
13
THE IMPACT OF WHOLE GENOME IN SILICO SCREENING FOR NUCLEAR RECEPTOR-BINDING SITES IN SYSTEMS BIOLOGY
Life Sciences Research Unit, University of Luxembourg, Luxembourg
13.1 INTRODUCTION
Each individual human gene is under the control of a large set of transcription factors that can bind upstream and downstream of its transcription start site (TSS) [1]. These sites typically arrange into collections of neighboring sites, the so-called modules or enhancers. Modules of transcription factors that act on focused genomic regions have been shown to be far more effective than individual factors on isolated locations and can act from large distances up to hundreds of thousands of base pairs. In an ideal case, such transcription factor modules can be identified by parallel and comparative analysis of their binding sites. Here, bioinformatics approaches can be of great help, in case they can predict the actions of the transcription factors precisely enough [2].
13.2 NUCLEAR RECEPTORS
Nuclear receptors (NRs) form a superfamily with 48 human members, of which most have the special property to be ligand-inducible [3, 4]. This property has attracted interest in the NR family as possible therapeutical targets. NRs are the best characterized representatives of approximately 3000 different mammalian proteins that are involved in transcriptional regulation in human tissues [5]. NRs modulate genes that affect processes as diverse as reproduction, development, inflammation, and general metabolism. They were first recognized as the receptors for the steroid hormones estradiol (ER α and β), progesterone (PR), testosterone (AR), cortisol (GR), and aldosterol (MR) for thyroid hormones (TR α and β) and for the biologically active forms of the fat-soluble vitamins A and D, all-trans retinoic acid (RAR α, β, and γ), and 1α, 25-dihydroxyvitamin D3 (VDR). This group of 12 NRs constitutes the classic endocrine NR subgroup. They can be defined functionally as being able to bind their specific ligand with kd of 1 nM or less [3]. The 36 remaining NRs are structurally related to the endocrine NRs but were orphans at the time of their cloning because neither their ligands nor their physiological functions were initially known [6]. During the past 17 years, however, natural and synthetic ligands have been identified for nearly half of these receptors. They now form the group of adopted orphan NRs [7]. Interestingly, most of the latter group of NRs have as their natural ligands dietary components, such as lipids or exogenously derived compounds, which are encountered in the micro- to millimolar concentration range. Subsequently, these receptors have activation thresholds (in terms of Kd) in the same molar range. This functionally separates them from the endocrine receptors. The current model of NR signaling is schematically depicted in Figure 13.1.
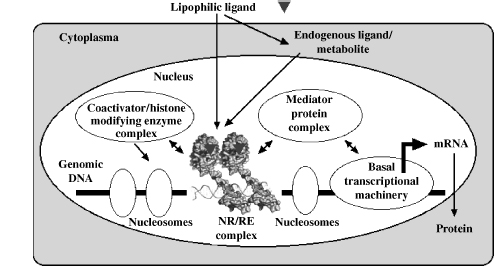
Figure 13.1 Molecular mechanism of NR action. NRs interact with discrete sequences in the proximity of genes to modulate transcription, a process that is governed by chromatin status and the availability of ligand and coregulators.
13.2.1 NRs as a Link between Nutrition Sensing and Inflammation Prevention
The interrelation of NRs, their diet-derived ligands, and metabolizing enzymes is a central issue in the new discipline nutrigenomics, which is the study of the impact of nutrient-derived compounds on the genome. It also encompasses the effects of food on physiological functions such as resistance to external assault from opportunistic pathogens [8]. For example, both nutritional overload and undernourishment have implications for immune function, and consequently, metabolism and immunity are closely linked [9]. Starvation and malnutrition can suppress immune function and increase susceptibility to infections, whereas obesity is associated with a state of aberrant immune activity and increasing risk for associated inflammatory diseases, including airway inflammation and fatty liver disease, a condition that impairs those organs' role in immunity [10].
One emerging concept in context of many diseases is the role of the immune system. Taking the nutrigenomics perspective and recognizing the lifestyle changes in the Western society, obesity can be taken as an example of a disease state characterized by a strong link with immune functions. Moreover, obesity is a strong risk factor to develop type 2 diabetes and atherosclerosis, making it one of the major health concerns of the industrialized world. In obesity, adipose tissue becomes inflamed both via infiltration by macrophages and as a result of adipocytes themselves producing inflammatory cytokines [11, 12]. Inflammation of adipose tissue is a crucial step in the development of peripheral insulin resistance [9]. In addition, in proatherosclerotic conditions, such as obesity and dyslipidemia, macrophages accumulate lipid to become foam cells in vessel wall plaques, where local inflammation is initiated. Inflammation itself is not problematic, if it is controlled and short term. During microbial infection, the inflammatory response defends the body while suppressing appetite and conserving fuel. An ill body is capable of defending itself by releasing adrenal steroids, mobilizing massive amounts of fuel, and finally suppressing inflammation once the pathogen is cleared. Concerning the latter aspect, the role of NRs is well-recognized. In fact, natural and synthetic GR ligands are used primarily as anti-inflammatory agents [13]. Other NRs, such as VDR, PPAR, RAR, and liver X receptor, also protect against inflammation. These receptors have the combined ability to manage energy and inflammation, indicating the important synergism between these two systems (Figure 13.2).
The duality between inflammatory and metabolic pathways is also highlighted by the overlapping biology and function of macrophages and adipocytes in obesity [9]. Gene expression of both cell types is highly similar; macrophages express many, if not the majority of “adipocyte” gene products, such as lipid-metabolizing and transporting proteins, while adipocytes can express many “macrophage” proteins, such as cytokines [14]. Inflammatory pathways can be initiated by extracellular mediators, such as cytokines and lipids, or by intracellular stresses, such as endoplasmic reticulum stress or excess reactive oxygen species production by mitochondria. NRs oppose these inflammatory pathways by promoting nutrient transport and metabolism and antagonizing inflammatory activity. In conditions of overnutrition, this becomes a particular challenge.
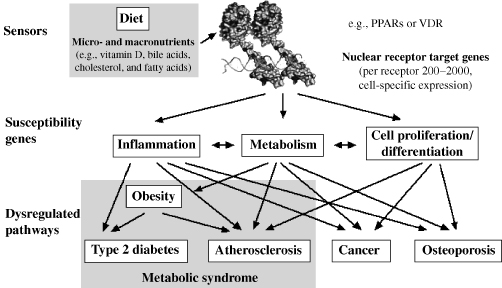
Figure 13.2 Potential interactions of NRs with complex processes. NR target genes regulated many physiological functions. When these pathways are dysregulated, they eventually result in disease states.
The commonality between distinct physiologic branches suggests that the NR superfamily should be investigated by systems biology approaches as an intact functional dynamic entity. Recent studies by the NURSA consortium (www.nursa.org) have provided evidence supporting this concept. They examined the expression of all NRs in both macrophages stimulated by the stress ligands, lipopolysaccharide, and IFNγ [15] and adipocytes induced by GR and PPARγ ligands [16]. In both cases, a subset of NRs was readily detectable with some of them, for example, VDR, rising in expression levels at various time points during the induction process. This implies new, presently overlooked roles for these NRs and their ligands. Furthermore, the discovery of increases in expression of certain NRs at intermediate and late time points indicates the importance of a multitude of NRs in the proper execution of complex processes. These and other observations suggest that larger organizational principles exist on the level of transcription factors and of enzyme complexes in the nucleus, the cytoplasm, and membranes that contain NRs or their target gene products. A molecular understanding of how the NR superfamily integrates important physiological aspects will provide a conceptual basis for the treatment of complex human diseases (Figure 13.2).
13.2.2 NRs and System Biology
Until recently, approaches to diseases and gene function, in general, tended to focus on one gene at a time. In the last 10 years, high-volume research approaches have allowed scientists to grasp the total information contained within a cell concerning transcriptional activity, protein content, and metabolites. One way to monitor and analyze these massive amounts of data is systems biology, which aims both to reduce experimental data to meaningful paradigms and also build up in silico testable hypotheses [17]. However, systems biology can do more than describing generic patterns within gene expression. The fine regulation of the NR network is specific to each human individual and depends on the constellation of regulatory small nucleotide polymorphisms (SNPs) in his/her genome [18]. It is envisaged that some of these regulatory SNPs will affect the binding of NRs to a subset of REs in their target genes. This could determine an individual's susceptibility to age-related diseases such as type 2 diabetes, atherosclerosis, cancer, and Alzheimer's disease.
Systems biology will also help to identify biomarkers (whether it be genes, proteins, or metabolites) for the early detection of these diseases [19]. NRs are able to integrate various central physiological actions in the human body. Therefore, NR signaling will benefit from an analysis with systems biology methodology on the level of (i) binding sites and changes of chromatin packaging in NR target genes, (ii) comparative mRNA expression of NR target genes in various human tissues, (iii) analysis of key NR target proteins and metabolites, and (iv) physiological consequences of NR signaling.
This review focuses on the PPAR subfamily of NRs studied within a Marie Curie research training network of 14 research teams (www.uku.fi/nucsys), which is funded by the FP6 program of the European Union.
13.3 THE PPAR SUBFAMILY
PPARs are adopted NRs that were initially described as the sensors for compounds that induce peroxisome proliferation in rodents [20], but now they are know to be important sensors of cellular levels of fatty acids and fatty acid derivatives that are mainly derived from the lipoxygenase and cyclooxygenase pathways [3]. Polyunsaturated fatty acids activate the three PPAR subtypes with relatively low affinity, whereas fatty acid derivatives show more binding selectivity [21]. PPARs are prominent players in the metabolic syndrome because of their role as important regulators of lipid storage and catabolism [22], but they also regulate cellular growth and differentiation and, therefore, have an impact on hyperproliferative diseases such as cancer [23]. Bioinformatics approaches to identify genomic targets of PPARs and important regulatory modules with colocalizing PPREs, as they will be described below, should have a major impact on understanding the role and potential therapeutic value of PPARs in complex disease.
The three PPAR subtypes α, β/δ, and γ are coexpressed in numerous cell types from either ectodermal, mesodermal, or endodermal origin, although their concentration relative to each other varies widely [24, 25]. PPARα is highly expressed in cells that have active fatty acid oxidation capacity, including hepatocytes, cardiomyocytes, enterocytes, and the proximal tubule cells of the kidney [26]. This PPAR subtype is a central regulator of hepatic fatty acid catabolism and glucose metabolism. Furthermore, it potently represses the hepatic inflammatory response by downregulating the expression of numerous genes such as various acute phase proteins. PPARα is the molecular target for the hypolipidemic fibrates, a group of drugs that are prescribed for their ability to lower plasma triacylglycerols and elevate plasma high-density lipoprotein levels. PPARβ/δ is expressed ubiquitously and often displays higher expression levels than PPARα and γ. It stimulates fatty acid oxidation in both adipose tissue and skeletal muscle, regulates hepatic very low density lipoprotein production and catabolism, and is involved in wound healing by governing keratinocyte differentiation [27]. PPARγ is expressed predominantly in adipose tissue and the immune system and exists as two distinct protein forms γ1 and γ2, which arise by differential TSSs and alternative splicing [26]. PPARγ is the master regulator of adipogenesis and regulates cell cycle withdrawal, as well as induction of fat-specific target genes that are involved in adipocyte metabolism [28]. PPARγ stimulates the expression of numerous genes that are involved in lipogenesis, including those for adipocyte fatty acid-binding protein, lipoprotein lipase, and fatty acid translocase. The general role for PPARγ in the regulation of lipid metabolism is underlined by the therapeutic utilization of the PPARγ ligands thiazolidinediones in obesity-linked type 2 diabetes [29].
Being transcription factors, the role that each PPAR subtype plays in different disease settings is reflected by the number and kind of target genes regulated in each tissue type. Current technologies allow researchers to address transcriptional regulation on a transcriptome-wide level using microarrays. A good example of a transcriptional approach in the PPAR field is the nutrigenomics initiative addressing PPARα target genes in liver and other metabolically active tissues [30, 31]. Integration of several microarray studies comparing wild-type and PPARα knockout mice, and high-fat diet-induced response to synthetic ligand response, identified several novel PPAR target genes functioning in hepatic lipid metabolism that had not been identified before despite much research dedicated on characterizing the role of PPARs in these pathways [32].
13.3.1 Global Datasets that Identify a Central Role for PPARs in Disease Progression
Within the field of top–down systems biology, emphasis is on network perturbations to understand disease. An important challenge will be to identify the critical networks where NRs play a role. Recently, two association studies that aimed to link gene expression profiles to clinical traits and identify underlying genetic loci suggest that a critical macrophage network is associated with clinical traits for metabolic syndrome in mouse and human [33, 34]. The mouse study identified associations of this network with glucose and insulin levels, blood pressure, aortic lesion formation, and obesity, whereas the human study focused on genetics of obesity only. PPARγ is one member of that network and the association of the expression level was validated by single gene perturbation in mouse. Furthermore, in a study of macrophage activation, where transcription factor expression profiles and target gene profiles were correlated and supported by binding site search data, PPARγ, retinoid X receptor (RXR) α, GR, and estrogen-related receptor-1 were differentially regulated [35]. The target genes found with TRANSFAC motif of PPARα–RXR (which according to our experience does not differ significantly from PPARγ-binding profile) were identified from a cluster with early and sustained induction profile, the members of which were suggested to maintain the response. In effect, such global and unbiased approaches also suggest a critical role for macrophages in determining key clinical phenotypes and an important role of PPARs in macrophage function as was already discussed earlier.
To understand more deeply the function of PPARs in cells, global binding site occupancy data will be needed to validate in silico predictions and to understand differences between tissues that vary on chromatin organization level. This type of data collection for transcription factors and chromatin modifications is the focus of the ENCODE project [57]. From the NR superfamily, RARα; and hepatocyte nuclear factor α (HNF4α) are represented. RARα is studied in connection with neutrophil differentiation together with other transcription factors, coregulators, and histone modification in retinoic acid stimulated cells (Affymetrix chromatin immunoprecipitation (ChIP)-chip track [36]). HNF4α is studied together with other key liver transcription factors and histone 3 acetylation that marks active regions in liver cells (Uppsala ChIP track [37]). ChIP-chip data exist also for ERα [38], and it is now recognized that distal binding sites are widely used by NRs and that their arrangement into modules with other transcription factors is being explored.
13.3.2 PPAR Response Elements
An essential prerequisite for the direct modulation of transcription by PPAR ligands is the location of at least one activated PPAR protein close to the TSS of the respective primary PPAR target gene. This is commonly achieved through the specific binding of PPARs to a DNA-binding site, a so-called PPRE, and DNA looping toward the TSS [39]. In detail, the DNA-binding domain of PPARs contacts the major groove of a double-stranded hexameric DNA sequence with the optimal AGGTCA core binding sequence. PPARs bind to DNA as heterodimers with RXR [40] (Figure 13.1). PPREs are, therefore, formed by two hexameric core-binding motifs in a direct repeat orientation with an optimal spacing of one nucleotide (DR1), where PPAR occupies the 5′-motif [41]. However, characterization of PPREs from regulated gene promoters has resulted in a large collection of PPREs that deviate significantly from this consensus sequence. An extensive binding data collection for PPARs was recently published [42], where more critical deviations and well-tolerated deviations from the consensus were identified. In the following paragraphs, we will focus on the importance of binding site prediction for the systems biology approach.
13.4 METHODS FOR IN SILICO SCREENING OF TRANSCRIPTION FACTOR-BINDING SITES
Statistically, a NR core-binding motif, such as RGKTSA (R = A or G, K = G or T, S = C or G), should be found, on average, in every 256 bp of genomic DNA. Furthermore, dimeric assemblies of such hexamers should show up as direct repeats every 65,536 bp and as everted repeats every 32,768 bp in a random sequence. Therefore, an in silico screen of the human genome would identify for every NR on an average of 50,000–100,000 putative REs. Since NR proteins have an abundance of at most a few thousand molecules per cell, a biologically realistic number of NR target genes per cell should be closer to this number. If we also consider the fact that many NR target genes appear to have more than one functional RE for any given NR, it could be expected that the real number of NR target genes in any cell type is much less than the number of NR molecules. These calculations make it obvious that not every putative NR-binding site is used in nature in any cell at any given time. Further background on the bioinformatics of NRs is summarized by Danielsen [43].
The specificity of PPARs for their binding sites allows constructing a model to describe the PPRE properties that can be used to predict potential binding sites in genomic sequences. For this, the PPAR-binding preference, often expressed as position weight matrix (PWM), has to be described on the basis of experimental data such as series of gel shift assays with a large number of natural binding sites [44–47]. However, PPAR–RXR heterodimers do not only recognize a pair of the consensus-binding motifs AGGTCA, but also a number of variations to it. Independent of the individual PWM description, this leads to a prediction of PPREs every 1000–10,000 bp of genomic sequence. This probably contains many false-positive predictions, which are mainly due to scoring methodology and the limitations that are imposed by the available experimental data. For example, the quantitative characteristics of a transcription factor, that is, its relative binding strength to a number of different binding sites, are neglected in a position frequency matrix, where simply the total number of observations of each nucleotide is recorded for each position. Moreover, in the past, there was a positional bias of transcription factor-binding sites upstream in close vicinity to the TSS. This would be apparent from the collection of identified PPREs but is in contrast with a multigenome comparison of NR-binding site distribution [48] and other reports on wide-range associations of distal regulatory sites [49]. Genome-wide approaches for the identification of NR REs and NR target genes are reviewed by Tavera-Mendoza et al. [50].
Internet-based software tools, such as TRANSFAC [51], screen DNA sequences with databases of matrix models. One approach used PWMs to describe the binding preferences of PPARs using all published PPREs [52]. The accuracy of such methods can be improved by taking the evolutionary conservation of the binding site and that of the flanking genomic region into account. Moreover, cooperative interactions between transcription factors, that is, regulatory modules, can be taken into account by screening for binding site clusters. The combination of phylogenetic footprinting and PWM searches applied to orthologous human and mouse gene sequences reduces the rate of false predictions by an order of magnitude but leads to some reduction in sensitivity [53]. Recent studies suggest that a surprisingly large fraction of regulatory sites may not be conserved but yet are functional, which suggests that sequence conservation revealed by alignments may not capture some relevant regulatory regions [54].
In effect, these approaches and tools are still insufficient, and there has to be a focus on the creation of bioinformatics resources that include more directly the biochemical restrains to regulate gene transcription. One important aspect is that most putative REs are covered by nucleosomes, so that they are not accessible to the respective transcription factor. This repressive environment is found in particular for those sequences that are either contained within interspersed sequences, are located isolated from transcription factor modules or lie outside of insulator sequences marking the border of chromatin loops [55]. This perspective strongly discourages the idea that isolated, simple PPREs may be functional in vivo. In turn, this idea implies that the more transcription factor-binding sites a given promoter region contains and the more of these transcription factors are expressed, the higher is the chance that the chromatin on this area of the promoter becomes locally opened.
The PAZAR information mall [56] is a tertiary database that is build on the resource of a multitude of secondary databases and provides a computing infrastructure for the creation, maintenance, and dissemination of regulatory sequence annotation. The unambiguous identification of the chromosome location for any given transcription factor-binding site using genomic coordinates allows to link the results from “big biology” projects, such as ENCODE [57], and other whole genome scans for histone modification and transcription factor association. Unfortunately, so far, only a few boutiques have been opened inside the PAZAR framework. In order to benefit from binding site predictions, it is still necessary to explore dedicated resources. For example, the well-known regulator of cell cycle progression, the transcription factor p53, has an own dedicated database (p53FamTaG) for integration of gene expression and binding site data [58].
13.5 BINDING DATASET OF PPREs AND THE CLASSIFIER METHOD
A general requirement for systems biology approaches is the existence of coherent and informative quantitative datasets. Approaches for NR RE predictions have been based on a collection of disparate binding data and in general lack quantitative comparison of different experimental results. To combine evidence from several publications for an efficient binding model has challenges, thus creating a demand for a coherent binding dataset. The recently published classifier method [42] used the in vitro binding preferences of the three PPAR subtypes on a panel of 39 systematic single nucleotide variations of the consensus DR1-type PPRE (AGGTCAAAGGTCA) [59] as an experimental dataset. Since then, similar datasets have been created for other transcription factors [60]. One way to utilize such a data type is to create an affinity matrix as was chosen for p53. However, initial trials to correlate binding strength of a multiple variant dataset to matrix scores were discouraging and an alternative more empirical, approach was chosen. The single nucleotide variants were sorted into three classes, where in class I the PPAR subtypes are able to bind the sequence with a strength of 75 ± 15 percent of that of the consensus PPRE, in class II with 45 ± 15 percent, and in class III with 15 ± 15 percent. Although the overall binding pattern of the three PPAR subtypes showed no major differences, some variations gave rise to a PPAR subtype-specific classification. Additional 130 DR1-type PPREs were sorted on the basis of counting increasing number of variations from the consensus and taking into account the single nucleotide variant binding strength. Those variants that alone decrease the binding only modestly (class I) could be combined with even three deviations from consensus still resulting in more than 20 percent binding relative to consensus. Other combinations resulted in faster loss of binding detailed in 11 categories, where such combinations still resulted in more than 1 percent relative binding. The in silico binding strength predictions of PPAR–RXR heterodimers were confirmed by gel shift assays for the six PPREs of the uncoupling protein 3 gene and showed a deviation of less than 15 percent, outperforming the affinity matrix and weight matrix that were created using the same datasets.
The main advantage, when comparing the classifier to PWM methods, is a clear separation between weak PPREs and those of medium and strong strength [42]. For the discovery of potential binding sites, this is extra information that could be especially of interest in processes considered context dependent, for example, for PPREs that reside in genomic context of transcription factor modules. Predicting the strength of PPAR binding can be a predictor of how prominent effect this receptor can have on a target gene. For example, if binding is easily competed by other transcription factors, the effect may not manifest in most tissues or it may manifest only in tissues expressing all transcription factors of a module containing the PPRE. As an example of the latter case, the insulin-like growth factor binding protein 1 gene has a weak PPRE located inside a well-conserved area (suggesting the presence of other transcription factorbinding sites) and was only in liver responsive to PPAR ligands [59]. In contrast, genes with strong PPREs, such as carnitine palmitoyltransferase 1A and angiopoietin-like 4, are PPAR responsive in many tissues (Heinäniemi et al., unpublished data).
13.6 CLUSTERING OF KNOWN PPAR TARGET GENES
The data added by binding strength analysis and by covering a larger regulatory region (±10 kB) was examined with all 38 human genes that are known to be primary PPAR targets together with their mouse ortholog. The clustering by predicted binding strength and evolutionary conservation of their PPREs resulted in four groups [42]. In general, clusters I–II contain genes that are well-conserved between human and mouse. Cluster I contains genes that carry multiple conserved PPREs, while genes in cluster II have only one or two strong or medium conserved PPRE in human, which are found in comparable strength and location in the mouse. Cluster III contains genes that have strong or medium PPREs in one species that are conserved only as weak PPREs in the other species. Finally, cluster IV contains more than 25 percent of all tested genes, which have the common property that they carry one or more PPREs, but none of them is conserved. These examples suggest that regulation of target gene can survive turnover of binding sites and might even benefit from it.
The clustering analysis indicated some useful features for whole genome PPRE screens. The presence of strong PPREs or more several medium strength PPREs within the 20 kB surrounding the annotated TSS of a gene was a strong indication for a PPAR target gene. In this way, 28 out of the 38 human genes would have been identified as PPAR targets. Similarly, for 29 of these 38 genes, the analysis of their murine ortholog would have come to the same conclusion. A combination of these two criteria (passing the threshold in either the human or mouse ortholog) would have identified 37 out of the 38 genes as PPAR targets. In this approach, full alignment is not required, just preservation of what could be called PPAR-binding potential. The more strong PPREs a gene has accumulated, the smaller the chances are that given all 250 human tissues, none of these sites would get accessible or be built into a regulatory module with other transcription factor-binding sites.
13.6.1 A Look at PPREs in their Genomic Context: Putative Target Genes and Binding Modules
In the paper described above, the gene-dense human chromosome 19 (63.8 MB, 1445 known genes) and its syntenic mouse regions (956 genes have known orthologs) were selected for an in silico screening based on the above explained criteria, that is, both species were investigated for medium and strong PPREs (based on a PPARγ prediction) [42]. Interestingly, 20 percent of genes of chromosome 19 contain a colocalizing strong PPRE, and additional 4 percent have more than two medium PPREs or a proximal medium PPRE. Experimentally, a complete evaluation of the selectivity of any such screen is complicated by the restricted expression profiles of the predicted genes, which prevents simple readouts from individual target tissues. When requiring the detection in human and mouse, 12.1 percent of genes from chromosome 19 were predicted as PPAR targets. As has been outlined in the previous discussion, a binding site screen will gain more power, when it can be integrated with other genomic screens, both experimental and bioinformatics. In a vision for future of targeting regulatory modules with colocalizing PPREs, a PPRE track provided by bioinformatics approaches can be compared against evidence of other regulatory modules provided by conservation analysis and screens for other transcription factors (Figure 13.3). Experimental data comparing regulation in a disease state versus normal cells can be visualized in the same context to detect overlap in functional binding sites. Given the high interest of the scientific community to better characterize binding profiles of different transcription factors and the improved experimental techniques to detect genome-wide binding events, such additional tracks combined with a PPRE-binding track could be available in near future. Importantly, these datasets will motivate studies that aim to integrate the knowledge with systems biology methodology in order to model NR function in healthy versus disease state in various human tissues.
13.7 CONCLUSION
The identification of genes showing a primary response to NRs and their ligands, the so-called NR regulome, can be used as a prediction of their therapeutic potential as well as their possible side effects. Methods incorporating both experimental- and informatics-derived evidence to arrive at a more reliable prediction of NR targets and binding modules can bring all available data together with the aim to predict outcome in a specific context. Taking the chromosome 19 in silico screening trial for PPREs as an example and extrapolating the results to the whole human genome, we suggest that approximately 10 percent of all human genes (an estimate of 2000–2500 genes) have the potential to be directly regulated by a specific NR by their RE content within 10-kB distance to their TSS. Translated to regulatory modules that colocalize with REs, an even larger number of genomic regions could be targeted by a given NR. In conclusion, in this chapter, we have addressed the identification of direct targets using genomic sequences and binding data. In parallel, we have discussed the potential of looking for NR REs inside regulatory modules foreseeing that in future, very likely, the emphasis will shift from target genes to target regulatory modules to alter a physiological response and from individual genes to whole genome response.
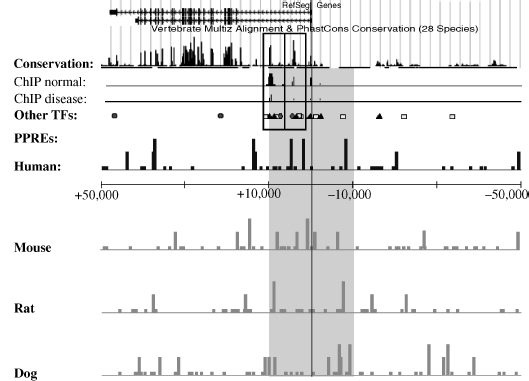
Figure 13.3 The superimposition of the PPRE tracks on other genome-wide datasets can reveal promising PPRE-containing binding modules for targeted therapy via PPAR activation. In this imaginary setting, transcription factor 1 (black triangles) is known to be one main regulator of the hypothetical gene X, and this regulation is altered in diabetes. Transcription factor 2 (represented by white squares) synergistically activates gene X but is lost in insulin-resistant beta cells. ChIP data comparing normal and disease state-binding profiles for this transcription factor reveal two main regulatory modules under normal conditions and a weaker binding in insulin-resistant samples due to loss of transcription factor 2. Combining these datasets with knowledge of PPAR binding and target genes will create hypothesis about their potential role in this disease setting. The gene is characterized by high enrichment of strong PPREs in several species providing strong evidence that PPARs can regulate this gene. A colocalizing PPRE in module 2 could enable PPARs to replace ranscription factor 2 in this module and to restore strong activation of the gene.
ACKNOWLEDGMENTS
Grants (all to C.C.) from the University of Luxembourg, the Academy of Finland, the Finnish Cancer Organization, the Juselius Foundation, and the EU (Marie Curie RTN NucSys) supported our research.
REFERENCES
1. S. Sperling. Transcriptional regulation at a glance. BMC Bioinform., 8 (Suppl 6):S2, 2007.
2. S.J. Jones. Prediction of genomic functional elements. Annu. Rev. Genomics Hum. Genet., 7:315–338, 2006.
3. A. Chawla, J. J. Repa, R. M. Evans, and D. J. Mangelsdorf. Nuclear receptors and lipid physiology: opening the X-files. Science, 294:1866–1870, 2001.
4. Nuclear-Receptor-Committee: a unified nomenclature system for the nuclear receptor, superfamily. Cell, 97:161–163, 1999.
5. J. M. Maglich, A. Sluder, X. Guan, Y. Shi, D. D. McKee, K. Carrick, K. Kamdar, T. M. Willson, and J. T. Moore. Comparison of complete nuclear receptor sets from the human, Caenorhabditis elegans and Drosophila genomes. Genome Biol., 2: RESEARCH0029, 2001.
6. V. Giguere. Orphan nuclear receptors: from gene to function. Endocr. Rev., 20:689–725, 1999.
7. R. Mohan and R. A. Heyman. Orphan nuclear receptor modulators. Curr. Top. Med. Chem., 3:1637–1647, 2003.
8. M. Müller and S. Kersten. Nutrigenomics: goals and strategies. Nat. Rev. Genet., 4:315–322, 2003.
9. K. E. Wellen and G. S. Hotamisligil. Inflammation, stress, and diabetes. J. Clin. Invest., 115:1111–1119, 2005.
10. G. S. Hotamisligil. Inflammation and metabolic disorders. Nature, 444:860–867, 2006.
11. P. Dandona, A. Aljada, and A. Bandyopadhyay. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol., 25:4–7, 2004.
12. W. Khovidhunkit, M. S. Kim, R. A. Memon, J. K. Shigenaga, A. H. Moser, K. R. Feingold, and C. Grunfeld. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J. Lipid Res., 45:1169–1196, 2004.
13. T. Rhen and J. A. Cidlowski. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N. Engl. J. Med., 353:1711–1723, 2005.
14. K. E. Wellen, R. Fucho, M. F. Gregor, M. Furuhashi, C. Morgan, T. Lindstad, E. Vaillancourt, C. Z. Gorgun, F. Saatcioglu, and G. S. Hotamisligil. Coordinated regulation of nutrient and inflammatory responses by STAMP2 is essential for metabolic homeostasis. Cell, 129:537–548, 2007.
15. G. D. Barish, M. Downes, W. A. Alaynick, R. T. Yu, C. B. Ocampo, A. L. Bookout, D. J. Mangelsdorf, and R. M. Evans. A nuclear receptor atlas: macrophage activation. Mol. Endocrinol., 19:2466–2477, 2005.
16. M. Fu, T. Sun, A. L. Bookout, M. Downes, R. T. Yu, R. M. Evans, and D. J. Mangelsdorf. A nuclear receptor atlas: 3T3-L1 adipogenesis. Mol. Endocrinol., 19:2437–2450, 2005.
17. S. Bornholdt. Systems biology. Less is more in modeling large genetic networks. Science, 310:449–451, 2005.
18. T. Pastinen and T. J. Hudson. Cis-acting regulatory variation in the human genome. Science, 306:647–650, 2004.
19. J. R. Idle and F. J. Gonzalez. Metabolomics. Cell Metab., 6:348–351, 2007.
20. I. Issemann and S. Green. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature, 347:645–650, 1990.
21. G. Krey, O. Braissant, F. L'Horset, E. Kalkhoven, M. Perroud, M. G. Parker, and W. Wahli. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-acitvated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol., 11:779–791, 1997.
22. T. M. Willson, P. J. Brown, D. D. Sternbach, and B. R. Henke. The PPARs: from orphan receptors to drug discovery. J. Med. Chem., 43:527–550, 2000.
23. L. Michalik, B. Desvergne, and W. Wahli. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat. Rev. Cancer, 4:61–70, 2004.
24. L. Michalik, B. Desvergne, C. Dreyer, M. Gavillet, R. N. Laurini, and W. Wahli. PPAR expression and function during vertebrate development. Int. J. Dev. Biol., 46:105–114, 2002.
25. A. L. Bookout, Y. Jeong, M. Downes, R. T. Yu, R. M. Evans, and D. J. Mangelsdorf. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell, 126:789–799, 2006.
26. J. P. Vanden Heuvel. The PPAR resource page. Biochim. Biophys. Acta., 1771:1108–1112, 2007.
27. L. Michalik, B. Desvergne, and W. Wahli. Peroxisome proliferator-activated receptors beta/delta: emerging roles for a previously neglected third family member. Curr. Opin. Lipidol., 14:129–135, 2003.
28. S. Heikkinen, J. Auwerx, and C. A. Argmann. PPARgamma in human and mouse physiology. Biochim. Biophys. Acta., 1771:999–1013, 2007.
29. J. Lehmann, L. B. Moore, T. A. Smith-Oliver, W. O. Wilkison, T. M. Willson, and S. A. Kliewer. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor g (PPARg). J. Biol. Chem., 270:12953–12956, 1995.
30. M. Bunger, G. J. Hooiveld, S. Kersten, and M. Müller. Exploration of PPAR functions by microarray technology: a paradigm for nutrigenomics. Biochim. Biophys. Acta., 1771:1046–1064, 2007.
31. C. Duval, M. Müller, and S. Kersten. PPARalpha and dyslipidemia. Biochim. Biophys. Acta., 1771:961–971, 2007.
32. M. Rakhshandehroo, L. M. Sanderson, M. Matilainen, R. Stienstra, C. Carlberg, P. J. de Groot, M. Müller, and S. Kersten. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res., 2007:26839, 2007.
33. Y. Chen, J. Zhu, P. Y. Lum, X. Yang, S. Pinto, D. J. MacNeil, C. Zhang, J. Lamb, S. Edwards, and S. K. Sieberts, et al. Variations in DNA elucidate molecular networks that cause disease. Nature, 452:429–435, 2008.
34. V. Emilsson, G. Thorleifsson, B. Zhang, A. S. Leonardson, F. Zink, J. Zhu, S. Carlson, A. Helgason, G. B. Walters, and S. Gunnarsdottir, et al. Genetics of gene expression and its effect on disease. Nature, 452:423–428, 2008.
35. S. A. Ramsey, S. L. Klemm, D. E. Zak, K. A. Kennedy, V. Thorsson, B. Li, M. Gilchrist, E. S. Gold, C. D. Johnson, and V. Litvak, et al. Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Comput. Biol., 4:e1000021, 2008.
36. S. Cawley, S. Bekiranov, H. H. Ng, P. Kapranov, E. A. Sekinger, D. Kampa, A. Piccolboni, V. Sementchenko, J. Cheng, A. J. Williams, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 116:499–509, 2004.
37. A. Rada-Iglesias, O. Wallerman, C. Koch, A. Ameur, S. Enroth, G. Clelland, K. Wester, S. Wilcox, O. M. Dovey, P. D. Ellis, et al. Binding sites for metabolic disease related transcription factors inferred at base pair resolution by chromatin immunoprecipitation and genomic microarrays. Hum. Mol. Genet., 14:3435–3447, 2005.
38. J. S. Carroll and M. Brown. Estrogen receptor target gene: an evolving concept. Mol. Endocrinol., 20:1707–1714, 2006.
39. S. Kersten, B. Desvergne, and W. Wahli. Roles of PPARs in health and disease. Nature, 405:421–424, 2000.
40. S. A. Kliewer, K. Umesono, D. J. Noonan, R. A. Heyman, and R. M. Evans. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature, 358:771–774, 1992.
41. A. Jpenberg, E. Jeannin, W. Wahli, and B. Desvergne. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem., 272:20108–20117, 1997.
42. M. Heinäniemi, J. O. Uski, T. Degenhardt, and C. Carlberg. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol., 8:R147, 2007.
43. M. Danielsen. Bioinformatics of nuclear receptors. Methods Mol. Biol., 176:3–22, 2001.
44. S. Mader, P. Leroy, J.- Y. Chen, and P. Chambon. Multiple parameters control the selectivity of nuclear receptors for their response elements. J. Biol. Chem., 268:591–600, 1993.
45. M. Schräder, K. Müller, M. Becker-André, and C. Carlberg. Response element selectivity for heterodimerization of vitamin D receptors with retinoic acid and retinoid X receptors. J. Mol. Endocrinol., 12:327–339, 1994.
46. M. Schräder, K. M. Müller, and C. Carlberg. Specificity and flexibility of vitamin D signalling. Modulation of the activation of natural vitamin D response elements by thyroid hormone. J. Biol. Chem., 269:5501–5504, 1994.
47. M. Schräder, M. Becker-Andre, and C. Carlberg. Thyroid hormone receptor functions as monomeric ligand-induced transcription factor on octameric half-sites. Consequences also for dimerization. J. Biol. Chem., 269:6444–6449, 1994.
48. X. Xie, J. Lu, E. J. Kulbokas, T. R. Golub, V. Mootha, K. Lindblad-Toh, E. S. Lander, and M. Kellis. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature, 434:338–345, 2005.
49. A. Barski, S. Cuddapah, K. Cui, T. Y. Roh, D. E. Schones, Z. Wang, G. Wei, I. Chepelev, and K. Zhao. High-resolution profiling of histone methylations in the human genome. Cell, 129:823–837, 2007.
50. L. E. Tavera-Mendoza, S. Mader, and J. H. White. Genome-wide approaches for identification of nuclear receptor target genes. Nucl. Recept. Signal., 4:e018, 2006.
51. V. Matys, E. Fricke, R. Geffers, E. Gossling, M. Haubrock, R. Hehl, K. Hornischer, D. Karas, A. E. Kel, O. V. Kel-Margoulis, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res., 31:374–378, 2003.
52. D. G. Lemay and D. H. Hwang. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J. Lipid Res., 47:1583–1587, 2006.
53. W. W. Wasserman, and A. Sandelin. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet., 5:276–287, 2004.
54. D. T. Odom, R. D. Dowell, E. S. Jacobsen, W. Gordon, T. W. Danford, K. D. MacIsaac, P. A. Rolfe, C. M. Conboy, D. K. Gifford, and E. Fraenkel. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet., 39:730–732, 2007.
55. J. L. Burns, D. A. Jackson, and A. B. Hassan. A view through the clouds of imprinting. FASEB J., 15:1694–1703, 2001.
56. E. Portales-Casamar, S. Kirov, J. Lim, S. Lithwick, M. I. Swanson, A. Ticoll, J. Snoddy, and W. W. Wasserman. PAZAR: a framework for collection and dissemination of cis-regulatory sequence annotation. Genome Biol., 8:R207, 2007.
57. ENCODE-Consortium. ENCODE (ENCyclopedia of DNA Elements) Project. Science, 306:636–640, 2004.
58. E. Sbisa, D. Catalano, G. Grillo, F. Licciulli, A. Turi, S. Liuni, G. Pesole, A. De Grassi, M. F. Caratozzolo, A. M. D'Erchia, et al. p53FamTaG: a database resource of human p53, p63 and p73 direct target genes combining in silico prediction and microarray data. BMC Bioinform., 8 (Suppl 1):S20, 2007.
59. T. Degenhardt, M. Matilainen, K. H. Herzig, T. W. Dunlop, and C. Carlberg. The insulinlike growth factor-binding protein 1 gene is a primary target of peroxisome proliferator-activated receptors. J. Biol. Chem., 281:39607–39619, 2006.
60. C. L. Wei Q. Wu, V. B. Vega, K. P. Chiu, P. Ng, T. Zhang, A. Shahab, H. C. Yong, Y. Fu, Z. Weng, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell, 124:207–219, 2006.
Elements of Computational Systems Biology Edited by Huma M. Lodhi and Stephen H. Muggleton Copyright © 2010 John Wiley & Sons, Inc.