
Characterization of fiber surface treatments in natural fiber composites by infrared and Raman spectroscopy
M.A. Mosiewicki, N.E. Marcovich and M.I. Aranguren, Institute of Materials Science and Technology (INTEMA), University of Mar del Plata, Argentina
Abstract:
The use of Fourier transform infrared (FTIR) and raman spectroscopies in the characterization of modified natural fibers and polymer interfaces in composite materials is reviewed. The changes in composition of the fibers owing to leakage of original components and/or incorporation of various species attached or adsorbed on the fiber surfaces, changes of morphology, as well as physical and chemical bonds developed at the polymer composite interfaces have been successfully characterized by these spectroscopic techniques. The potential and complementary use of these techniques is also discussed.
4.1 Introduction
The incorporation of natural fibers as filler and/or reinforcing materials in polymeric matrices is an increasingly growing area of importance in industrial and academic fields. One of the major problems found in the research and development of these materials has been to reach good compatibility between the two main phases, fibers and matrices. It has been established that the mechanical performance of the composites depends not only on the properties of the principal components but also on the nature and strength of the interface, that is responsible for the load transfer from the matrix to the fibers. For this reason, numerous strategies have been developed to improve the fillers surface wettability by resins or thermoplastics, to increase the adherence of the matrix to the filler and the corresponding interfacial strength. These strategies include the surface modification of the fillers, the incorporation of coupling agents or, less frequently, the modification of the matrix. Thus, techniques that allow investigation of these changes on filler surfaces or directly (although with increasing difficulties) on the composite interfaces are important tools for establishing correlations between interfaces characteristics and composite properties.
Among available surface techniques, probably the most commonly utilized is Fourier transform infrared (FTIR) spectroscopy. All materials absorb infrared radiation and the frequency intervals at which absorption bands appear are associated with the vibrational modes of specific functional groups. Large bodies of infrared data presented in tables and charts of software libraries are accessible to the users and are the results of years of careful and continuous work by a very large number of scientists. For a vibrational motion to be IR active, the dipole moment of the molecule must change, the higher the magnitude of this change, the higher the intensity of the band. Because a functional group is usually associated with more than one absorption band (more than one vibrational mode) and different functional groups may absorb in overlapping regions, it is always wise not to base conclusions on variations of a single band, but to look for confirmation in other non-overlapping FTIR regions or in supporting results from other techniques. Additionally, a group absorbs in a relatively narrow range of frequencies, but the position of a given absorption band can be shifted owing to interferences or perturbations from the surrounding atoms. Although, this may complicate the interpretation of the spectra, it can provide further information regarding possible interactions at the composite interface.
Although at present it is used less frequently, Raman spectroscopy is also based on the vibrational motion of functional groups, detecting changes in polarizability of the molecule. This polarizability decreases with increasing electron density, increasing bond strength and decreasing bond length. This last characteristic has been cleverly utilized to detect extension ratios in fibers/microfibers embedded in different composites, as discussed in detail in chapter 13.
In this way, infrared and Raman spectroscopies are techniques that could be used to provide complementary information about the chemical nature of materials, enabling, in particular, the investigation of the composition of surfaces and/or interactions developed at interfaces of composite materials.
4.2 Methods and techniques
4.2.1 Infrared spectroscopy
Although the infrared portion of the electromagnetic spectrum covers 14000–10 cm − 1, the majority of analytical applications use the mid-infrared range, approximately 4000–400 cm− 1. Many different techniques are currently available and only those related to the scope of this chapter will be summarized below:
Transmission
Even with the development of new FTIR techniques, transmission remains the most popular technique for samples that can be prepared in transparent form (diluted powdery samples pressed in KBr mixtures are included in this definition of transparent samples). In this technique, the sample-beam geometry corresponds to the beam passing through the sample, while the transmitted energy is collected in the detector (Fig. 4.1a). Because of its quantitative capabilities, this technique is generally preferred. Information on a modified surface can be obtained by spectral subtraction of the unmodified sample from the modified sample, to remove absorption from the bulk material.
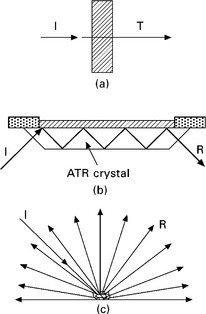
Attenuated total reflection (ATR)
Attenuated total reflection (ATR) is based on the multiple internal reflection of the beam incident on the sample (Fig. 4.1b). This technique is useful for studying the surface of the materials (up to a depth of 0.5–5 μm) that are put into contact with the crystal (usually ZnSe or monolithic diamond crystals). To obtain good surface contact may be a major problem for some samples. If the materials are rough and hard, the contact with the crystal may only be a point or line; increasing the pressure of the material on the crystal is the usual solution although repeatability may become an issue, as well as the useful life of the crystal. Soft flexible materials more readily provide contact and thus repeatability in the measurements.
Diffuse reflectance infrared spectroscopy
In Fourier transform diffuse reflectance infrared spectroscopy (DRIFT), the IR beam is reflected and dispersed on the matt surface of the sample (Fig. 4.1c). The emerging radiation leaves the sample in all directions, but with appropriate optical setups is directed towards the detector. The intensity of the infrared peaks is expressed in Kubelka–Munk units, in which case the spectra look similar to transmission spectra shown as absorbance. The Kubelka–Munk theory was developed for purely diffuse radiation, similarly to Beer’s Law for transmission.
Usually, highly absorbing zones are also reflecting regions and some samples may produce regions of flat appearance that correspond to strongly absorbing regions in the transmission spectra. Improving the spectra may require further dilution of the sample in dry KBr powder.
4.2.2 Raman spectroscopy
when a sample is irradiated with a monochromatic radiation of high intensity, such as solid lasers, a fraction of the radiation is scattered and the shifts produced with respect to the wavelength of the incident beam depend on the chemical structure of the molecules. Although Raman spectra and infrared spectra somewhat resemble each other (the peaks corresponding to vibrational modes active in both spectroscopies have the same energy shifts, that is, they appear at the same wavenumbers); the differences are also important, the relative intensity of the peaks differs and in some instances some peaks may not appear. Differences with infrared spectroscopy are not surprising because the mechanisms involved are also different, change in dipolar moment for FTIR-active groups and change in polarizability for Raman-active groups, and these differences make them complementary rather than competitive techniques. In particular, raman spectroscopy is very useful for samples containing water, because water does not cause interference in raman, as it strongly does in infrared spectroscopy. Another useful advantage is that glass or quartz cells can be used instead of salt crystals, which are environmentally unstable and fragile. As for FTIR spectroscopy, various methods are available in Raman spectroscopy whose selection will depend on the sample characteristics and the specific aims of the characterization.
4.3 Analysis of natural fibers and surface treatments
4.3.1 Characterization of cellulosic materials
Lignocellulosic fibers consist of bundles of hollow cellulose fibrils. Their cell walls are reinforced with spirally oriented cellulose in a hemicellulose and lignin matrix. Therefore, the cell wall is a composite structure of lignocellulosic material reinforced by helical microfibrillar bands of cellulose. The composition of the external surface of the cell wall is a layer of lignaceous material and waxy substances that bond the cell to its adjacent neighbors (Li et al., 2000; Jacob et al., 2005). These components are the ones to be considered in the study of the interaction with polymer matrices. Thus, to discuss their spectra and modifications or their interactions with different polymers, one should begin by identifying their corresponding absorption bands. Table 4.1 summarizes some FTIR peak assignments for lignocellulosic materials (Marcovich et al., 1996a; Bessadok et al., 2009; Jayaramudu et al., 2009).
Table 4.1
Peak assignments of the infrared spectra of lignocellulosic materials
| Band position (cm− 1) | Assignment |
| 3550–3650 | O–H stretching in free or weakly H-bonded hydroxyls |
| 3200–3400 | O–H stretching in H-bonded hydroxyls |
| 2840–2940 | C–H stretching region |
| 2725 | Overtone of interacting C–O stretch and O–H deformation |
| 2568 | Same |
| 1720–1740 | C = O stretching in carbonyl |
| 1625–1660 | Adsorbed water molecules in noncrystalline cellulose |
| ~ 1600 | Aromatic skeleton ring vibration and vibrations owing to adsorbed water |
| ~ 1505 | Aromatic skeleton ring vibration |
| 1450–1475 | C–H deformation and CH2 (sym.) + OH deformation |
| 1400–1430 | C–H deformation (methoxyl group in lignin) |
| ~ 1370 | C–H deformation (symmetric) |
| ~ 1327 | Syringyl ring breathing with C–O stretching (lignin) and CH2 wagging in cellulose |
| 1250–1260 | Guaiacyl ring breathing with C–O stretching (lignin) |
| 1240–1245 | C–O bond of the acetyl group in xylan and hemicellulose |
| ~ 1230 | Phenolic O–H deformation (lignin) – syringyl structure |
| 1160–1230 | C–O stretching of ester groups |
| 1150–1160 | C–O–C stretching (antisymmetrical) in cellulose and aromatic C–H CH2 wagging in cellulose |
| 1098–1120 | Skeletal vibration involving C–O stretching of the p glycosidic linkages |
| ~ 1060 | C–OH stretching vibration |
| 1036 | Aromatic C–H in plane deformation, guaiacyl and C–O deformation primary alcohol in lignin and C–O stretching in cellulose |
| 1003 | Skeletal vibration and C–O stretching in cellulose |
| 890–900 | Antisymmetrical stretching owing to β linkage in cellulose |
| 830 | Aromatic C–H out of plane vibration owing to lignin |
On the other hand, Fig. 4.2 is a good example of the information that can be obtained from different techniques and modes applied to a non-treated cellulose paper (Proniewicz et al., 2001). The ATR technique (Fig. 4.2a) is a non-destructive method that requires no sample preparation. The results are collected from a region between the surface and a depth of about 0.5–5 μm depending on the sample characteristics. Intimate contact between the surfaces of the sample and the crystal is required, thus information can vary with sample topology, because of the interfering signal of interstitial trapped air and owing to the beam penetration in the sample.
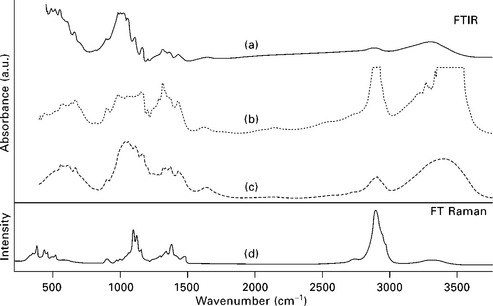
4.2 Vibrational spectra of non-treated cellulose in the range 200–3800 cm− 1: (a) ATR technique; (b) transmission through sample; (c) transmission through KBr pellet; (d) FT Raman. Reprinted from Proniewicz et al., 2001, with permission from Elsevier.
The older method of transmission FTIR can be used on a neat sample if it is transparent or thin enough to ensure a relatively large percentage of the beam goes through the sample. In Fig. 4.2 b and c the differences found by using pellets prepared from the neat sample and that diluted with KBr pellets are shown. Thus, when the sample is too concentrated or too thick (Fig. 4.2b), some high absorbent regions show essentially zero transmission. That is the case for the 2600–3500 cm− 1 region, corresponding to the C–H and O–H stretching vibrations, and 1000–1300 cm− 1 region, which comprises vibrations from the C–C stretching and the COH and CCH deformations. Usually, samples are diluted with KBr and pressed into a pellet to eliminate the problem of highly absorbent samples. However, this method is destructive and time consuming because the cellulose has to be in the form of a fine powder and well dispersed into the salt in order to avoid spectral artifacts. When sampling plant fibers, the pressing step can also have the added disadvantage of destroying the cell walls, exposing internal groups, in particular hydroxyl groups that were originally not available. Thus, a difference can be expected in the -OH bands if other not so invasive methods are utilized.
Similarly, Marcovich et al. (1996a) discussed the effect of sample concentration when using transmission and DRIFT modes for studying sawdust samples. They found that the opacity of the pellets limited the use of the transmission technique to weight concentrations equal or lower than 2% by weight in KBr. However, the DRIFT technique applied to neat samples gave good spectra with detailed band structure. Moreover, Fig. 4.3 illustrates that the frequency at which the bands appear is independent of the methods used, but the intensity depends strongly on the technique selected for collecting the spectra or preparing the sample. For example, the strong absorption band at 3000–3500 cm− 1 (corresponding to –OH absorption) is highly dependent on the sample preparation, as well as the spectroscopic method selected: it appears with higher intensity in the transmission spectra, for which a KBr pellet was prepared by pressing. Although DRIFT requires also grinding of the sample (as in transmission), it is not necessary to apply pressure hence the band appears with much lower intensity. Similarly, the peak at about 1650 cm− 1 corresponding to absorbed water appears clearly in transmission but not so much in DRIFT. Finally, the region between 900 and 1250 cm− 1 looks similar for the diluted samples under the two FTIR methods, although the DRIFT trace of the neat sample shows distortion of the main absorption band (around 1100 cm− 1).
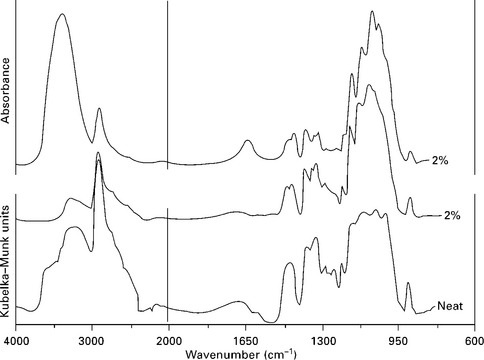
4.3 Transmission and DRIFT spectra of cellulose. Transmission spectrum was obtained at 2 (wt%) of cellulose in KBr. DRIFT spectra were obtained from 2% and neat samples. Reprinted from Marcovich et al., 1996a, with permission from Brill.
The FT Raman technique displays a more direct correlation between band and bonds. However, it is not very sensitive to hydrogen bonds, as can be seen from the very low intensity of the band in the region of 3000 to 3500 cm− 1 (Fig. 4.2d). Similarly the band at around 1625 cm− 1, which appears in the infrared spectra and is assigned to the bending motions of water molecules, is almost missing in the Raman spectra.
4.3.2 Characterization of modified fibers (and nanofibers)
As already mentioned, the high-modulus, low-cost, lightweight characteristics associated with natural fibers make them attractive as polymer reinforcements. However, the performance of a composite depends not only on the properties of the reinforcement and matrix, but also on the bonding between them. A poor fiber–matrix interaction derived from polar fibers and, typically, non-polar polymers leads to composites with poor mechanical properties and low durability when exposed to aggressive environments. Thus, to improve interfacial adhesion between reinforcement and polymer, the fiber surface is frequently modified or coupling agents are included in the composite formulation. The hydrophilization of non-polar polymeric matrices is also possible, but most often treatments are performed on the reinforcements (Gauthier et al., 1998a; Nuñez et al., 2003). Lignocellulosic materials contain surface hydroxyl groups that can be bonded to molecules that have functional groups selected to improve polymer compatibility. The use of FTIr and raman spectroscopies in the analysis of the most frequently used chemical modifications is summarized below.
4.4 Chemical treatments
4.4.1 Alkaline treatment
The alkaline treatment, also called mercerization, involves the immersion and soaking of the fibers in an alkaline medium, typically a rather concentrated NaOH aqueous solution. This treatment removes practically all non-cellulose components except waxes (Idicula et al., 2006; Luz et al., 2008). Owing to the dissolution of lignin by alkali, some pores are formed on the fiber surface. The loss of the cementing material of the vegetable fibers produces some fibrillation because of the breakage and separation of the fiber bundles (Aranguren and Reboredo, 2007). These changes increase the contact area between the fiber and the matrix (Idicula et al., 2006).
The effects of the alkaline treatment on fabrics of Polyalthia cerasoides tree, palm fibers, sisal fibers, hemp and kenaf fibers, henequen fibers and wood flour were addressed by Alawar et al. (2009), Jayaramudu (2009), Marcovich et al. (2005) and Rahman (2009), Rong et al. (2001), Sgriccia et al. (2008), among others. Using FTIR and chemical analysis, these studies confirmed the reduction of the hemicellulose and lignin contents after fiber mercerization. The major change in the FTIR spectra of the fibers owing to the alkaline treatment was the reduction (or sometimes the complete disappearance) of the peak centered at 1730–1740 cm− 1, attributed to the carbonyl group (C==O) stretching. This disappearance is a consequence of the extraction of hemicellulose and lignins and/or the formation of ionic carboxylates in the incompletely extracted samples, in which instance the corresponding peak appears at lower frequencies (1590 cm− 1). It was also established that the peak located at 1240 cm− 1, assigned to the C–O bond of the acetyl group in xylan, separated into two smaller peaks at 1260 and 1230 cm− 1. The first one corresponds to vibrations in the guaiacyl structure of the lignin and the second to the syringyl structure (Marcovich et al., 2005; Reddy et al., 1990; Roy et al., 1991). Since NaOH modification happens to increase the amount of accessible polar hydroxyl groups from the cellulosic fibers (Marcovich et al., 1998; 1999), the water sensitive regions of the FTIR spectrum become useless to detect other changes. In this sense, FT Raman spectroscopy results are very attractive because water does not interfere with the sample signal. This advantage was interestingly used by Jähn and coworkers (2002) to study the extent of the polymorphic transformation of cellulose I into cellulose II as a function of the alkali concentration. They detected the polymorphic transformation of the cellulosic fine structure of the flax fibers in vivo by analyzing the FT Raman spectra at frequencies below 1500 cm− 1, as denoted in Fig. 4.4. The Raman lines characterizing the cellulose modification I are denoted by an asterisk whereas the typical Raman lines of modification II are denoted by a cross. Particularly, in the range of the methylene bending vibrations (1400–1475 cm− 1) the spectra show the differences between the conformational arrangements of the side chains of the anhydroglucopyranose residues of cellulose. They indicate the simultaneous presence of two stereochemically non-equivalent –CH2OH groups in cellulose I (1476 and 1455 cm− 1). In cellulose II, only one type of –CH2OH group is present (1460 cm− 1) and the two scissoring vibrations of the methylene groups merge into one signal. on the other hand, the Raman lines at 1120 and 1098 cm− 1 assigned to the skeletal vibrational modes vs (C–O–C) and vas (C–O–C) of the β(1 → 4) glycosidic linkages of the β-D-glucopyranosyl units of cellulose can serve as characteristic marker bands for multicomponent systems such as cellulosic plant tissues. In particular, the vibrational mode vs (C–O–C) at 1120 cm− 1 is superimposed on skeletal modes of non-cellulosic carbohydrates (Himmelsbach and Akin, 1998) and lignin components (Agarwal and Atalla, 1986; Agarwal et al., 1995). For that reason, the intensity and the band shape of this cellulosic Raman line, which is also coupled with the C–C stretching mode of the breathing vibration of the glucopyranose rings at 1153 cm− 1, is strongly affected. In contrast, the Raman line at 1098 cm− 1 assigned to the asymmetric stretching mode vas (C–O–C), is not influenced by non-cellulosic carbohydrates. Taking into account the previous considerations, the changes in the C–H stretching region could be studied and related to the effect of the alkali treatment on the molecular structures of cellulosic plant fibers.
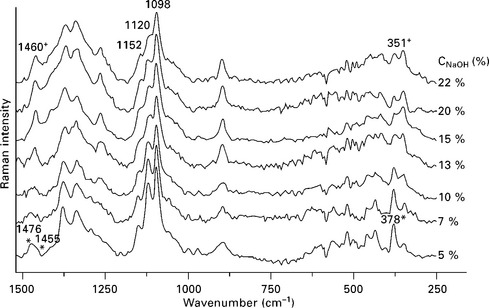
4.4 FT Raman spectra of a series of NaOH treated flax fiber bundles in the frequency range below 1500 cm− 1. Raman lines characterizing the most common polymorphic modifications are shown: cellulose | (*) and cellulose || (+). Reprinted from Jähn et al., 2002, with permission from Elsevier.
4.4.2 Esterification
The esterification of natural fibers using various anhydrides has been studied extensively. The type of lignocellulosic material used as a substrate in the esterification reaction is of vital importance, because its three main components, i.e. lignin, hemicelluloses and cellulose exhibit different reactivity towards the anhydrides. The acetylation reaction is one of the most frequently informed in literature (Adebajo and Frost, 2004; Bessadok et al., 2009; Liu et al., 1995; Luz et al., 2008; Rong et al., 2001; van Hazendonk et al., 1996; Zafeiropoulos et al., 2002). For example, Luz et al. (2008) reported the changes observed in the FTIR spectra for cellulose from sugarcane bagasse and sugarcane bagasse without hemicellulose after attaching acetyl groups to the OH groups of the fibers. The authors reported the appearance of a band at 1758 cm− 1, corresponding to the C = O carbonyl peak from bonded acetyl groups. Furthermore, after fiber acetylation, there was a decrease in OH band intensity, which was shifted from 3400 to 3700 cm− 1 owing to the reduction of hydroxyl groups available to form hydrogen bonds and the higher energy of the remaining free-OH groups. Similar effects were found by Zhang and coworkers (2008), who performed a mechanochemical acetylation of cellulose powder (solid-state at ambient temperature) using acetic anhydride. They demonstrated that the reaction occurred under those conditions and essentially on the cellulose surface.
Tserkia and coworkers (2005) modified flax, hemp and wood fibers by acetylation and propionylation, and confirmed the esterification reaction by ATR-FTIR. The treatment with both anhydrides led to the appearance/increase of the absorbance in the regions 1735–1737 cm− 1 (stretching vibration of the C = O group in ester bonds) and 1162–1229 cm− 1 (C–O stretching) in all cases. A reduction of the strong absorption between 3400 and 3600 cm− 1 (OH groups) was also noticed after fiber esterification.
Marcovich and coworkers (1996a, 1996b, 1997, 2005) showed that the DRIFT technique was effective in assessing surface modifications on wood flour resulting from esterification with maleic anhydride (MAN), even under mild conditions (room temperature without catalyst). The formation of new ester bonds owing to the reaction of the OH groups in the wood with the anhydride was confirmed by the increased intensity of the 1710–1740 cm− 1 band (attributed to both the C = O groups in acid and ester groups attached to the filler) and the appearance of a new peak at 850 cm− 1 related to the absorption of cis C = C conjugated to the carbonyl group in MAN (Bellamy, 1975). The similar intensity of the peaks corresponding to ester and acid carbonyls indicated that only one of the formed acid groups reacts with the filler surface and the other remains unreacted, in agreement with other studies (Ly et al., 2008; Kishi et al., 1988; Felix and Gatenholm, 1991; Maldas and Kokta, 1992). Samples esterified under stronger conditions (xylene reflux temperatures) showed an increase in the absorbance of the band at 1740 cm− 1 that was also a function of the reaction time. This observation corresponds to the increase in the concentration of ester bonds.
Wood flour was also esterified with a commercial alkenyl succinic anhydride (ASA) (Acha et al., 2003; Marcovich et al., 2005, Fig. 4.6). The FTIR spectrum of the treated sample revealed the usual increase in the intensity of the carbonyl absorption at 1735 cm− 1, owing to ester formation, and the absence of the anhydride absorption bands at 1600–1800 cm− 1. An increase in the CH stretching bands (2900–3000 cm− 1) was also observed owing to the CH groups of the attached ASA molecule. Additionally, a new peak at 1650 cm− 1 appeared, which corresponds to the unsaturation of the attached ASA. As is the case with many unsaturated fatty acids, in which the double bond is located approximately in the middle of the molecule, the 1650 cm− 1 absorption peak, characteristic of C = C absorption, did not appear in the FTIR spectrum of the neat ASA and had to be identified using Raman spectroscopy (strong peak). However, it clearly appeared in the spectra of the modified wood flour, because the reaction broke the symmetry of the molecule.
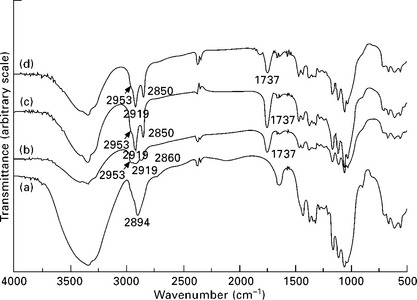
4.6 FTIR spectra (KBr pellets) of ramie cellulose whiskers: (a) unmodified; surface modified with (b) hexanoyl chloride; (c) lauroyl chloride; and (d) stearoyl chloride, after Soxhlet extraction with acetone. Reprinted from Junior de Menezes et al., 2009, with permission from Elsevier.
The attachment of longer chains on cellulose whiskers and flax strands is also well exemplified by Junior de Menezes et al. (2009) and Cañigueral et al. (2009), respectively. The cellulose was modified by grafting organic acid chlorides with aliphatic chains of different lengths, and thus, their spectra showed the expected peak at 1737 cm− 1 and an increase in the signals at 2953, 2919 and 2850 cm− 1 ascribed to the presence of grafted alkane chains (Fig. 4.6). The concomitant decrease of the magnitude of the broad band around 3300 cm− 1 for modified whiskers compared with unmodified ones is attributed to the partial disappearance of OH groups, confirming the success of the grafting reaction with organic acid chlorides. On the other hand, the flax strands were modified with alkyl ketene dimer and the resulting spectrum showed the same signals discussed previously added to the peak at 1700 cm− 1, which corresponds to the ketone group of the hydrolyzed reactive.
Regarding commercial coupling agents, the use of maleated polypropylene (MAPP) or maleated polyethylene (MAPE) copolymers to improve the interface between cellulosic fibers and PP or PE matrices is a common practice and has been frequently reported in the scientific literature (Acha et al., 2006; Bledzki et al., 2005; Felix and Gatenholm, 1991; Gassan and Bledzki, 1997; Hristov et al., 2004; Ichazo et al., 2001; Joly et al., 1996; Lu et al., 2005; Miguez Suarez et al., 2003). Just to include an example, Lu et al. (2005) demonstrated by FTIR spectroscopy and ESCA (electron spectroscopy for chemical analysis) the chemical linkage formed between the wood fibers and several MAPE coupling agents through esterification and proposed that succinic and half succinic esters were the two primary covalent bonding products of the reaction. Their results are discussed further in 4.4.5.
4.4.3 Silanization
Researchers at Grenoble have been involved in a series of studies (Abdelmouleh et al., 2002, 2004, 2005 and 2007) aiming at understanding the silane–cellulose system. The main advantages of using silane coupling chemicals is that at one end, they bear alkoxysilane groups capable of reacting with the OHrich surface of the fibers, whereas the other end can be tailored to suit a specific matrix. One of the studies (Abdelmouleh et al., 2007) illustrates the grafting of the coupling agent through initial physical adsorption of the hydrolyzed silane followed by a curing process at 120 °C under an inert atmosphere. Figure 4.7 presents the DRIFT spectra corresponding to the cellulose samples treated with γ-methacryloxypropyltrimethoxy silane (MPS), before and after heat treatment. In both cases, the spectrum corresponding to untreated cellulose was subtracted to investigate the band issuing from the silane moiety. The two spectra show different bands at 1713 and 1636 cm− 1, which are associated with the stretching vibrations of the C = O and C = C groups in the acrylic moieties, respectively. The broad shape of the C = O peak suggests that this group is interacting by hydrogen bonding with the surface hydroxyl groups. The broad intense bands around 1200 and 1135 cm− 1 were assigned to the stretching of the –Si–O–cellulose and –Si–O–Si– bonds, respectively (Felix and Gatenholm, 1991; Navoroj et al., 1984). The strong increase in the intensity of these bands after the heat treatment suggested that the grafting of silane onto cellulose as well as the intermolecular condensation between adjacent adsorbed –Si–OH groups were substantially enhanced. The peaks near 1100 and 1080 cm− 1 are related to residual unhydrolyzed Si–OCH3 groups and their small intensity indicates that most of the silane adsorbed under these conditions was hydrolyzed. The large band around 1015 cm− 1, present in the spectrum of the uncured sample, was attributed to –Si–OH groups. This band disappeared after the heat treatment and was replaced by a wide band around 1040 cm− 1, characteristic of –Si–O–Si– moieties. These peak assignments are in agreement with those reported in other studies dealing with glass surfaces treated with the same coupling agents (Navoroj et al., 1984). However, FTIR spectroscopy applied to the study of silanized natural fibers has not always been reported as a successful analysis technique. Sgriccia et al. (2008) treated hemp and kenaf fibers with silane using gentler conditions than the Grenoble group and were unable to identify the characteristics of silane moieties on the surface of the fibers by FTIR spectroscopy.
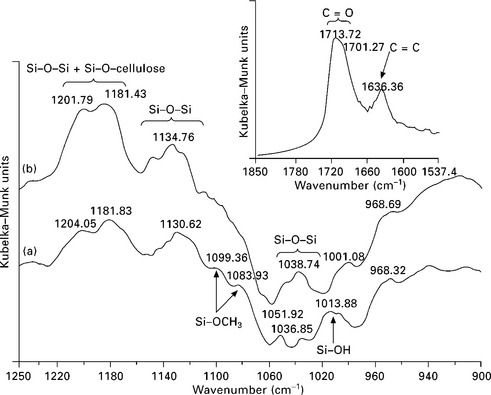
4.7 DRIFT subtraction spectra (MPS treated fibers – untreated fibers): (a) before and (b) after treatment (900–1250 cm− 1 region). Inset: 1850–1540 cm− 1 region after treatment. Reprinted from Abdelmouleh et al., 2007, with permission from Elsevier.
Other studies (Herrera-Franco and Aguilar-Vega, 1997; Valdez-Gonzalez et al., 1999a, 1999b) have also used FTIR spectroscopy and ESCA to confirm the reaction of a silane coupling agent with henequen fibers and found that the fiber–matrix interaction became stronger when the fibers were previously treated with alkali. The pretreatment eliminated cementitious components and lead to a rougher topology and larger accessible surface for reaction.
4.4.4 Urethane bonds
The reactivity of lignocellulosic fibers towards isocyanates has been frequently used as a compatibilization procedure in polymeric based composites (Kokta et al., 1990; Ly et al., 2008; Maldas et al., 1989a,b). As an example, Joly and coworkers (1996) demonstrated the success of the chemical modification by showing the presence of two new bands at 3343 and 1705 cm− 1, attributed to the formation of urethane groups and associated with their NH and CO vibrations, respectively, after modifying Avicel cellulose, flax and ramie fibers with aliphatic isocyanates. More recently, Ly and coworkers (2008) modified cellulose fibers and kraft softwood pulps with both 1,4-phenylene diisocyanate (PDI) and methylene-bis-diphenyl diisocyanate (MDI). The FTIR spectra of the Soxhlet extracted modified fibers revealed a new band at 2275 cm− 1, corresponding to the isocyanate function and a peak at 831 cm− 1, associated with the presence of disubstituted aromatic ring. The small size and rigidity of the isocyanate molecules selected exclude the possibility of bridging two different fibers, which explained the presence of isocyanate functions. However, the expected urethane linkage signal was masked by a large broad signal from absorbed water (~1650 cm− 1). Gandini et al. (2001) used TDI (toluene di-isocyanate) to treat cellulose and also confirmed the reaction by FTIR spectroscopy.
In a comprehensive experiment, Marcovich et al. (2006) treated cellulose nanocrystals (prepared from acid hydrolysis of microcystalline cellulose Avicel) with a polymeric MDI, followed by Soxhlet extraction with toluene to remove unreacted isocyanate. Because of the large area available for reaction in the cellulose nanocrystals, the modifications were distinctively observed in the FTIR spectra. The spectrum of the treated (unwashed) crystals showed new peaks corresponding to the absorption of isocyanate (2270 cm− 1) and urethane (1720 cm− 1). The spectrum of the extracted crystals showed that there was no change in the 1720 cm− 1 peak, because the urethane bonds were formed by reaction with the OH in the surface of the cellulose crystals. On the other hand, a large, but incomplete reduction of the isocyanate band was reported, which results from the removal of non-attached MDI, and the presence of unreacted isocyanate in the attached moieties.
4.4.5 Acrylate derivatives
Kalia and coworkers (2008) reported the preparation of graft copolymers of flax fibers with methyl acrylate (MA) using a redox system. FTIR spectrum of original flax showed a broad peak at 3422.8 cm− 1 owing to bonded –OH groups and at 2918.8, 1653.5 and 1058.7 cm− 1 arising from –CH2, C–C and C–O stretching, respectively. However, for the graft copolymer, an additional peak at 1731.2 cm− 1 owing to the > C = O group of MA was observed, suggesting MA graft copolymerization onto flax fiber through covalent linkages.
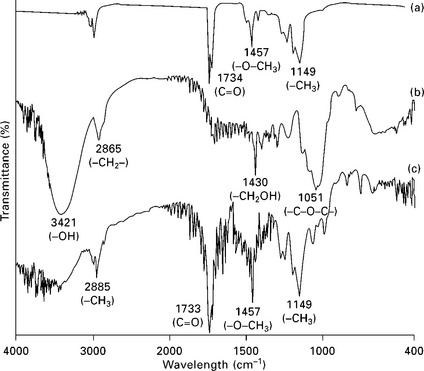
Bessadok et al. (2009) treated agave (Americana L.) fibers with acrylic acid (AA), among other reactives. Owing to the introduction of new CH and CH2 groups of the acrylic acid, the spectra of the treated agave fibers presents stronger bands at 2960, 2900 and 2855 cm− 1 than that of the untreated fibers. At the same time the new ester groups resulting from the acidic AA treatment are highlighted by the band at 1730 cm− 1 characteristic of the carbonyl (C = O) group. Analagously, Sangthong et al. (2009) treated sisal by admicellar polymerization with a poly(methyl methacrylate) film coating (Fig. 4.8). Figure 4.9 shows the FTIR spectra of pure PMMA, untreated sisal fiber, and admicellar-treated sisal fiber. The admicellar-treated sisal fiber spectrum, shows the peaks at 1734 cm− 1 (C = O) and 1457 cm− 1 (–O–CH3), which are the characteristic peaks of pure PMMA whereas the spectrum of bare sisal fiber, shows the characteristic peaks of cellulose at 3421 cm− 1 (–OH) and 1430 cm− 1 (–CH2OH). `These results confirm that the sisal fiber surface was successfully coated with PMMA by admicellar polymerization.
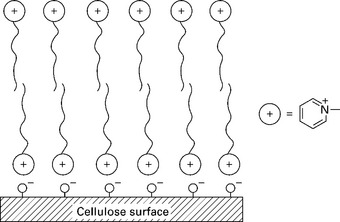
4.8 The ion pairing mechanism between the cellulose anions and the pyridinium cations in admicelle formation. Reprinted from Sangthong et al., 2009, with permission from Elsevier.
4.4.6 Enzymatic treatment
In some instances, the outermost surface of a lignocellulosic reinforcement is covered with hydrophobic noncellulosic components, i.e. lipophilic extractives and silica. This is true for wheat straws (WSs), stalks, husks, grasses, cereals and others. These materials impede uniform and efficient fiber–matrix adhesion and are responsible for the nonwetting behavior of these kinds of fillers when they are used for the production of particleboards (Han et al., 2001). Jiang and co-workers (2009) demonstrated that lipases from Candida cylindracea could effectively remove the hydrophobic lipophilic extractives and silica from the outer surface of untreated wheat straw, and increased the hydroxyl group content in the outer surface. They found, in the FTIR spectrum of the wheat straw outer surfaces, two strong and sharp peaks at 2916 and 2850 cm− 1 assigned to the asymmetric and symmetric stretching, respectively, of the CH2 group comprising the majority of the aliphatic fractions of waxes. Meanwhile, the small sharp band at 718 cm− 1 being characteristic of the –(CH2)n – (n ≥ 6) in plane deformations rocking (Bio-Rad, 2005) was observed. After enzymatic treatment, the sharp peaks at 2916 and 2850 cm− 1 significantly decreased and they turned into a weak rounded band, now associated with the contribution of the CH2– group in lignin and polysaccharides (Fang et al., 2002; Moran et al., 2008). The peak at 718 cm− 1, meantime, was reduced to a very weak shoulder. The two peaks at 790 and 970 cm− 1, attributed to the Si–C stretching vibration and Si–O vibration, respectively (Das et al., 2007; Frost and Mendelovici, 2006), were reduced to two corresponding weak shoulders after the lipase treatment indicating that the silicon-containing components were also removed concurrently during the modification. Additionally, the intensity of several typical peaks assigned to lignin and polysaccharides (cellulose and hemicellulose) increased significantly after treatment: the band at 896 cm− 1, characteristic of β-glucosidic linkages between the sugar units (Gupta et al., 1987), the infrared bands at 1422, 1600, 1502 cm− 1, assigned to methoxyl group in lignin, the aromatic skeletal vibrations and the aromatic skeletal vibrations coupled with C–H in plane deformations, and the peaks at 1245 cm− 1 (C–O stretching of acetyl group present in the lignin moiety as well as in hemicellulose, Subramanian et al., 2005) and 1160 cm− 1 (C–O–C antisymmetric bridge in hemicelluloses and cellulose and to aromatic C–H deformation of the syringyl and guaiacyl units in lignin) were revealed. Moreover, the broad band associated with hydrogen bonded hydroxyl groups at 3320 cm− 1 and the band at 1648 cm− 1, attributable to the bending of adsorbed water (Brandrup and Immergut, 1989; Lojewska et al., 2005), increased significantly, illustrating that the lipase treatment increased the amount of accessible hydroxyl groups and thus, of water absorption owing to the higher hydrophilicity of the WS outer surface.
4.4.7 Irradiation
Kato et al. (1999) treated different types of cellulose fibers with vacuum ultraviolet (VUV) irradiation in order to ascertain the efficiency of this technique in terms of oxidation capacity and compared their results with other common oxidation techniques. They monitored the evolution of the surface chemical composition by FTIR spectroscopy through the intensity variations of the band at 1720–1740 cm− 1 (stretching vibration of carbonyl) and showed that VUV irradiation induced surface oxidation as efficiently as chromic and nitric acids, with the additional advantage of neither causing losses in bulk mechanical properties nor coloring the cellulose fibers. Strlic et al. (2003) studied the effect of neodymium-doped yttrium aluminium garnet (Nd:YAG) laser cleaning at 1064 nm on the surface of Whatman cellulose, rag paper, cotton, linen, silk and wool. They used FTIR diffuse reflectance spectroscopy in order to establish the chemical changes occurring at the surface of these materials. The peaks affected by laser treatment were the vibration at 1063 cm− 1, associated with C–O stretching, the band at 1111 cm− 1 related with skeletal ring asymmetric stretching and that at 1413 cm− 1 associated with scissoring, whereas the bands at 1492 and 875 cm− 1 are new and cannot be associated with any of the usual bands appearing in FTIR spectra of pure cellulose. They concluded that chemical modifications of the surface were typical of the thermal degradation of cellulosic materials.
The application of γ-irradiation to cellulose and its derivatives has been extensively studied, but mostly in terms of the degradation mechanisms taking place (Takács et al., 1999). The effects of this treatment are shown in FTIR spectra by the appearance of a carbonyl band with increasing intensity as the duration of the treatment increased, attributed to terminal oxidized functions (carbonyl and carboxyl moieties) borne by the degraded cellulose fragments (Földváry et al., 2003; Takács et al., 1999).
4.5 Interfaces in polymer composites
Following the development of the field of composite materials, the need to assess and understand interface interactions has become increasingly important as the information can be further used in tailoring and improving material properties. The previously discussed studies utilized FTIR and Raman spectroscopies to investigate the efficiency of fiber modifications, which were aimed to affect interface interactions (De la Orden et al., 2007). However, although covalent bonding between filler and matrix has been frequently invoked in the literature (Bledzki and Gassan, 1999; Gauthier et al., 1998b; Kamdel et al., 1991; Li et al., 2000) to explain changes in the macroscopic properties, most frequently the interfacial reaction was not directly confirmed, because of the obvious complexity of accessing the composite interface. To the best of our knowledge, the first study that proved an interfacial reaction between fibers and polymeric matrix was that by Zadorecki and Flodin (1985). They synthesized two coupling agents based on trichloro-s-triazine with various terminal unsaturated groups in order to improve the bonding between cellulose fibers and an unsaturated polyester matrix. They proved both the covalent reaction of the coupling agent onto cellulose and the copolymerization of the treated fibers with styrene by FTIR and XPS (x-ray photoelectron spectroscopy) techniques. In the same context, De la Orden et al. (2007) presented an interesting FTIR spectroscopic analysis of composites made from bleached eucalyptus kraft pulp (92% cellulose) sprayed with polyethylenimine (PEI) as reactive coupling agent and PP matrix. Figure 4.10 shows the DRIFT spectra corresponding to composites with and without PEI as coupling agent (graphs a and b, respectively). The difference spectrum (curve c) reveals a negative band centered at 1715 cm− 1 (C = O stretching vibration). The study suggested that during the high temperature processing of the polyolefin composites, carbonyl and carboxyl groups are generated owing to oxidation of cellulose and PP. These groups further react with the amine groups of the coupling agent (PEI) present in the composites. Thus, the absorption of the C = O band disappears, and the formation of secondary amides explains the appearance of new bands, amide I at 1657 cm− 1 and amide II at 1567 cm− 1. The formation of Schiff bases (imines) absorbing at 1660 cm− 1 was also considered as a possible contribution to the bands observed in this region for the PEI-containing composites.
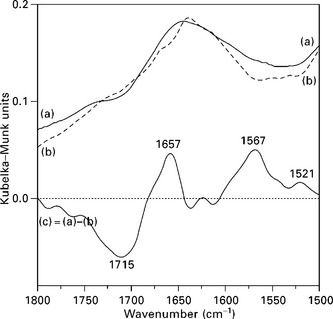
4.10 DRIFT spectra of PP composites with PEI-treated fibers (a) and untreated fibers (b). Graph (c) is the difference spectrum, (a – b). Reprinted from De la Orden et al., 2007, with permission from Elsevier.
In order to investigate the participation of cellulose in the formation of the observed covalent bonds, composite samples were subjected to Soxhlet extraction with refluxing xylene to extract the fibers and remove all the components not covalently bonded to them. Again, spectral subtraction was performed between the spectra of the fibers extracted from the composites with and without coupling agent. The results agree with the observations made on the bulk composites, that is, the absence of the carbonyl/carboxyl band and the presence of amide and/or imine groups in the extracted fibers from PEI-containing composites. At the same time, these results also confirm that the PEI became covalently attached to the fibers.
Analogously, Demjén et al. (1999) also reported a covalent reaction occurring between amine functionalities of aminosilanes and carboxyl groups produced in the thermal oxidation of PP during processing.
Methacrylic anhydride was used by Hill and Cetin (2000) to attach wood flour to polystyrene. Using FTIR spectroscopy they demonstrated that methacrylic anhydride modified wood flour was able to form covalent bonds with styrene monomers via free radical polymerization. The authors reported the appearance of a weak bond at 1657 cm− 1 in the FTIR spectrum of the grafted wood flour, owing to the attached unsaturation of the anhydride. This band substantially decreased after reaction with styrene, and weak absorption peaks appeared at 700–760 cm− 1 owing to the phenyl group of styrene. The reactions were also confirmed by weight gain, saponification of the grafted flour and 13C NMR.
Further to this, Marcovich et al. (2005) investigated the co-reaction between maleic anhydride previously attached to wood flour and an unsaturated polyester resin, through their co-reaction with styrene. They used a DRIFT technique in order to enhance the signal from surface chemical groups. They investigated the co-reaction of the grafted wood flour with styrene by mixing treated and untreated wood flour with styrene and hot-pressing the mixture (as was done with the styrene–unsaturated polyester composites). The untreated flour–styrene mixture led to a material that could be easily converted into powder by hand pressure. This material was extracted with refluxed toluene leaving a solid residue undistinguishable by DRIFT analysis from the original flour. However, the material obtained from MAN-treated wood flour and styrene could only be broken using a hammer. DRIFT spectra of the toluene-extracted material (Fig. 4.11) clearly showed the disappearance of the 1650 cm− 1 band corresponding to the anhydride unsaturations, which reacted with styrene. This study as well as that of Hill and Cetin (2000) showed that the unsaturated anhydrides grafted to wood flour by esterification are active for radical co-reaction with styrene and styrene containing resins.
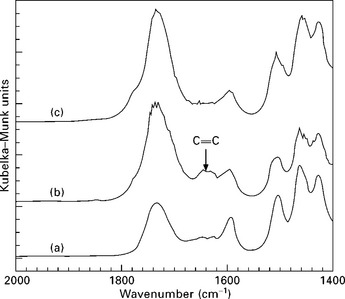
4.11 DRIFT spectra of (a) untreated wood flour, (b) MAN-esterified wood flour and (c) the same esterified wood flour after being treated with styrene and extracted with toluene.
More recently, Krouit and coworkers (2008) reported the grafting of polycaprolactone chains onto the surface of cellulose fibers via click-chemistry under heterogeneous conditions. Cellulose esters were prepared by reacting Avicel cellulose with undecynoic acid, in order to prepare cellulose substrate bearing multiple C ≡ C-terminated hairs and in parallel, polycaprolactonediol (PCL) was converted to an azidoderivative. Finally, modified cellulose was reacted with azido-PCL under heterogeneous conditions through ‘click chemistry’, which was followed by a Soxhlet extraction. After the initial modification, the FTIR spectrum of the treated cellulose fibers (Fig. 4.12) showed a new band at 1740 cm− 1, characteristic of the ester O–C = O moiety. The fingerprint of C ≡ C bands at 2100–2260 cm− 1 was not observed owing to the low concentration of these surface groups with respect to the absorption of the bulk cellulose. Moreover, the C ≡ C–H signal expected to appear at 3300 cm− 1, was found to be overlapped by the peak corresponding to hydroxyl group. However, the reaction could be confirmed by elemental analysis and XPS, which indicated a low degree of substitution, because the reaction was mainly confined to the surface of the cellulose particles.

4.12 Comparative FTIR spectra of (a) cellulose, (b) cellulose undecynoate, and (c) cellulose grafted with PCL. Reprinted from Krouit et al., 2008, with permission from Elsevier.
The click-chemistry step reaction with the azido-PCL was monitored in situ by FTIR spectroscopy through the reduction of the azido band (~2100 cm− 1). The FTIR analysis also revealed an increasing intensity of the ester band around 1740 cm− 1, which indicated that the grafting of the matrix on the cellulosic fibers was successfully achieved (Fig. 4.12c). In this case, XPS and weight gain (20%) confirmed the FTIR analysis.
In polyolefin composites, the use of coupling agents is the usual method for improving compatibility. Lu et al. (2005) investigated the interface in composites made from wood fiber and high density polyethylene (HDPE), in the presence of different coupling agents, a maleated polyethylene (MAPE) and a maleated polypropylene (MAPP). The composites were prepared in a one step reactive process in which the wood fiber, polyolefin, coupling agent and peroxide initiator were fed into an intensive mixer. The melt was cooled, ground and molded in a heated press. To investigate the composition of the interface, the prepared composites were extracted by a Soxhlet procedure with hot xylene and dried before being analyzed by transmission FTIR, using KBr pellets at approximately 1 wt% dilution. The most interesting features in the FTIR spectra obtained from the unextractable composite samples (solid residue consisting of the fibers and attached polymer/copolymer) are the shoulders appearing in the ester carbonyl region and the large increase of the absorption bands in the C-H region (~2900 cm− 1). These characteristics are an indication that the coupling agent remained attached to the surface of the fiber after the xylene extraction. The authors additionally proposed that succinic and half succinic ester were the two types of covalent products that bonded the wood fiber to the polymer matrix at the interface. The formation of a half ester was also reported as the main product of reaction in the modification of wood flour with maleic anhydride according to Marcovich et al. (2005), who characterized the treated wood flour using DRIFT.
Analogously, Hristov and Vasileva (2003) used also FTIR to investigate the effect of a maleated PP, added as coupling agent, on the interface of a wood fiber–PP composite. They also reported that the coupling agent was located at the interface and covalently attached to the wood fibers by esterification.
Reduced hydrophilicity was the main effect reported by Colom et al. (2003). They studied two different coupling agents added to aspen fibers–HDPE composites (a maleated polyethylene and a γ-methacryloxypropyl trimethoxy silane). The effect of the chemicals on the interface was inferred from the FTIR spectra of composites prepared with 40 wt% of aspen fibers. Comparison of the spectra, showed that the peak at ca. 1635 cm− 1 (corresponding to the vibrational absorption of water) appeared with a relatively lower intensity in the composites that incorporated coupling agents. This peak appeared in the spectra of the composites because of the hydrophilic nature of the incorporated fibers, but its intensity was much reduced by the protective effect of the coupling agents that surround the lignocellulosic reinforcement.
4.6 Summary
The various examples presented in this chapter on the use of FTIR and Raman spectroscopies applied to vegetable-based composites are not meant to provide an exhaustive review of the subject, but rather to give a glimpse into the different approaches to identify interfacial interactions.
FTIR spectroscopy is by far the most used of the two techniques, because of the easiness of use, the amount of information returned and also the long tradition in analytical chemistry. Different modes allow different geometries of the samples to be used and thus access to slightly different aspects of the chemistry being analyzed, (for example, different depths of beam incidence into the samples) or simply allow selection of the most suitable sample preparation through the selection of the FTIR mode to be used (transmission, attenuated or diffuse reflectance and others).
Because a functional group is associated with multiple vibrational absorptions, the spectra of composites, whose individual components can absorb also in overlapping regions, are difficult to analyze. Given the complexity of the spectra of unmodified lignocellulosic materials, it is not wise to base conclusions on the variations of a single band, but to look for confirmation in other non-overlapping regions or, as has been shown to be the case in many of the presented examples, in supporting results from other techniques.
Although less used up to the present, Raman was shown to be a valuable technique in confirming chemical changes occurring during modification of vegetable materials. In this respect, the fact that water does not interfere with Raman absorption is presented as a characteristic to take advantage of when working with natural fibers, which are well known for being very hygroscopic in nature.
It is clear that many interesting contributions to the identification of interfacial interactions in natural fiber composites are the result of ingenious experiments by many researchers, who utilized model/simplified matrices to identify a particular covalent interfacial bond or took advantage of extracting fibers from soluble matrix-composites to investigate the nature of any attached material. However, the direct observation of the interfacial bonds by FTIR and Raman spectroscopy is not possible, at least at the moment. The ever increasing improvement of dedicated equipment and the possibility of using microscopy associated to these techniques (for example, confocal Raman microscopy) may offer in a near future new tools to enable direct access closer to the interface region of lignocellulosic composites.
4.7 References
Abdelmouleh, M., Boufi, S., Belgacem, M.N., Duarte, A.P., Ben Salah, A., Gandini, A. Modification of cellulosic fibres with functionalised silanes: development of surface properties. Int J Adh Adhes. 2004; 24:43–54.
Abdelmouleh, M., Boufi, S., Belgacem, M.N., Dufresne, A. Short natural-fibre reinforced polyethylene and natural rubber composites: effect of silane coupling agents and fibres loading. Comp Sci Technol. 2007; 67:1627–1639.
Abdelmouleh, M., Boufi, S., Belgacem, M.N., Dufresne, A., Gandini, A. Modification of cellulose fibres with functionalized silanes: effect of the fiber treatment on the performance of cellulose-thermoset composites. J Appl Polym Sci. 2005; 98:974–984.
Abdelmouleh, M., Boufi, S., Ben Salah, A., Belgacem, M.N., Gandini, A. Interaction of silane coupling agents with cellulose. Langmuir. 2002; 18:3203–3208.
Acha, B.A., Aranguren, M.I., Marcovich, N.E., Reboredo, M.M. Composites from PMMA modified thermosets and woodflour. Polym Eng Sci. 2003; 43(5):999–1010.
Acha, B.A., Reboredo, M.M., Marcovich, N.E. Effect of coupling agents on the thermal and mechanical properties of PP-jute fabric composites. Polym Int. 2006; 55(9):1104–1113.
Adebajo, M.O., Frost, R.L. Infrared and 13C MAS nuclear magnetic resonance spectroscopic study of acetylation of cotton. Spectrosc Acta Part A: Mol Biomol Spectrosc. 2004; 60(1–2):449–453.
Agarwal, U.P., Atalla, R.H. In-situ Raman microprobe studies of plant cell walls: macromolecular organization and compositional variability in the secondary wall of Picea mariana (Mill.) B.S.P. Planta. 1986; 169:325–332.
Agarwal, U.P., Atalla, R.H., Forsskahl, I. Sequential treatment of mechanical and chemimechanical pulps with light and heat: a Raman spectroscopic study. Holzforschung. 1995; 49:300–312.
Alawar, A., Hamed, A.M., Al-Kaabi, K. Characterization of treated date palm tree fiber as composite reinforcement. Composites: Part B. 2009; 40:601–606.
Aranguren, M.I., Reboredo, M.M. Plant based reinforcements for thermosets matrices, processing and properties. In: Bhattacharyya D., Fakirov S., OH, Cincinnati, eds. Engineering biopolymers: homopolymers, Blends and Composites. Hanser Gardner Publications, 2007.
Bellamy, L.J. The infrared spectra of complex molecules, 3rd, UK: Chapman and Hall, 1975. [Vol 1].
Bessadok, A., Langevin, D., Gouanvé, F., Chappey, C., Roudesli, S., Marais, S. Study of water sorption on modified agave fibres. Carb Polym. 2009; 76:74–85.
Bio-Rad Laboratories Informatics DivisionThe Sadtler handbook of infrared spectra. Bio-Rad Laboratories, Inc, 2005.
Bledzki, A.K., Gassan, J. Composites reinforced with cellulose based fibres. Prog. Polym. Sci.. 1999; 24:221–274.
Bledzki, A.K., Letman, M., Viksne, A., Rence, L. A comparison of compounding processes and wood type for wood fibre-PP composites. Composites Part A: Appl Sci Manuf. 2005; 36(6):789–797.
Brandrup, J., Immergut, E.H. Polymer handbook, 3, NewYork, USA: Wiley & Sons, 1989.
Cañigueral, N., Vilaseca, F., Méndez, J.A., López, J.P., Barberà, L., Puig, J., Pèlach, M.A., Mutjé, P. Behavior of biocomposite materials from flax strands and starch-based biopolymer. Chem Eng Sci. 2009; 64:2651–2658.
Colom, X., Carrasco, F., Pagès, P., Canavate, J. Effects of different treatments on the interface of HDPE/lignocellulosic fiber composites. Comp Sci Technol. 2003; 63(2):161–169.
Das, G., Bettotti, P., Ferraioli, L., Raj, R., Mariotto, G., Pavesi, L., Soraru, G.D. Study of the pyrolysis process of an hybrid CH3SiO1.5 gel into a SiCO glass. Vib Spectrosc. 2007; 45:61–68.
De la Orden, M.U., González Sánchez, C., González Quesada, M., Martínez Urreaga, J. Novel polypropylene-cellulose composites using polyethylenimine as coupling agent. Composites: Part A. 2007; 38:2005–2012.
Demjén, Z., Pukánszky, B., Nagy, J., Jr. Possible coupling reactions of functional silanes and polypropylene. Polymer. 1999; 40:1763–1773.
Fang, J.M., Fowler, P., Tomkinson, J. Preparation and characterization of methylated hemicelluloses from wheat straw. Carbohydr Polym. 2002; 47:285–293.
Felix, J.M., Gatenholm, P. The nature of adhesion in composites of modified cellulose fibers and polypropylene. J Appl Polym Sci. 1991; 42:609–620.
Földváry, C., Takács, E., Wojnárovits, L. Effect of high-energy radiation and alkali treatment on the properties of cellulose. Rad. Phys. Chem.. 2003; 67:505–508.
Frost, R.L., Mendelovici, E. Modification of fibrous silicates surfaces with organic derivatives: an infrared spectroscopic study. J Colloid Interface Sci. 2006; 294:47–52.
Gandini, A., Botaro, V., Zeno, E., Bach, S. Activation of solid polymer surfaces with bifunctional reagents. Polym Int. 2001; 50(1):7–9.
Gassan, J., Bledzki, A.K. The influence of fiber-surface treatment on the mechanical properties of jute-polypropylene composites. Composites Part A. 1997; 28A:1001–1005.
Gauthier, H., Coupas, A.-C., Villemagne, P., Gauthier, R. Physicochemical modifications of partially esterified cellulose evidenced by inverse gas chromatography. J. Appl. Polym. Sci.. 1998; 69:2195–2203.
Gauthier, R., Joly, C., Coupas, A.C., Gauthier, H., Escoubes, M. Interfaces in polyolefin/cellulosic fiber composites: chemical coupling, morphology, correlation with adhesion and aging in moisture. Polym Comp. 1998; 19(3):287–300.
Gupta, S., Madan, R.N., Bansal, M.C. Chemical composition of Pinus caribaea hemicellulose. Tappi J. 1987; 70:113–116.
Han, G., Umenura, K., Zhang, M., Honda, T., Kawai, S. Development of highperformance UF-bonded reed and wheat straw medium-density fiberboard. J Wood Sci. 2001; 47:350–355.
Herrera-Franco, P.J., Aguilar-Vega, M. Effect of fiber treatment on the mechanical properties of LDPE–henequen cellulosic fiber composites. J. Appl. Polym. Sci.. 1997; 65:197–207.
Hill, C.A.S., Cetin, N.S. Surface activation of wood for graft polymerization. Int J Adhes Adhes. 2000; 20:71–76.
Himmelsbach, D.S., Akin, D.E. Near-infrared Fourier-transform Raman spectroscopy of flax (Linum usitatissimum L.) stems. J Agric Food Chem. 1998; 46:991–998.
Hristov, V.N., Lach, R., Grellmann, W. Impact fracture behavior of modified polypropylene/wood fiber composites. Polym Test. 2004; 23(5):581–589.
Hristov, V., Vasileva, S. Dynamic mechanical and thermal properties of modified poly(propylene) wood fiber composites. Macrom Mater Eng. 2003; 288(10):798–806.
Ichazo, M.N., Albano, C., González, J., Perera, R., Candal, M.V. Polypropylene/wood flour composites: treatments and properties. Comp Struct. 2001; 54(2–3):207–214.
Idicula, M., Boudenne, A., Umadevi, L., Ibos, L., Candau, Y., Thomas, S. Thermophysical properties of natural fibre reinforced polyester composites. Comp Sci Technol. 2006; 66:2719–2725.
Jacob, M., Joseph, S., Pothan, L.A., Thomas, S. A study of advances in characterization of interfaces and fiber surfaces in lignocellulosic fiber-reinforced composites. Compos Interfaces. 2005; 12(1–2):95–124.
Jähn, A., Schröder, M.W., Füting, M., Schenzel, K., Diepenbrock, W. Characterization of alkali treated flax fibres by means of FT Raman spectroscopy and environmental scanning electron microscopy. Spectrosc Acta Part A. 2002; 58:2271–2279.
Jayaramudu, J., Jeevan Prasad Reddy, D., Guduril, B.R., Varada Rajulu, A. Properties of natural fabric polyalthia cerasoides. Fibers Polymers. 2009; 10(3):338–342.
Jiang, H., Zhang, Y., Wang, X. Effect of lipases on the surface properties of wheat straw. Ind Crops Prod. 2009; 30:304–310.
Joly, C., Kofman, M., Gauthier, R. Polypropylene/cellulosic fiber composites chemical treatment of the cellulose assuming compatibilization between the two materials. JMS: Pure Appl. Chem. A. 1996; 33:1981–1996.
Junior de Menezes, A., Siqueira, G., Curvelo, A.P.S., Dufresne, A. Extrusion and characterization of functionalized cellulose whiskers reinforced polyethylene nanocomposites. Polymer. 2009; 50:4552–4563.
Kalia, S., Kaith, B.S., Sharma, S., Bhardwaj, B. Mechanical properties of flax-g-poly(methyl acrylate) reinforced phenolic composites. Fibers and Polymers. 2008; 9(4):416–422.
Kamdel, D.P., Riedel, B., Adnot, A., Kaliaguine, S. ESCA spectroscopy of poly(methyl methacrylate) grafted onto wood fibers. J. Appl. Polym. Sci. 1991; 43:1901–1912.
Kato, K., Vasilets, V.N., Fursa, M.N., Meguro, M., Ikada, Y., Nakamae, K. Surface oxidation of cellulose fibers by vacuum ultraviolet irradiation. J Polym Sci A: Polym Chem. 1999; 37:357–361.
Kishi, H., Yoshioka, M., Yamanoi, A., Shiraishi, N. Composites of wood and polypropylenes I. Mokuzai Gakkaishi. 1988; 34(2):133–139.
Kokta, B.V., Maldas, D., Daneault, C., Beland, P. Composites of polyvinyl chloridewood fibers. I. Effect of isocyanate as a bonding agent. Polymer Plast Technol Eng. 1990; 29(1–2):87–118.
Krouit, M., Bras, J., Belgacem, M.N. Cellulose surface grafting with polycaprolactone by heterogeneous click-chemistry. Eur Polym J. 2008; 44:4074–4081.
Li, Y., Mai, Y.-W., Ye, L. Sisal fibre and its composites: a review of recent developments. Comp Sci Technol. 2000; 60:2037–2055.
Liu, F.P., Wolcott, M.P., Gardner, D.J., Rials, T.G. Characterization of the interface between cellulosic Ffibers and a thermoplastic matrix. Compos Interfaces. 1995; 2(6):419–432.
Lojewska, J., Miskowiec, P., Lojewski, T., Pronienwicz, L.M. Cellulose oxidative and hydrolytic degradation: in situ FTIR approach. Polym Degrad Stab. 2005; 88:512–520.
Lu, J.Z., Negulescu, I.I., Wu, Q. Maleated wood-fiber/high-density-polyethylene composites: coupling mechanisms and interfacial characterization. Compos Interfaces. 2005; 12(1–2):125–140.
Luz, S.M., Del Tio, J., Rocha, G.J.M., Gonfalves, A.R., Del’Arco, A.P., Jr. Cellulose and cellulignin from sugarcane bagasse reinforced polypropylene composites: effect of acetylation on mechanical and thermal properties. Composites: Part A. 2008; 39:1362–1369.
Ly, B., Thielemans, W., Dufresne, A., Chaussy, D., Belgacem, M.N. Surface functionalization of cellulose fibres and their incorporation in renewable polymeric matrices. Comp Sci Technol. 2008; 68:3193–3201.
Maldas, D., Kokta, B.V. Performance of hybrid reinforcements in PVC composites. IV. Use of surface-modified glass fiber and different cellulosic materials as reinforcements. Int J Polym Mater. 1992; 17:205–214.
Maldas, D., Kokta, B.V., Daneault, C. Influence of coupling agents and treatments on the mechanical properties of cellulose fiber–polystyrene composites. J Appl Polym Sci. 1989; 37(3):751–775.
Maldas, D., Kokta, B.V., Daneault, C. Thermoplastic composites of polystyrene: effect of different wood species on mechanical properties. J Appl Polym Sci. 1989; 38(3):413–439.
Marcovich, N.E., Aranguren, M.I., Reboredo, M.M., Chemical modification of lignocellulosic materials. The utilization of natural fibers as polymer reinforcement, in Lignocellulosic–plastics composites. A.L. Leão, F.X. Carvalho, E. Frollini. San Pablo, Universidade de Sao Paulo – Universidade Estadual Paulista, 1997.
Marcovich, N.E., Bellesi, N.E., Auad, M.L., Nutt, S.R., Aranguren, M.I. Cellulose micro/nanocrystals reinforced polyurethane. J Mater Res. 2006; 21(4):870–881.
Marcovich, N.E., Reboredo, M.M., Aranguren, M.I. FTIR spectroscopy applied to woodflour. Composite Interfaces. 1996; 4(3):119–132.
Marcovich, N.E., Reboredo, M.M., Aranguren, M.I. Sawdust modification: maleic anhydride treatment. Holz als Roh-und Werkstoff European Journal for Wood and Wood Industries. 1996; 54(3):189–193.
Marcovich, N.E., Reboredo, M.M., Aranguren, M.I. Mechanical properties of woodflour unsaturated polyester composites. J Appl Polym Sci. 1998; 70:2121–2131.
Marcovich, N.E., Reboredo, M.M., Aranguren, M.I. Moisture diffusion in polyester–woodflour composites. Polymer. 1999; 40(26):7313–7320.
Marcovich, N.E., Reboredo, M.M., Aranguren, M.I. Lignocellulosic materials and unsaturated polyester matrix composites: interfacial modifications. Compos Interfaces. 2005; 12(1–2):3–24.
Miguez Suarez, J.C., Coutinho, F.M.B., Sydenstricker, T.H. SEM studies of tensile fracture surfaces of polypropylene–sawdust composites. Polym Test. 2003; 22(7):819–824.
Moran, J.I., Alvarez, V.A., Cyras, V.P., Vazquez, A. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose. 2008; 15:149–159.
Navoroj, S., Culler, R., Koenig, J.L., Ishida, H. Structure and adsorption characteristics of silane coupling agents on silica and E-glass fiber; dependence on pH. J Colloid Interf Sci. 1984; 97(2):309–317.
Nuñez, A.J., Sturm, P.C., Kenny, J.M., Aranguren, M.I., Marcovich, N.E., Reboredo, M.M. Mechanical characterization of PP – woodflour composites. J App Polym Sci. 2003; 88(6):1420–1428.
Proniewicz, L.M., Paluszkiewicz, C., Weselucha-Birczynska, A., Majcherczyk, H., Baranski, A., Konieczna, A. FT-IR and FT-Raman study of hydrothermally degradated cellulose. J Mol Struct. 2001; 596:163–169.
Rahman, M.M. UV-cured henequen fibers as polymeric matrix reinforcement: studies of physico-mechanical and degradable properties. Mater Design. 2009; 30:2191–2197.
Reddy, S.S., Bhaduri, S.K., Sen, S.K. Infrared spectra of alkali treated jute stick. J Appl Polym Sci. 1990; 41:329–336.
Rong, M.Z., Zhang, M.Q., Liu, Y., Yang, G.C., Zeng, H.M. The effect of fiber treatment on the mechanical properties of unidirectional sisal-reinforced epoxy composites. Comp Sci Technol. 2001; 61:1437–1447.
Roy, A.K., Sen, S.K., Bag, S.C., Pandey, S.N. Infrared spectra of jute stick and alkali-treated jute stick. J Appl Polym Sci. 1991; 42:2943–2950.
Sangthong, S., Pongprayoon, T., Yanumet, N. Mechanical property improvement of unsaturated polyester composite reinforced with admicellar-treated sisal fibers. Composites: Part A. 2009; 40:687–694.
Sgriccia, N., Hawley, M.C., Misra, M. Characterization of natural fiber surfaces and natural fiber composites. Composites: Part A. 2008; 39:1632–1637.
Strlic, M., Kolar, J., Vid-Simon, S., Marincek, M. Surface modification during Nd:YAG (1064 nm) pulsed laser cleaning of organic fibrous materials. Appl Surface Sci. 2003; 207:236–245.
Subramanian, K., Kumar, P.S., Jeyapal, P. Characterization of ligno-cellulosic seed fiber from Wrightia tinctoria plant for textile applications: an exploratory investigation. Eur Polym J. 2005; 41:853–861.
Takács, E., Wojnárovits, L., Borsa, J., Földváry, C., Hargittai, P., Zöld, O. Effect of γ-irradiation on cotton-cellulose. Rad Phys Chem. 1999; 55:663–666.
Tserkia, V., Matzinosa Kokkoub, P., Panayiotoua, C. Novel biodegradable composites based on treated lignocellulosic waste flour as filler. Part I. Surface chemical modification and characterization of waste flour. Composites: Part A. 2005; 36:965–974.
Valdez-Gonzalez, A., Cervantes-Uc, J.M., Olayo, R., Herrera-Franco, P.J. Chemical modification of henequén fibers with an organosilane coupling agent. Compos, Part B: Eng. 1999; 30:321–331.
Valdez-Gonzalez, A., Cervantes-Uc, J.M., Olayo, R., Herrera-Franco, P.J. Effect of fiber surface treatment on the fiber–matrix bond strength of natural fiber reinforced composite. Compos, Part B: Eng. 1999; 30:309–320.
van Hazendonk, J.M., Reinerink, E.J.M., de Waard, P., van Dam, J.E.G. Structural analysis of acetylated hemicellulose polysaccharides from fibre flax (Linum usitatissimum L.). Carbohydr Res. 1996; 291:141–154.
Zadorecki, P., Foldin, P. Surface modification of cellulose fibers. I. Spectroscopic characterization of surface-modified cellulose fibers and their copolymerization with styrene. J Appl Polym Sci. 1985; 30(6):2419–2429.
Zafeiropoulos, N.E., Williams, D.R., Baillie, C.A., Matthews, F.L. ‘Engineering and characterization of the interface in flax fibre/polypropylene composite materials. Part I. Development and investigation of surface treatments’, Compos, Part A: Appl Sci Manuf. 2002; 33(8):1083–1093.
Zhang, W., Zhang, X., Liang, M., Lu, C. Mechanochemical preparation of surface-acetylated cellulose powder to enhance mechanical properties of cellulose-filler-reinforced NR vulcanizates. Compos Sci Technol. 2008; 68:2479–2484.