental models, including isolated mitochondria, permeabilized muscle fibers, and intact muscles (Chen et al., 2015; Figueiredo et al., 2008; Hempenstall et al., 2012; Lanza et al., 2012). Research that evaluated human muscle mitochondrial function in vivo by nuclear magnetic resonance found that rates of mitochondrial OXPHOS in muscles of the elderly is 40% less compared with rates in young individuals with matched lean body mass and fat mass (Petersen et al., 2003). Mitochondrial efficiency defined as the amount of ATP produced per unit of consumed oxygen also decreased with aging (Lanza et al., 2012). Lanza et al. normalized the mitochondrial function with mitochondrial DNA copy numbers and indicated that the decreased mitochondrial content plays the key role in the age-related decrease of mitochondrial function in muscle cells (Lanza et al., 2012). Collectively, ATP generation capacity in muscles decreases with aging. The age-related decrease of energy generation capacity is associated with the decreases of mitochondrial number and efficiency, mitochondrial DNA mutations, downregulation of proteins in TCA cycles and ETC, and reduced mitochondrial respiration.
Impacts of the Age-Related Changes in Skeletal Muscle Metabolism
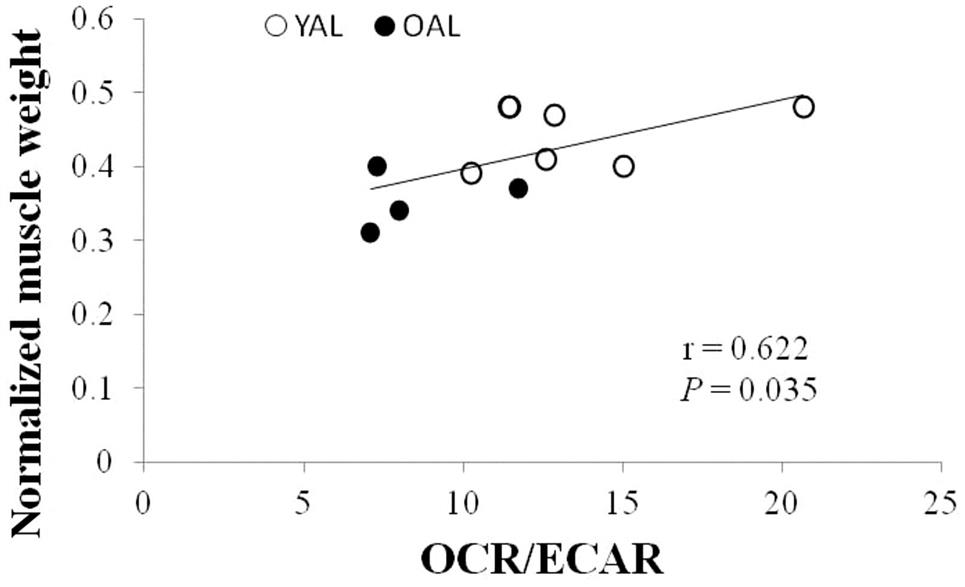
The age-related changes of skeletal muscle metabolism have multiple facets of influences on skeletal muscles, including skeletal muscle mass, exercise performance, fuel metabolism, and muscular insulin resistance. It has been shown that the cellular metabolic pattern associates with muscle mass. The greater the ratio of mitochondrial OXPHOS to glycolysis, the greater the muscle mass (Chen et al., 2015) (Fig. 28.3). Similarly, functional annotation analysis revealed that the downregulation of genes in the mitochondrial energy metabolic pathway (e.g., tricarboxylic acid cycle and oxidative phosphorylation) is associated with age-related muscle loss (Ibebunjo et al., 2013). Stein et al. also indicated that the shifts of muscle metabolism away from fat oxidation toward glycolysis contributes to disuse-induced loss of muscle mass because the metabolic changes precede the decrease of muscle mass (Stein and Wade, 2005). ATP deficiency likely mediates the metabolic shift and muscle atrophy. Glycolysis is less efficient than OXPHOS in terms of ATP generation, so muscles that depend more on glycolysis over OXPHOS may suffer ATP deficiency, which is a trigger for protein degradation and the inhibition of protein synthesis (Sandri, 2008). Another possible mechanism for the link between mitochondrial dysfunction and the loss of muscle mass is oxidative stress. Mitochondria are major sites of free radical generation; the dysfunction of mitochondria results in greater release of free radicals (Marzetti et al., 2013). The increased free radical generation can result in oxidative modification of proteins, which increases the susceptibility of proteins for degradation (Jackman and Kandarian, 2004).
Reduced exercise performance is another result of metabolic changes of skeletal muscles. Santanasto et al. investigated the relationship between skeletal muscle mitochondrial function and fatigability in the elderly during exercise. They found that individuals with higher fatigability during exercise have lower oxidative capacity in their skeletal muscles (Santanasto et al., 2015). Similarly, Gouspillou et al. compared muscle energetics (e.g., the response of energy supply to the changes of energy demand) by modular control analysis and magnetic resonance spectroscopy measurements between young and old rats. They found that the increased energy supply in response to muscle contractions was significantly reduced in the muscles of old rats. Mitochondrial dysfunction is found to be the main contributor to the reduced energetics in aging muscles (Gouspillou et al., 2014). Collectively, mitochondria, being the powerhouses of skeletal muscles, influence the exercise performance of individuals. The impaired mitochondrial function and the metabolic shifts away from mitochondrial respiration toward glycolysis reduce the exercise performance of the elderly.
Shifts in fuel metabolism and increased muscular insulin resistance (e.g., reduced muscular glucose uptake during insulin stimulation) are the results of metabolic changes in skeletal muscles. It was found that mitochondria in the muscles of the elderly are impaired in their ability to shift the metabolism from lipid to glucose oxidation under insulin stimulation. The impairment in the shifting of substrate utilization may result in muscle insulin resistance and an increase of intramyocellular lipid content in the elderly (Petersen et al., 2015). In agreement with this, studies that assessed peripheral insulin resistance by hyperinsulinemic-euglycemic clamp reported that peripheral insulin resistance is greater in the elderly compared to young individuals with matched lean body mass and fat mass (Fink et al., 1983; Petersen et al., 2003). Using glucose and glycerol tracer infusions, Petersen indicated that skeletal muscle is the main tissue responsible for the increased peripheral body insulin resistance of the elderly (Petersen et al., 2003). Taken together, the changes of skeletal muscle metabolism with aging impair the glucose uptake of skeletal muscles and further influence the glucose homeostasis of the elderly.
Mechanisms of Age-Related Changes in Skeletal Muscle Metabolism
Increased oxidative stress and blunted mitochondrial biogenesis are the most proposed mechanisms underlying the age-related changes of skeletal muscle metabolism. Increased oxidative stress and mitochondrial DNA mutations contribute to the age-related mitochondrial dysfunction in skeletal muscles. It has been shown that the release of superoxide from mitochondria and the hydrogen peroxide level in mitochondria are greater in the muscles of old animals than those in young animals (Lanza et al., 2012; Mansouri et al., 2006; Xu et al., 2010). The accumulation of oxidative modified proteins and markers of DNA damage in muscles are also greater in older animals (Chen et al., 2008; Lanza et al., 2012; Li et al., 2012; Mansouri et al., 2006; Xu et al., 2010). Research that identified the specific mitochondrial proteins that are targets of oxidative modification found citrate synthase is one target of oxidants (Li et al., 2012). Oxidative modification impairs protein functions. For example, the activity of mitochondrial protein cytochrome c oxidase was found to be negatively correlated with oxidant 4-hydroxy-2-nonenal concentration (Chen et al., 2001). The ability to shift substrate utilization of mitochondria is also affected by oxidative stress. The study of Anderson et al. found that lowering the mitochondrial oxidative stress improved insulin resistance in animals fed a high-fat diet (Anderson et al., 2009). Upregulation of uncoupling proteins is a mechanism for aging muscles to protect mitochondria from oxidative damages (Bellanti et al., 2013). Uncoupling proteins, although it reduces the generation of ROS, increases the proton leak and reduces the mitochondrial efficiency of muscles (Anderson and Weindruch, 2010). Taken together, oxidative stress in skeletal muscles increases with aging. The increased oxidative stress results in dysfunctions of mitochondrial enzymes and mitochondrial function (Johnson et al., 2013; Peterson et al., 2012).
Regulators of mitochondrial biogenesis change with aging. The content of AMPK and phosphorylated AMPK in muscles is greater in middle-aged animals compared to young animals (Chen et al., 2015). The upregulation of AMPK in muscles of middle-aged animals is likely a compensatory adaptation for the impaired AMPK function. The compensatory adaptation observed in middle-aged animals disappears when animals are older. The levels of AMPK content and phosphorylation in muscles of old animals are shown significantly decreased than those in young animals (Koltai et al., 2012). In addition, the response of AMPK to stimuli is blunted in old animals. Reznick et al. compared muscle AMPK activity and its responsiveness between young and old animals. They found that basal AMPK activity in muscles is similar between young and old animals, but the responsiveness of AMPK in the muscles of old animals is significantly decreased than that of young animals. Muscle AMPK activity was found increased in young rats in response to acute stimulation by exercise and 5ʹ-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (an activator of AMPK), however the AMPK activity in the muscles of old animals does not change in response to the acute stimuli (Reznick et al., 2007). Findings on the changes of PGC-1α in relation to aging are inconsistent. Some studies reported decreased PGC-1α expression in aging muscles (Hempenstall et al., 2012; Ibebunjo et al., 2013; Koltai et al., 2012), while other studies reported no changes (Chen et al., 2015). Hempenstall investigated the expression of PGC-1α in different cellular localization and found that the expression of total PGC-1α in muscles decreased with aging, but the level of PGC-1α in the nucleus is greater in old animals (Hempenstall et al., 2012). Because PGC-1α is translocated into nucleus under stress conditions (Anderson et al., 2008), the increased nuclear localization of PGC-1α in aging muscles suggests these muscles are under energy deficit.
Caloric Restriction Alters Skeletal Muscle Metabolism
Scientists have devoted much time to discover the “pill” that can extend both health span and life span. To date, caloric restriction is the only nonpharmaceutical and nongenetic strategy that has been shown to improve health, delay the onset of age-related pathologies on a range of model organisms, and possibly extend the life span of animals (Fontana et al., 2010).
One remarkable effect of CR is improving whole-body glucose homeostasis. Golman et al. reported that adult onset of caloric restriction lowered the incidence of impaired glucose homeostasis in rhesus monkeys (Colman et al., 2009). Similarly, Civitarese et al. found that 25% of CR for six months improved the whole-body insulin sensitivity of young overweight individuals (Civitarese et al., 2007). The improvement of whole-body glucose homeostasis is partly due to the CR-related changes of skeletal muscle metabolism. Insulin signaling of skeletal muscles in adult monkeys with four years of CR diet was found upregulated more than that in monkeys fed ad libitum, characterized by greater content and activation of the insulin receptor substrate (IRS), IRS-associated PI3-kinase, and glucose transporter GLUT4 (Wang et al., 2009). Together with the improvement of insulin sensitivity and glucose uptake, the metabolic pattern in skeletal muscles is also modified with CR. It has been shown that CR reprograms the cellular metabolism from glycolysis to OXPHOS in the muscles of middle-aged rats (Chen et al., 2015). This CR-induced metabolic reprogramming in skeletal muscles is likely a result of improved mitochondrial function that reverses the age-related increase of dependency on glycolysis as the energy source.
Mitochondrial function in skeletal muscles improves with CR. Morphologically, CR decreases the age-related increased number of muscle fibers with abnormal ETC enzymes (McKiernan et al., 2011). CR reduces the increased number of abnormal mitochondria in aging muscles (Jang et al., 2012; Lanza et al., 2012). Mitochondrial pyruvate carrier, a transmembrane protein responsible for the transportation of pyruvate from cytosol into the mitochondrial matrix, shows increased content in the muscles of middle-aged rats with CR than those in middle-aged rats fed ad libitum (Chen et al., 2015). Functionally, CR improves the age-related reduced oxidative capacity of mitochondria (Hempenstall et al., 2012; Lanza et al., 2012). In addition, CR improves decreased mitochondrial efficiency in aging muscles (Lanza et al., 2012). In human research, it was shown that 30% of CR reversed the age-related transcriptional changes of mitochondrial ETC and oxidative phosphorylation in the muscles of middle-aged individuals (Mercken et al., 2013).
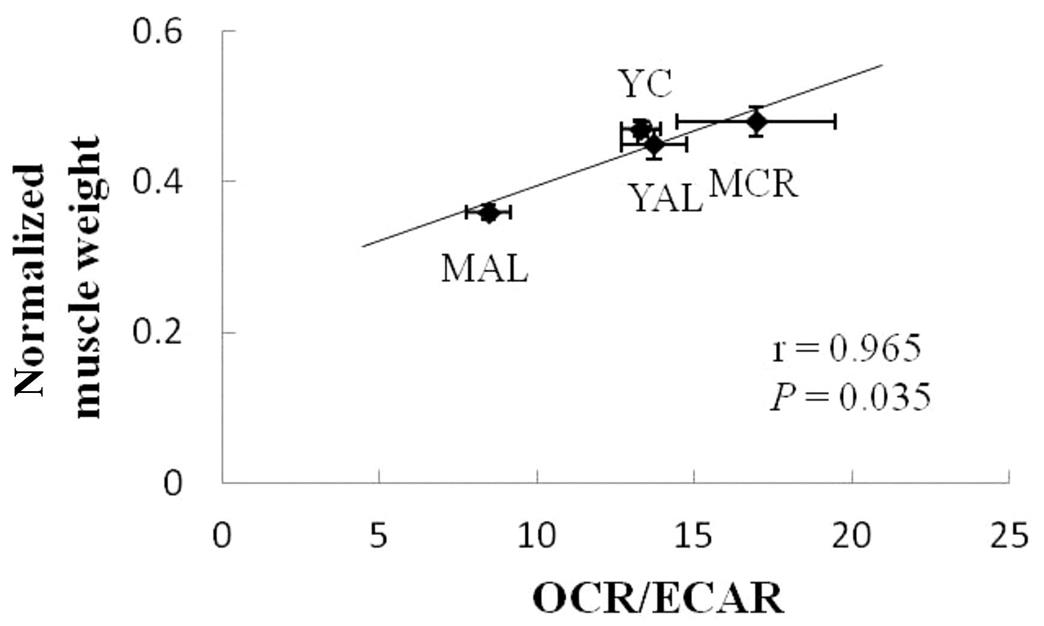
CR-induced improvement of cellular metabolism not only enhances the insulin sensitivity of skeletal muscles but also benefits the size of skeletal muscles. CR was shown to attenuate age-related muscle loss in adult monkeys and rats (Colman et al., 2009; Kim et al., 2008; McKiernan et al., 2011). CR-induced decrease of protein degradation contributes to the beneficial effects of CR on muscle size. Adult monkeys with CR showed lower proteasome content and activity in skeletal muscles compared to monkeys fed ad libitum (Wang et al., 2009). In addition, apoptosis decreases in the muscles of animals with CR (Wang et al., 2009). The study of Chen et al. investigated the relationship between the cellular metabolic pattern and muscle mass. They found that the effects of CR on muscle mass are highly correlated with the effects of CR on cellular metabolism. Muscles that show greater increases in the ratio of the oxygen consumption rate to the cellular glycolytic rate have greater increases of muscle weight per body weight (Chen et al., 2015) (Fig. 28.4).
Mechanisms of the CR-Induced Alteration in Skeletal Muscle Metabolism
Increased antioxidant capacity and reduced oxidative stress are key mechanisms underlying the beneficial effects of CR on aging skeletal muscles. CR increases catalase activity and manganese superoxide dismutase (MnSOD) expression in the muscles of animals (Jang et al., 2012; Joseph et al., 2013; Lanza et al., 2012). Generation of mitochondrial reactive oxygen species (ROS) in skeletal muscles decreases with CR (Jang et al., 2012). Age-related increases of oxidative damage, including DNA damage and the accumulation of oxidative modified proteins, are shown to decrease with CR (Lanza et al., 2012; Li et al., 2012). The CR-induced upregulation of the protective system is likely via the activation of AMPK that activates forkhead box O (FOXO), a transcriptional factor that regulates a spectrum of stress-resistance genes, including antioxidants (Canto and Auwerx, 2011; Mercken et al., 2013). Mercken et al. compared the effect of aging and chronic CR on skeletal muscles among young individuals with Western diets, middle-aged individuals with Western diet, and middle-aged individuals with 4–20 years of calorie-restricted diet. They found that CR upregulated the expression of FOXO (Mercken et al., 2013). The expression of MnSOD in muscles is also greater in middle-aged individuals with CR than in middle-aged individuals with Western diets (Mercken et al., 2013).
Regarding mitochondrial biogenesis, many studies found that CR upregulates the regulators of mitochondrial biogenesis (Civitarese et al., 2007; Hempenstall et al., 2012; Joseph et al., 2013; Mercken et al., 2013; Miller et al., 2012). The gene expressions of SIRT-1, AMPK, and PGC-1α in muscles were found upregulated in middle-aged individuals with CR than in middle-aged individuals with Western diets (Mercken et al., 2013). Similarly, Civitarese et al. found that gene expressions of TFAM, SIRT-1, and PGC-1α were upregulated in young overweight individuals receiving six months of 25% CR compared to that of the control group (Civitarese et al., 2007). Although most studies found that regulators of mitochondrial biogenesis increase with CR, the effects of CR on mitochondrial content is still being debated. For example, Hempenstall et al. found that even though total and nuclear protein content of PGC-1α increased in skeletal muscles with CR, mitochondrial content did not increase in muscles, irrespective of short-term (one month) or long-term (18 months) of CR (Hempenstall et al., 2012). The research of Civitarese et al., however, found that mitochondrial DNA content increased together with the increased expression of regulators of mitochondrial biogenesis (Civitarese et al., 2007). Because many studies found that CR improves mitochondrial functions but does not increase mitochondrial content in skeletal muscles (Hempenstall et al., 2012; Lanza et al., 2012), it is thought that CR-induced improvement of mitochondrial function is via the protection from oxidative damages but not the enhancement of mitochondrial biogenesis (Hancock et al., 2011; Hempenstall et al., 2012; Lanza et al., 2012).
Conclusion
Caloric restriction is a diet strategy for healthy aging. It delays the onset of many age-related pathologies, including diabetes and cardiovascular diseases. Regarding skeletal muscles, age-related muscle loss is partly due to increased oxidative stress and the shift of metabolic patterns away from mitochondrial respiration and toward glycolysis. Because metabolic reprogramming and the upregulation of cellular protective systems are important features of CR, evidence supports the beneficial effects of CR on aging skeletal muscles.