
CHAPTER 5
Chemical Binding of Five-Membered and Six-Membered Aromatic Molecules
FRANKLIN (FENG) TAO AND STEVEN L. BERNASEK
5.1 INTRODUCTION
Understanding the mechanism of reaction of five- and six-membered aromatic organics with semiconductor surfaces at the atomic level provides the foundation for further chemical attachment of multilayer organic materials to these semiconductor surfaces. These chemical reactions and related biofunctionalization of semiconductor surfaces is important for the development of sensing technology. This chapter focuses on the reaction mechanism of these representative categories of organic molecules. It is organized by starting from simple five-membered ring molecules, benzene, six-membered aromatic molecules with one heteroatom, and ending at six-membered aromatic molecules with two heteroatoms. For each category of these molecules, their reaction mechanisms on different semiconductor surfaces are reviewed and compared in order to understand the electronic and structural factors that affect the chemical functionalization.
5.2 FIVE-MEMBERED AROMATIC MOLECULES CONTAINING ONE HETEROATOM
Thiophene, furan, and pyrrole are three representative five-membered ring aromatic molecules with one heteroatom, as shown in Fig. 5.1. Their chemical binding on Si(111)-(7×7), Si(100), and Ge(100) have been extensively studied. Each of the three molecules exhibits different reaction mechanisms in the modification and functionalization of semiconductor surfaces due to the participation of their heteroatoms.
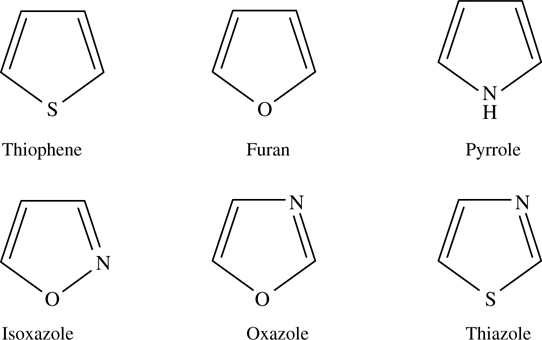
FIGURE 5.1 Molecular structure of thiophene, furan, pyrrole, isoxazole, oxazole, and thiazole.
5.2.1 Thiophene, Furan, and Pyrrole on Si(111)-(7×7)
Compared to the homogeneous distribution of electronic density on benzene, the electron density on the molecular ring of thiophene is unevenly distributed due to its heteroatom, sulfur. The electron density of the HOMO is mostly concentrated at the α-position, suggesting a highly nucleophilic nature for the C1 and C4 atoms. Thus, they are expected to interact with electrophilic dangling bonds on adatoms of Si (111)-(7×7). Both experimental techniques and theoretical approaches reveal that thiophene can be chemically bound to one adatom–rest atom pair via two Si–C sigma bonds at the C1 and C4 atoms through a [4+2]-like addition [1–3]. STM studies [1,4] show the higher reactivity of the center-adatom in contrast to the corner-adatom due to its geometric arrangement in a unit cell and a smaller steric strain induced by thiophene bonded at the center adatom site. In addition, the faulted half-unit cell exhibits higher reactivity than the unfaulted half, due to a relatively higher electrophilicity of the adatom sites on the faulted subunit.
In contrast to thiophene, furan (with an oxygen heteroatom) exhibits a significantly different reaction mechanism on interaction with Si(111)-(7×7) (Fig. 5.2). Both high-resolution electron energy loss spectroscopy (HREELS) and TDS experiments revealed two chemisorbed states (β1 and β2) at both low temperature (110 K) and room temperature [5,6]. β1 is assigned to a [4+2]-like adduct linked to an adatom–rest atom pair through two Si–C sigma bonds (Fig. 5.2a). β2 is a di-sigma-linked dimerized furan formed through a di-radical mechanism (Fig. 5.2). Both adstates are formed from the same intermediate, a mono–sigma complex. Due to the different adsorption energy of the two products, the product distribution exhibits strong temperature dependence. Distinctly different from thiophene and furan, pyrrole is chemically bonded on Si(111)-(7×7) through a dissociation of the N–H bond [7]. It forms silicon-based pyrroyl and Si–H species. Compared to the dissociation at the N–H bond, the [4+2]-like addition is expected to be thermodynamically and kinetically unfavorable for pyrrole.
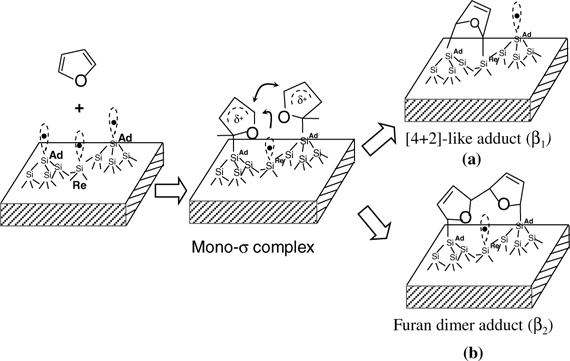
FIGURE 5.2 Scheme showing the mechanism for the formation of [4+2]-like adduct (βl) (a) and dimerized product (β2) (b) of furan on Si(111)-(7×7).
Theoretical simulations show that there is a common reaction pathway for thiophene, benzene, and 1,3-butadiene on Si(111)-(7×7) [2]. Isolated reactants initially form a mono–sigma bonded radical-like intermediate via a transition state. Passing another transition state, the mono–sigma intermediate converts into the final [4+2]-like adduct. The stability of the intermediate depends on the height of the barrier between the intermediate and final [4+2]-like adduct. This barrier increases from cis-1,3-butadiene, benzene to thiophene because the increased separation between C4 and the rest atom due to the decreased C1–C4 distance in their mono–σ intermediates, results in a higher energy transition state. Following this dependence, furan may have a more stable intermediate due to its smaller C1–C4 distance (2.20 Å) in contrast to benzene and thiophene.
Based on the theoretical simulation of conjugated dienes including 1,3-butadiene, benzene, and thiophene [2], all of them initially adsorb onto electrophilic adatoms by the electrostatic interaction with a nucleophilic Cα atom to form a mono–sigma-bonded intermediate. The unstable mono–sigma-bonded intermediate has two possible reaction channels: binding to an adjacent rest atom (Fig. 5.2a) or coupling with an adjacent mono–sigma-bonded complex (Fig. 5.2b). The mono–sigma-bonded complex of furan is expected to be more stable than that of benzene or thiophene, due to its smaller C1 and C4 separation, lower aromaticity, or lower resonance energy. This suggests the possible concurrent operation of these two reaction channels for furan on Si(111)-(7×7). At a temperature of 110 K, the mono–σ-bonded furan intermediate is thermally stabilized and a high population of this intermediate can be built up on the surface. Two adjacent radical-like mono–sigma-bonded species can readily couple to each other to form a new C–C bond, giving rise to the β2 state (Fig. 5.2b), the dimerized furan on Si(111)-(7×7). Figure 5.2 is the scheme of the two reaction channels for furan on Si(111)-(7×7). In fact, the formation of the more stable dimer complex (β2 state) for furan on Si(111)-(7×7) was further supported by a PM3 semiempirical calculation [5]. The calculation results show that the resulting dimerized furan complex (β2) is about 23 kcal/mol more stable than the [4+2]-like adduct (β1).
There is no theoretical simulation for the pathways of N–H dissociation of pyrrole on Si(111)-(7×7). On the basis of theoretical calculations of the dissociation pathways of this molecule on Si(100) [8], N–H dissociation on Si(111)-(7×7) could occur through two possible pathways, including a direct pathway through an initial binding at the nitrogen atom and an alternative pathway through an initial binding at the Cα atom and a subsequent isomerization. Notably, this chemical binding through N–H dissociation does not break the aromaticity of pyrrole in contrast to loss of aromaticity of products in the addition reactions of benzene, thiophene, and furan. The preserved aromatic π conjugation allows a weak π–π electronic coupling between two adjacent pyrroyl groups bonded on the surface. This weak electronic coupling makes the next molecule preferentially bind to an unreacted site adjacent to the bonded pyrroyl ring, evidenced in the observation of molecular wire-like binding behavior at low exposure (Fig. 5.3) [7].
5.2.2 Thiophene, Furan, and Pyrrole on Si(100) and Ge(100)
Both thiophene and furan molecularly chemisorb on Si(100) through formation of Si–C sigma bonds. Both experimental studies and theoretical calculations show that the chemisorbed thiophene molecules have similar multiple binding configurations as benzene on Si(100) [9–12]. Compared to the [4+2]-like addition on Si(111)-(7×7) [1,4], thiophene forms multiple products, including 2,3-dihydrothiophene-like species and 2,5-dihydrothiophene-like species through di-sigma binding, as well as twist bridge-like and tight bridge-like species with tetra-sigma binding [9–11]. For furan on Si(100) [10,12], no dimerized complex was observed, in contrast to the coexistence of [4+2]-like adduct and dimerized complex on Si(111)-(7×7) [5]. Furan is chemisorbed on Si(100) through a [4+2]-like addition pathway [10,12]. This is probably due to the absence of a reactive site on Si(100), similar to two neighboring adatom–adjacent rest atom sites with a reasonable separation on Si(111)-(7×7), which allows the electronic coupling of two adjacent mono–sigma complexes. The chemical binding of thiophene and furan on Si(100) was investigated by hybrid density functional (B3LYP) calculation in combination with a cluster model approach [12]. The calculations show that the [4+2]-like addition is barrier-less and favorable over the [2+2]-like addition, since the barriers of a [2+2]-like addition for thiophene and furan are 2.6 and 1.2 kcal/mol, respectively.

FIGURE 5.3 STM image of Si(111)-(7×7) with the chemisorbed pyrrole showing the formation of molecular chain-like structure on this surface.
Compared to thiophene and furan, pyrrole exhibits a major N–H dissociation pathway as on Si(111)-(7×7) [7] and a minor C–H dissociation pathway [13] on Si(100) [7,8,13,14]. A direct dissociation pathway (Fig. 5.4a) and an alternative dissociation channel (Fig. 5.4b) through N–H cleavage were theoretically simulated [8,15,16]. DFT calculations suggest that this reaction takes place via a barrierless addition of the pyrrole molecule at the Cα position to a Si=Si dimer, followed by N–H dissociation and isomerization to form a pyrroyl group bonded to Si(100) through a Si-N covalent linkage [8,15,16]. Basically, this reaction can be divided into three steps [8] as shown in Fig. 5.4b. The first step is the molecular adsorption of gaseous pyrrole onto the silicon surface through one of its Cα atoms to form a stable adsorbate (iii in Fig. 5.4b) (–6.6 kcal/mol referred to the reactants) without a barrier. In fact, this step is similar to the electrophilic attack of thiophene and furan at a Cα atom to form a mono–sigma intermediate for [4+2]-like addition on Si(111)-(7×7). The second step is the dissociation of the intermediate formed in the first step to give C4H4N•ads+Hads through a five-membered ring transition state (iv in Fig. 5.4b) with a barrier of 3.9 kcal/mol. The last step is an isomerization of the dissociated intermediate to form final product C4H4N–Si (vi in Fig. 5.4b) (–52.6kcal/mol) through a transition state with a barrier of 11.5 kcal/mol. Notably, this reaction has no overall barrier for the dissociation of pyrrole to form C4H4N–Si and H–Si species. Thus, the alternative dissociation channel is more kinetically favorable than a direct dissociative adsorption of pyrrole (Fig. 5.4a) by the interaction of the lone pair electrons of the nitrogen atom with the unoccupied orbital of the Si=Si dimer, as the direct pathway has an overall energy barrier of 6.4 kcal/mol. In addition, compared to the dissociation channel through the formation of an intermediate binding at Cα, the [2+2]- and [4+2]-like additions of pyrrole on Si(100) are not favorable, either kinetically or thermodynamically.
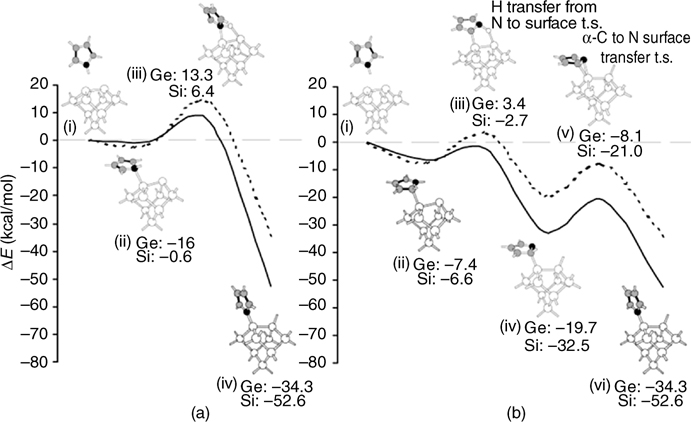
FIGURE 5.4 Critical points on the potential energy surfaces of the N–H dissociation reaction of pyrrole on Si(100) and Ge(100) through direct pathway by an initial binding at nitrogen atom (a) and alternative pathway by an initial binding at Cα (b). Adapted from Ref. 8.
Interestingly, thiophene chemisorbs onto Ge(100) through the formation of a Ge···S dative bond at a coverage lower than 0.25 monolayer (ML) and through the [4+2]-like addition at higher coverage at room temperature [11,17,18]. In contrast to the [4+2]-like addition of thiophene on Si(100), the [4+2]-like addition reaction on Ge(100) has a higher activation barrier for the transition states [18], as the major contribution to the barrier, the energy difference between the buckled and symmetric dimers of Ge(100) (6.9kcal/mol), is larger than that for Si(100) (3.2kcal/mol) [19]. Alternatively, dative bonding is barrierless, suggesting that Ge···S dative bonding is kinetically favorable [18]. Thus, dative-bonded thiophene is formed at low coverage. At high coverage, the [4+2]-like addition similar to the formation of 2,5-dihydrothiophene on Si(100) is found on Ge(100). The formation of dative bonds on semiconductor surfaces will be further reviewed in Chapter 8.
Pyrrole dissociates on Ge(100) without [4+2]-like or [2+2]-like additions [8,20]. STM studies revealed three dissociation pathways for this molecule [20]. In the first dissociation pathway, pyrrole is chemically bound to two dangling bonds of two adjacent dimers in a dimer row on Ge(100) through both Ge–N and Ge–C covalent bonds via the dissociation at both N–H and Cα–H of one molecule. Molecular aromaticity is still retained upon dissociation in this pathway. Therefore, it appears as a bright protrusion in the STM image (A in Fig. 5.5b). In the second and third pathways, pyrrole forms a tilted species through N–H dissociation; the dissociated pyrrolyl interacts with two adjacent Ge=Ge dimer rows through both a strong Ge–N bond and a relatively weak Ge–Cβ interaction. The only difference between the second and third pathways is the bonding of the dissociated hydrogen atom. In the second pathway, the hydrogen atom diffuses out of the region of interest; therefore the bonded pyrrolyl-like product is present as a complete hexagonal flower-like bright protrusion in the STM image (B in Fig. 5.5b). However, in the third pathway the dissociated hydrogen atom bonds to the other Ge atom of the Ge=Ge dimer interacting with the dissociated pyrrolyl, making the product observed as a flowerlike image with a dark site contributed from the bonded hydrogen atom (C in Fig. 5.5b). Notably, all three pathways involve at least two Ge=Ge dimers for each molecule as the arrangement of two adjacent dimers makes them accessible to one molecule simultaneously. The aromaticity of the products from the second and third pathways is weakened to some extent because the molecular Cβ atom interacts with the Ge atom of the adjacent dimer row. DFT calculations show the first pathway forms the most stable product [21].

FIGURE 5.5 (a) Occupied state STM image of Ge(100) with the chemisorbed thiophene. (b) Occupied state STM image of Ge(100) with the chemisorbed pyrrole.
Pyrrole does not react with Ge(100) through [4+2]- or [2+2]-like addition. Similar to the higher barrier for the transition state of [4+2]-like addition of thiophene on Ge(100) compared to that on Si(100), it is expected that the energy barrier for [4+2]-like addition of pyrrole on Ge(100) is higher than that on Si(100). DFT calculation [21] shows that [4+2]-like addition is not favorable thermodynamically and kinetically, in contrast to N–H dissociation via initial attachment to the Cα atom. Recent theoretical studies [8] show that N–H dissociation on Ge(100) forms a product bonded to Ge(100) through a Ge–N bond with an adsorption energy of – 34.3 kcal/mol, through a direct dissociation pathway with an energy barrier of 14.9 kcal/mol (Fig. 5.4a) or through an alternative barrierless dissociation channel (Fig. 5.4b).
5.3 FIVE-MEMBERED AROMATIC MOLECULES CONTAINING TWO DIFFERENT HETEROATOMS
Isoxazole, oxazole, and thiazole are three representative five-membered ring aromatic molecules containing two different heteroatoms (Fig. 5.1). They can be considered as aromatic molecules formed by replacing two carbon atoms of benzene with an oxygen or sulfur atom, and one carbon atom of benzene with a nitrogen atom. The oxygen/sulfur atom and the nitrogen atom contribute two and one electrons, respectively, for the formation of an aromatic π-conjugation of 4n+2 electrons in the ring. In the three molecules, each nitrogen atom has sp2 hybridization and contributes one electron of the unhybridized 2p orbital into the formation of the six-electron π-conjugation. The lone pair localized in one sp2 hybridized orbital of the nitrogen atom can be donated to form a dative bond. Thus, the electronic structure of the nitrogen atom in each of the 3 five-membered ring aromatic molecules is different from that of nitrogen in pyrrole. Compared to the two heteroatoms separated by carbon in oxazole and thiazole, the two heteroatoms in isoxazole are adjacent. The different geometric arrangement of heteroatoms in oxazole and isoxazole results in a different distribution of electron density on the aromatic ring. In addition, the different heteroatoms in oxazole and thiazole also induce a slightly different distribution of electron density. Chemical binding of the three molecules on Si (111)-(7×7) was studied using HREELS.
Isoxazole has two heteroatoms with different electronic structures. Similar to pyridine, the lone pair of the nitrogen atom in isoxazole does not participate in the formation of the aromatic π-conjugation, though the lone pair is slightly withdrawn by its neighboring oxygen atom. Therefore, the nitrogen atom of isoxazole has an electron density higher than the oxygen atom and all the carbon atoms of this molecule. It can act as an electron donor to form a dative bond with an electron-deficient adatom of Si(111)-(7×7) [22] (Fig. 5.6a). Oxazole is an isomer of isoxazole. The difference between the two isomers is the geometry of the nitrogen atom on the five-membered ring. For isoxazole, the nitrogen atom is adjacent to the oxygen atom. However, it is separated by one carbon atom from the oxygen atom in oxazole. The electron-withdrawing effect of oxygen for the nitrogen atom in oxazole is weaker than that for the case of isoxazole. Therefore, the nitrogen atom of oxazole has an electron density similar to that of pyridine, implying capability for a significant donation of electron density. Similar to oxazole, thiazole also exhibits the capability of donating electron density to form a Si···N dative bond. These molecules exhibit different pathways on Si(111)-(7×7) as shown in Fig. 5.6c.
The lone pair in an unhybridized p orbital of the oxygen atom of oxazole, which contributes to the formation of aromatic π-conjugation, in fact plays a different role in the formation of the aromatic π-conjugation than the two electrons in the two p orbitals of two carbon atoms of a pyridine molecule. To some extent, oxazole may be considered as a hybrid of furan and pyridine. In fact, oxazole can be chemically attached to Si(111)-(7×7) through both the covalent addition channel similar to thiophene [4] and furan [5], as well as the dative-bond addition similar to pyridine [23]. Oxazole carries out the addition reaction at low temperature through a [2+2]-like addition at N and Cα atoms, instead of a [4+2]-like addition. This is determined by its characteristic electronic structure. It is understandable if we consider this addition reaction as a step-wise reaction mechanism as schematically shown in Fig. 5.6d. The attack of a nitrogen atom on an adatom forms an intermediate with a lower energy (Fig. 5.6d2) than other possible intermediates because the nitrogen atom and silicon adatom are electron-rich and electron-deficient, respectively, and geometrically the adatom is at a favorable outward position in contrast to the inward rest atom. In addition, the interaction of the radical of an electron-rich rest atom with a Cα atom with lower electron density due to the electron-withdrawing effect of its adjacent oxygen atom (Fig. 5.6d3), will facilitate the subsequent formation of a Si–C sigma bond. Therefore, [2+2]-like addition is a kinetically favorable pathway for oxazole at low temperature. However, a [4+2]-like addition at two Cα atoms is not kinetically favorable for oxazole due to a high barrier for an initial binding at the Cα atom, in contrast to an initial binding at the N atom in the [2+2]-like addition. For pyridine, [2+2]-like addition is not kinetically favorable over [4+2]-like addition, as both transition states are formed through an initial binding at the N atom. Thus, pyridine is chemisorbed on Si(111)-(7×7) through a thermodynamically favorable [4+2]-like addition mechanism at C1 and C4 in addition to the formation of a Si···N dative bond.
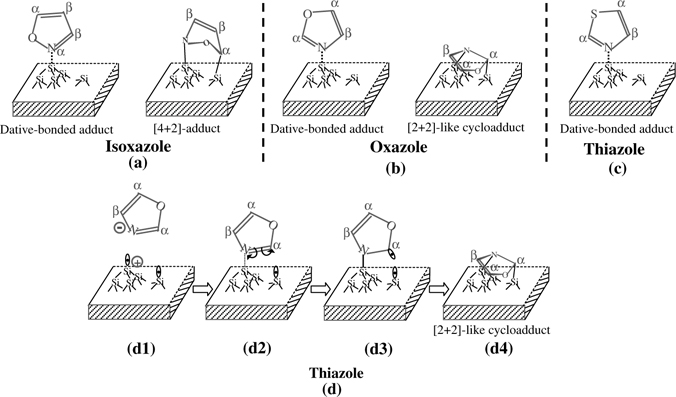
FIGURE 5.6 Binding modes of isoxazole (a), oxazole (b), and thiazole (c) on Si(111)-(7×7) at low temperature. (d) Schematic showing pathway for the formation of [2 + 2]-like adduct of oxazole on Si(111)-(7×7) at low temperature.
For isoxazole, an isomer of oxazole, its electron-rich nitrogen atom can form an intermediate with the electron-deficient adatom of Si(111)-(7×7). Its Cα atom has lower electron density than the Cβ atom due to the strong electron-withdrawing effect of the oxygen atom on its adjacent Cα atom. It can interact with the radical of the rest atom of this surface, forming a Si–C sigma bond [22]. Thus, the low electron density and favorable geometric arrangement of the Cα atom of isoxazole makes [4+2]-like addition thermodynamically and kinetically favorable (Fig. 5.6b).
The chemisorption of thiazole on Si(111)-(7×7) (Fig. 5.6c) is different from the simultaneous dative-bonded addition and [4+2]-like or [2+2]-like addition for isoxazole and oxazole at low temperature (Fig. 5.6a and b). This difference can be understood in the contrast of their electronic structures. For both isoxazole and oxazole, the Cα atom has a low electron density due to the electron-withdrawing effect of the electronegative oxygen atom. Thus, for isoxazole the binding between the electron-rich N atom and the electron-deficient Cα atom and the adatom–rest atom pair of Si(111)-(7×7) (Fig. 5.6b) is thermodynamically and kinetically favorable; for oxazole the attachment of the electron-rich N atom and the electron-deficient Cα to the adatom–rest atom pair is kinetically favorable at low temperature. Compared to isoxazole and oxazole, the Cα atom of thiazole has a relatively high electron density due to the absence of a strong electron-withdrawing effect from the sulfur atom with a relatively lower electronegativity than the oxygen atom. Thus, referring to the [2+2]-like addition of oxazole at low temperature (Fig. 5.6b), for thiazole a chemical binding between the nitrogen atom and the Cα atom of thiazole and the adatom–rest atom pair to form a [2+2]-like adduct is neither kinetically nor thermodynamically favorable. Alternatively, dative-bond addition between the electron-rich nitrogen atom of thiazole and the electron-deficient adatom site is kinetically favorable at low temperature (Fig. 5.6c). Therefore, the dative-bonded thiazole is the major product at low temperature.
5.4 BENZENE
5.4.1 Different Binding Configurations on (100) Face of Silicon and Germanium
Benzene is the prototype molecule for aromatic systems. Using both vibrational EELS and TDS, Taguchi et al. explored the chemical binding of benzene on Si(100) at 300 K early on [24]. Two chemisorption states with desorption peaks at ~432–460 and ~500K were identified [24,25]. They are attributed to benzene adsorbed on defect-free and near-defect regions, respectively. Several binding configurations were proposed for the molecularly chemisorbed benzene on Si(100) [24–29]. Two disigma binding modes were initially proposed for benzene adsorbed on the defect-free region [24]. They are schematically shown in Fig. 5.7a and b. One is formed via two Si–C covalent linkages at C1 and C4 atoms, producing a 1,4-cyclohexadiene-like product (Fig. 5.7a) and the other involving the bonding at C5 and C6, giving rise to a 1,3-cyclohexadiene-like product (Fig. 5.7b). Besides the two di-sigma binding modes, four tetra-sigma binding modes involving two pairs of Si=Si dimers in each case giving three bridge-like configurations as shown in Fig. 5.7d,e, and f were proposed [26–29]. Figure 5.7c is a tetra-sigma binding mode involving two dangling bonds from two adjacent dimer rows. A symmetric bridge binding configuration was proposed as shown in Fig. 5.7d [27,28]. Figure 5.7e and f are asymmetric twisted-bridge and tight-bridge binding modes on two adjacent dimers in a dimer row [28].
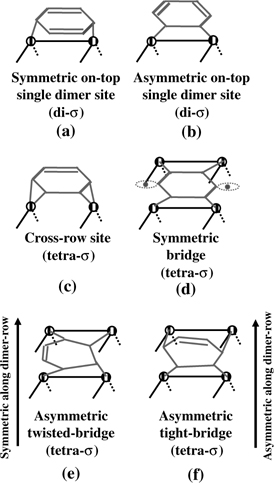
FIGURE 5.7 Six possible binding configurations of benzene molecules chemisorbed on Si(100).
STM and FTIR experimental techniques [28,30] revealed the coexistence of symmetric on-top single dimer binding (Fig. 5.7a), asymmetric twisted-bridge binding (Fig. 5.7e), and asymmetric tight-bridge binding (Fig. 5.7f) on Si(100). Notably, the on-top di-sigma bound molecules can be converted into asymmetric bridge bound molecules by thermal promotion, suggesting that asymmetric tetra-sigma bridge binding is thermodynamically more favorable than the on-top di-sigma binding [28,29].
Compared to the di-sigma binding of benzene on Si(100), benzene weakly binds to Ge(100). TDS revealed two desorption peaks for benzene on Ge(100) at 234 and 252K [25,31], corresponding to molecular adsorption at terrace and step sites, respectively. However, the adsorbed benzene on Si(100) desorbs at ~432–460 and ~500 K [24,25], corresponding to the chemisorbed benzene at terrace and step sites, respectively. DFT calculations [31] rationalized the experimentally observed difference in the adsorption of benzene on Ge(100) and Si(100). The calculated adsorption energies for benzene on Ge(100) and Si(100) are 1.4 and 20.0 kcal/mol, respectively. This significant difference in adsorption energy could partially result from the larger Ge–C bond length compared to the Si–C bond length in the adsorbed benzene. In fact, a similar difference in molecular adsorption energy on the two semiconductor surfaces has been seen for other organic molecules [8].
5.4.2 Di-Sigma Binding on Si(111)-(7×7)
On Si(111)-(7×7), early investigation using vibrational EELS with low resolution indicated that benzene chemisorbs on this surface at room temperature through a π-interaction [32]. Recent studies clearly show that benzene chemically binds to Si (111)-(7×7) with two Si–C sigma bonds formed through a [4+2]-like pericyclic addition mechanism [33,34]. DFT calculations show that the [4+2]-like addition is both thermodynamically and kinetically preferred [2,6,35]. The binding site, an adatom and its adjacent rest atom, where the C1 and C4 atoms terminate on an adjacent adatom–rest atom pair, was confirmed in recent STM studies [34,36]. Compared to the multiple binding configurations of benzene on Si(100), the di-sigma bonded 1,4-cyclohexadiene-like product is the only one seen on Si(111)-(7×7). This difference results from the accessible multiple reactive sites on Si(100). The difference among these binding sites on Si(100) that have different geometric and even electronic structures is mainly their spatial arrangement. Alternatively, in general Si(111)-(7×7) has only one reactive site, consisting of an adatom and its adjacent rest atom, which forms a binding configuration.

FIGURE 5.8 STM image of Si(111)-(7×7) with the chemisorbed benzene molecules. F and U represent the faulted and unfaulted unit cells, respectively. Adapted from Ref. 45.
Figure 5.8 is an STM image of benzene on Si(111)-(7×7). Clearly, each half unit cell contains at most three reacted adatoms appearing as dark features, which do not align in a straight-line. This observation provides evidence for a nondissociated, di-sigma binding of benzene on an adatom–rest atom pair on Si(111)-(7×7) [34,36]. Each unit cell of the Si(111)-(7×7) has six corner-adatoms and six center-adatoms. On average, each center-adatom–rest atom pair has more opportunity to react with benzene in contrast to a corner-adatom–rest atom pair. This is because each center-adatom faces two adjacent rest atoms in contrast to only one for the corner-adatom due to the geometric arrangement of adatoms on Si(111)-(7×7). A similar difference in binding density was also observed for the binding of thiophene on Si(111)-(7×7) by STM [1].
5.5 SIX-MEMBERED HETEROATOM AROMATIC MOLECULES
Pyridine is a tertiary amine with an aromatic ring. Compared to pyrrole, the nitrogen atom of pyridine has significantly different electronic density and structure though the nitrogen atoms of both molecules are sp2 hybridized. In pyridine, the sp2 hybridization of the nitrogen atom is different from that of pyrrole. For pyrrole, the three sp2-hybridized orbitals are equivalent, forming two N–C bonds and one N–H bond. In pyridine, the nitrogen atom is inequivalently sp2 hybridized (Fig. 5.9f). The remaining unhybridized p orbital of the nitrogen atom has only one electron that combines with the other five p orbitals of the carbon atoms to form an aromatic sextet. Two sp2 orbitals with one electron in each orbital form two sigma bonds with two Cα atoms. Notably, one sp2 orbital has a lone pair that does not participate in the aromatic π-conjugation system. Thus, compared to the nitrogen atom of pyrrole, the nitrogen atom of pyridine is electron rich. More importantly, the lone pair on the nitrogen atom of pyridine is localized on this atom (Fig. 5.9b). Thus, the lone pair could be donated to the electron-deficient adatom of the Si(111)-(7×7) or the buckled down atoms of Si=Si or Ge=Ge dimers to form a dative bond.
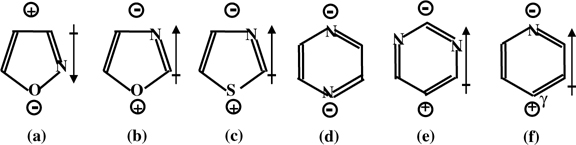
FIGURE 5.9 Polarity of isoxazole (a), oxazole (b), thiazole (c), pyrazine (d), pyrimidine (e), and pyridine (f).
Pyridine forms both the dative-bonded adduct and the [4+2]-like adduct on Si(111)-(7×7) [23] and Si(100) [37,38] at low temperature. At room temperature, the dative-bonded pyridine desorbs or partially converts into the [4+2]-like adduct [23,37]. There is no dative-bonded adduct of pyridine formed on the two silicon single crystal surfaces at room temperature. Compared to Si(100), pyridine forms a nearly complete dative-bonded monolayer on Ge(100) at room temperature [38,39]. The difference in reaction mechanisms of pyridine on silicon and germanium surfaces reflects the trend of increased electron affinity along the periodic table from Si(100) to Ge(100).
The reasons for the absence of a [4+2]-like addition for pyridine on Ge(100) could be both thermodynamic and kinetic. From the viewpoint of thermodynamics, the relatively weak Ge–N sigma bond in contrast to Si–N sigma bond results in a lower adsorption energy for [4+2]-like addition on Ge(100) than on Si(100). The weak Ge–N bond stems from the large strain in the [4+2]-like adduct due to the larger Ge–Ge bond length of the Ge=Ge dimer. In addition, the larger energy difference between the buckled Ge=Ge dimer and the symmetric Ge=Ge dimer [19] results in a higher energy barrier for the transition state of the [4+2]-like addition pathway on Ge(100) than on Si(100); therefore from the point of kinetics pyridine prefers the dative-bonding pathway on Ge(100), which is barrierless.
5.6 SIX-MEMBERED AROMATIC MOLECULES CONTAINING TWO HETEROATOMS
Pyrazine (Fig. 5.9d) is a six-membered aromatic molecule containing two nitrogen atoms at two opposite ends of the aromatic ring. Recent studies show that pyrazine can be chemically bound to Si(100) and Si(111)-(7×7) via a [4+2]-like addition through binding the two para-nitrogen atoms to a Si=Si dimer on Si(100) [40] or a pair of adjacent adatom–rest atoms on Si(111)-(7×7) [41]. This indicates that the carbon atoms of the ring are not directly involved in any chemical binding with the two surfaces.
Pyrimidine (Fig. 5.9e) is an isomer of pyrazine. Compared to pyrazine, the two nitrogen atoms are not at opposite positions on the aromatic ring. It has two nitrogen atoms at α and γ positions, which have electronic structure similar to that of pyridine. Interestingly, pyrimidine forms a [4+2]-like adduct on Si(111)-(7×7) [42], but a dative-bonded product on Ge(100) [43]. The absence of the [4+2]-like addition on Ge(100) could be for the same reasons as the absence of the [4+2]-like adduct for pyridine on Ge(100).
5.7 ELECTRONIC AND STRUCTURAL FACTORS OF THE SEMICONDUCTOR SURFACES FOR THE SELECTION OF REACTION CHANNELS OF FIVE-MEMBERED AND SIX-MEMBERED AROMATIC RINGS
Thiophene forms a dative bond on Ge(100) but not on Si(100) because [4+2]-like addition on Ge(100) is unfavorable due to the higher energy barrier mainly contributed by the larger energy difference between the buckled and symmetric Ge=Ge dimers [44]. On the other hand, the strong electron affinity in terms of a larger amount of transferred electron density in the formation of the buckled Ge=Ge dimer than that in the Si=Si dimer, makes the buckled-down Ge atom more electron-deficient and therefore forms a relatively more stable dative-bonded product.
Interestingly, pyrrole carries out N–H dissociation on both Si(100) and Ge(100) surfaces as the N–H bond of the molecule can dissociate through an alternative pathway in which the electron-deficient substrate atom is initially attached to the electron-rich Cα atom of pyrrole rather than the nitrogen atom. An additional five-membered ring transition state is involved for transferring the initial binding at the Cα to the N atom [8,15]. This transition state has a lower energy than the first one. In the first transition state, the adsorption of pyrrole at the Cα atom is facilitated in this aromatic molecule due to its capability for delocalizing the π-electron on the aromatic ring. Thus, the first transition state has a lower barrier. The overall process of the alternative dissociation pathway is barrierless. However, for pyrrolidine and 3-pyrroline, a transition state formed via initial attachment at the carbon atom could be highly unstable as it results in a pentavalent carbon due to the absence of electronic delocalization in the nonaromatic rings. Thus, the two nonaromatic molecules cannot process N–H dissociation through the alternative pathway in terms of an initial binding at the carbon atom adjacent to the nitrogen atom. For a potential direct dissociation pathway for pyrrolidine and 3-pyrroline on Ge(100), the barriers are 10.2 and 8.2 kcal/mol higher than the same barriers on Si(100), suggesting that the N–H dissociation on Ge(100) is suppressed [8]. Thus, the two molecules adopt a kinetically favorable pathway on Ge(100), formation of dative bonds. However, they follow a thermodynamically favorable pathway on Si(100), N–H dissociation, though the formation of dative bonds on Si(100) for these molecules is also barrierless.
Furan dimerizes on Si(111)-(7×7), but not on Si(100) and Ge(100) as a reasonable arrangement of reactive sites accessible for a subsequent coupling of two mono-sigma bonded intermediates is absent on the (100) surfaces. Alternatively, it bonds to Si(100) through a [4+2]-like addition reaction.
Pyridine, pyrazine, and pyrimidine all have nitrogen atoms in the aromatic ring, which are electron-rich with a formal lone pair. In the case of these molecules, the electronic structure of the semiconductor surface controls the reaction channel for adsorption. In each case, donation of the lone pair electrons to the electron-deficient Si(111)-(7×7) adatom, or the buckled down Si or Ge atoms of the dimer rows on the (100) surfaces controls the reaction pathway. Differences in reactivity for these molecules on the elemental semiconductor surfaces follow the differences in electron affinity for the elemental surfaces.
REFERENCES
1. Cao, Y.; Yong, K. S.; Wang, Z. H. et al. J. Chem. Phys. 2001, 115 (7), 3287.
2. Lu, X.; Wang, X. L.; Yuan, Q. H. et al. J. Am. Chem. Soc. 2003, 125 (26), 7923.
3. Lu, X.; Lin., M. C. Int. Rev. Phys. Chem. 2002, 21 (1), 137.
4. Cao, Y.; Yong, K. S.; Wang, Z. Q. et al. J. Am. Chem. Soc. 2000, 122 (8), 1812.
5. Cao, Y.; Wang, Z. H.; Deng, J. F. et al. Angew. Chem. Int. Ed. 2000, 39 (15), 2740.
6. Wang, Z. H.; Cao, Y.; Xu., G. Q. Chem. Phys. Lett. 2001, 338 (1), 7.
7. Yuan, Z. L.; Chen, X. F.; Wang, Z. H. et al. J. Chem. Phys. 2003, 119 (19), 10389.
8. Wang, G. T.; Mui, C.; Tannaci, J. F. et al. J. Phys. Chem. B 2003, 107 (21), 4982.
9. (a) Isobe, N.; Shibayama, T.; Mori, Y. et al. Chem. Phys. Lett. 2007, 443 (4–6), 347. (b) Qiao, M. H.; Cao, Y.; Tao, F. et al. J. Phys. Chem. B 2000,104 (47), 11211. (c) Jeong, H. D.; S Lee, Y.; Kim., S. J. Chem. Phys. 1996, 105 (12), 5200.
10. (a) Qiao, M. H.; Tao, F.; Cao, Y. et al. J. Chem. Phys. 2001, 114 (6), 2766. (b) Lee, H. K.; Kim, K. J.; Kang, T. H. et al. Surf. Sci. 2008, 602 (4), 914.
11. Rousseau, G. B. D.; Dhanak, V.; Kadodwala., M. Surf. Sci. 2001, 494 (3), 251.
12. Lu, X.; Xu, X.; Wang, N. Q. et al. J. Phys. Chem. B 2001, 105 (41), 10069.
13. Cao, X. P.; Coulter, S. K.; Ellison, M. D. et al. J. Phys. Chem. B 2001, 105 (18), 3759.
14. (a) Qiao, M. H.; Cao, Y.; Deng, J. F. et al. Chem. Phys. Lett. 2000,325 (5–6), 508. (b) Kim, K.; Han, J.; Kang, T. H. et al. J. Electron Spectrosc. Rel. Phenom. 2005, 144, 429. (c) Qiao., M. H.; Tao., F.; Cao, Y. et al. Surf Sci. 2003, 544 (2–3), 285. (d) Barteau., M. A. Chem. Rev. 1996, 96, 1413.
15. Luo, H. B.; Lin., M. C. Chem. Phys. Lett. 2001, 343 (3–4), 219.
16. Seino, K.; Schmidt, W. G.; Furthmuller, J. et al. Surf. Sci. 2003, 532, 988.
17. (a) Lee, H.; Jeon, S. M.; Kim, H. D. et al. J. Phys.: Condens. Matter 2008, 20 (13), 135006. (b) Jeon, S. M.; Jung, S. J.; Lim, D. K. et al. J. Am. Chem. Soc. 2006, 128 (19), 6296.
18. Jeon, S. M.; Jung, S. J.; Kim, H. D. et al. J. Phys. Chem. B 2006, 110 (43), 21728.
19. Kruger, P.; Pollmann., J. Phys. Rev. Lett. 1995, 74 (7), 1155.
20. (a) Kim, D. H.; Choi, D. S.; Kim, A. et al. J. Phys. Chem. B 2006, 110 (15), 7938. (b) Kim, D. H.; Choi, D. S.; Kim, A. et al. Jpn. J. Appl. Phys. Part 1 2006, 45 (3B), 2148.
21. Kim, D. H.; Choi, D. S.; Hong, S. et al. J. Phys. Chem. C 2008, 112 (19), 7412.
22. Tao, F.; Bernasek., S. L. J. Am. Chem. Soc. 2007, 129 (15), 4815.
23. Tao, F.; Lai, Y. H.; Xu., G. Q. Langmuir 2004, 20 (2), 366.
24. Taguchi, Y.; Fujisawa, M.; Takaoka, T. et al. J. Chem. Phys. 1991, 95 (9), 6870.
25. Fink, A.; Menzel, D.; Widdra., W. J. Phys. Chem. B 2001, 105 (18), 3828.
26. (a) Craig., B. I. Surf. Sci. 1993, 280(3), L279. (b) Wolkow, R. A.; Lopinski, G. P.; Moffatt., D. J. Surf. Sci. 1998, 416 (3), L1107.
27. Jeong, H. D.; Ryu, S.; Lee, Y. S. et al. Surf. Sci. 1995, 344 (3), L1226.
28. Lopinski, G. P.; Fortier, T. M.; Moffatt, D. J. et al. J. Vac. Sci. Technol. 1998, 16 (3), 1037.
29. Lopinski, G. P.; Moffatt, D. J.; Wolkow., R. A. Chem. Phys. Lett. 1998, 282 (3–4), 305.
30. Hofer, W. A.; Fisher, A. J.; Lopinski, G. P. et al. Surf. Sci. 2001, 482, 1181.
31. Cho, J. H.; Kim, K. S.; Morikawa., Y. J. Chem. Phys. 2006, 124 (2), 024716.
32. Taguchi, Y.; Fujisawa, M.; Nishijima., M. Chem. Phys. Lett. 1991, 178 (4), 363.
33. (a) Cao, Y.; Wei, X. M.; Chin, W. S. et al. J. Phys. Chem. B 1999, 103 (27), 5698. (b) Tomimoto, H.; Sekitani, T.; Sumii, R. et al. Surf. Sci. 2004, 566, 664.
34. (a) Kawasaki, T.; Sakai, D.; Kishimoto, H. et al. Surf. Inter. Anal. 2001, 31 (2), 126. (b) Yong, K. S.; Yang, S. W.; Zhang, Y. P. et al. Langmuir 2008, 24 (7), 3289. (c) Horn, S. A.; Patitsas., S. N. Surf. Sci. 2008, 602 (2), 630.
35. (a) Petsalakis, I. D.; Polanyi, J. C.; Theodorakopoulos., G. Israel J. Chem. 2005, 45 (1–2), 111. (b) Li, Y. C.; Wang, W. N.; Cao, Y. et al. Acta Chim. Sinica 2002, 60 (4), 653.
36. Tomimoto, H.; Takehara, T.; Fukawa, K. et al. Surf. Sci. 2003, 526 (3), 341.
37. (a) Tao, F.; Qiao, M. H.; Wang, Z. H. et al. J. Phys. Chem. B 2003, 107 (26), 6384. (b) Miwa, J. A.; Eves, B. J.; Rosei, F. et al. J. Phys. Chem. B 2005, 109 (43), 20055.
38. Kim, H. J.; Cho., J. H. J. Chem. Phys. 2004, 120 (17), 8222.
39. (a) Hong, S.; Cho, Y. E.; Maeng, J. Y. et al. J. Phys. Chem. B 2004, 108 (39), 15229. (b) Cho, Y. E.; Maeng, J. Y.; Kim, S. et al. J. Am. Chem. Soc. 2003, 125 (25), 7514.
40. (a) Huang, H. G.; Huang, J. Y.; Ning, Y. S. et al. J. Chem. Phys. 2004, 121 (10), 4820. (b) Lu, X.; Xu, X.; Wu, J. M. et al. New J. Chem. 2002, 26 (1), 160. (c) Shimomura, M.; Ichikawa, D.; Fukuda, Y et al. Phys. Rev. B 2005, 72 (3),
41. Huang, H. G.; Wang, Z. H.; Xu., G. Q. J. Phys. Chem. B 2004, 108 (33), 12560.
42. Huang, H. G.; Huang, J. Y.; Wang, Z. H. et al. Surf. Sci. 2007, 601 (5), 1184.
43. Lee, J. Y.; Jung, S. J.; Hong, S. et al. J. Phys. Chem. B 2005, 109 (1), 348.
44. (a) Wolkow., R. A. Phys. Rev. Lett. 1992, 68 (17), 2636.(b) Weakliem, P. C.; Carter., E. A. J. Chem. Phys. 1992, 96 (4), 3240. (c) Kubby, J. A.; Griffith, J. E.; Becker, R. S. et al. Phys. Rev. B 1987, 36 (11), 6079.
Functionalization of Semiconductor Surfaces, First Edition
Edited by Franklin (Feng) Tao and Steven L. Bernasek.
© 2012 John Wiley & Sons, Inc. Published 2012 by John Wiley & Sons, Inc.