
Chapter 13. Miscibility, Solubility, and Other Phase Equilibria
In the previous chapters we applied the theory of phase equilibrium to a very important and very common problem in chemical engineering: vapor-liquid equilibrium. This is not the only type of phase equilibrium that is encountered in practice. Some liquids have limited miscibility in each other. When mixed, they form two liquid phases and give rise to liquid-liquid equilibrium (LLE). When such a system is brought to boil, it forms a third phase, vapor, and the thermodynamic problem is one of vapor-liquid-liquid equilibrium (VLLE). Limited solubility is also encountered in mixtures of gases with liquids (oxygen in water, for example) and of solids in liquids (glucose in water). Another problem of industrial and biological relevance is osmosis. In this case partial equilibrium is established between two liquids via a semipermeable membrane that restricts the passage of one component. These problems may seem unrelated but they have a common thread: they are all governed by the basic principle that requires the chemical potential (or the fugacity) of a component distributed in various phases to be the same in all phases. In this chapter we apply the principles of thermodynamics to such problems. The objectives in this chapter are to:
1. Apply the minimization of the Gibbs free energy as a criterion to determine the limits of mutual miscibility of liquids.
2. Use Henry’s law to calculate the fugacity of a dissolved gas and calculate phase equilibrium in gas-liquid systems.
3. Calculate equilibrium through a semipermeable membrane and analyze separation processes that are based on osmotic effects.
13.1 Equilibrium between Partially Miscible Liquids
Liquids exhibit partial miscibility when their interactions show strong positive deviations from ideality. This indicates that cross interactions are unfavorable to such an extent that full miscibility is not possible. When a mixture of two liquids forms two separate liquid phases, each phase contains both components, though the composition is generally different in each phase. The situation is schematically shown in Figure 13-1, in which the two phases have been identified by the superscripts I and II. Each phase is saturated in both components so that if small additional amounts are added of either component, these will be distributed among the two liquids in such a way that the composition in each liquid will remain unchanged. The distribution of components is governed by the general-phase equilibrium criterion that requires the fugacity of a component to be the same in both phases. Using the low-pressure approximation of the fugacity of component in solution, the equilibrium criterion becomes:
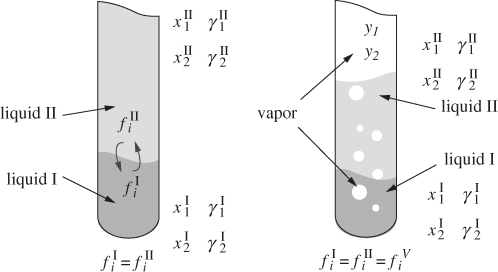
Figure 13-1: Schematic of liquid-liquid equilibrium (LLE) and vapor-liquid-liquid (VLLE) equilibrium between partially miscible liquids.
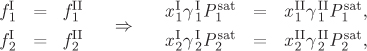
where ![]() is the mol fraction of component i in phase I,
is the mol fraction of component i in phase I, ![]() is its activity coefficient,
is its activity coefficient, ![]() is the saturation pressure at the temperature of the system, and similarly for phase II. Because the compositions in each liquid are different, the activity coefficients are also different. The saturation pressures are of course the same since both phases are at the same temperature. Thus the equilibrium criterion simplifies to
is the saturation pressure at the temperature of the system, and similarly for phase II. Because the compositions in each liquid are different, the activity coefficients are also different. The saturation pressures are of course the same since both phases are at the same temperature. Thus the equilibrium criterion simplifies to

These equations express the conditions for liquid-liquid equilibrium (LLE). If the activity coefficients are known, the compositions of the two-liquid phases can be obtained by solving the system of the two equations.
When a two-liquid system is brought to boil, the vapor constitutes a third phase, which coexists with the two liquids. The equilibrium criterion at this point applies to all three phases, and thus we have,

These equations fix the equilibrium compositions of the three phases. As a consequence of these relationships, and as long as both liquid phases are present, the system boils as an azeotrope: the boiling temperature and the composition of all phases remain constant and the only change is that the vapor phase increases at the expense of the liquid. When one of the two liquids boils off completely, the system consists of a single liquid and a vapor and behaves as usual, namely, its boiling temperature and the composition of the two phases vary continuously as more liquid is converted into vapor.
To perform calculations with partially miscible liquids we need expressions for the activity coefficients. These may be calculated by any of the models discussed in Chapter 12, with the notable exception of the Wilson model, which is not capable of describing partially miscible liquids. If the activity coefficients are known, LLE and VLLE calculations are fundamentally no different from the VLE calculations discussed in the previous chapter.
G12 = 0.96556, G21 = 0.514543.
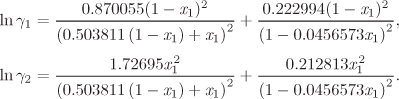
To obtain the mutual solubility of methanol and carbon disulfide, we solve eq. (13.1) for ![]() and
and ![]() using these activity coefficients. This requires a numerical solution and it is best done using a mathematical package. The solution is
using these activity coefficients. This requires a numerical solution and it is best done using a mathematical package. The solution is
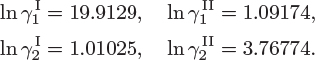
Comments The numerical solution of eq. (13.1) requires some attention. These equations are always satisfied at ![]() but such solutions are trivial and do not represent the composition of true coexisting phases. Trivial solutions must be identified and rejected, whenever the numerical method happens to converge to one of them.
but such solutions are trivial and do not represent the composition of true coexisting phases. Trivial solutions must be identified and rejected, whenever the numerical method happens to converge to one of them.
13.2 Gibbs Free Energy and Phase Splitting
In practice it is useful to know before mixing whether we will obtain a homogeneous solution, or whether phase splitting will occur. One way to answer this question is suggested in the previous example: if eq. 13.1 has a nontrivial solution, the system phase separates into two liquids whose compositions are given by the solution to these equations. If no solution exists, then the liquids form a homogenous solution at all compositions. Alternatively, the determination of phase splitting can be done by examining the Gibbs energy of mixing. Recall that the equilibrium state of a system at fixed pressure and temperature has the minimum possible Gibbs free energy. With this in mind, we analyze the problem of phase splitting as follows. We form 1 mol of solution by mixing x1 mol of liquid component 1 and x2 mol of liquid component 2 (x1 + x2 = 1). If components are fully miscible at this composition, the result is a single liquid with composition (x1, x2). The Gibbs free energy of this single-phase system is
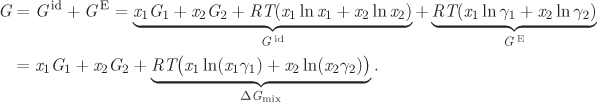
Choosing the reference state for each component to be the pure liquid (i.e., G1 = G2 = 0), the Gibbs energy of a single phase system simplifies to
If the liquids are only partially miscible, then two phases are formed, phase I with composition (![]() ,
, ![]() ), and phase II with composition (
), and phase II with composition (![]() ,
, ![]() ). The number of moles in each phase are related by the mass-balance equations in the form of the familiar lever rule:
). The number of moles in each phase are related by the mass-balance equations in the form of the familiar lever rule:
Notice that nI + nII = 1 because we are working with a total of 1 mol. As an extensive property, the Gibbs free energy of the two-phase system is the sum of the two phases:
where GI and GII are the molar Gibbs free energies of each liquid phase. These are given by eq. (13.3) using the composition of the corresponding phase.
To decide whether the system forms a single phase or two liquid phases we compare the Gibbs free energy for each case:
• If G < GI+II, the single-phase system is more stable;
• If G > GI+II, the two-phase system is more stable.
To demonstrate the application of this criterion, we reanalyze the system methanol/ carbon disulfide of Example 13.1. The Gibbs free energy of the solution is calculated from eq. (13.3) using the NRTL equation with the constants given in Example 13.1 and is plotted in Figure 13-2 as a function of the mol fraction of methanol.
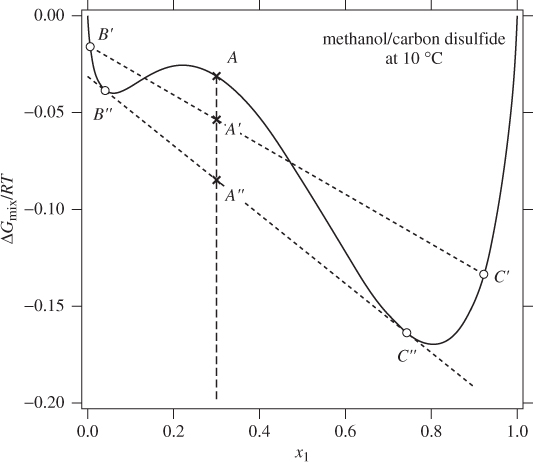
Figure 13-2: Graphical determination of phase splitting. Of all states along the dashed vertical line, A″ is the most stable one because it has the lowest possible Gibbs free energy. This state corresponds to a mixture of two liquid phases, B″ and C″.
Suppose that we mix 0.3 mol of methanol with 0.7 mol of carbon disulfide. If the system forms a single phase, its state would be represented by point A on the Gibbs line. If it forms two phases, say, liquids B′ and C′, the overall state is represented by point A′. Points B′ and C′ lie on the Gibbs curve (both states are a single-phase liquid) and their composition satisfies the condition xB′ < xA′ < xC′. Point A′ lies on the straight line that connects points B′ and C′. The molar Gibbs energy that corresponds to this point (read off the vertical axis at point A′) is the overall molar Gibbs energy of the two-phase system.1 Comparing the two states, state A′ has lower Gibbs energy. According to the Gibbs inequality, state A′ is more stable than A. Liquid states B′ and C′ were selected arbitrarily in this trial. Many other two-liquid states can be constructed in this manner in such a way that the overall state has even lower Gibbs energy than point A′. This can be done systematically by moving point B′ and C′ lower. This procedure converges to points B″ and C″ whose distinguishing feature is that they form a straight line that is tangent to the Gibbs curve at both contact points. All other lines that connect any two points to the left and to the right of point A lie above line B″ C″ and thus correspond to higher molar Gibbs energy. We conclude then that state A″, consisting of a mixture of liquids B″ and C″, is the most stable among all single or two-phase systems that can be constructed. Accordingly, state C″ is identified as liquid phase II (methanol saturated in carbon disulfide) and state B″ with liquid phase I (carbon disulfide saturated in methanol).
1. To convince yourself of this, look at eq. (13.4) and notice that GI+II moves on a straight line between GI and GII as nI is varied between 1 and 0.
The procedure described here represents the graphical solution of eq. (13.1) (the compositions of points B″ and C″ are the same as those obtained by solving eq. [13.1]). Its main advantage is that the graphical procedure allows us to determine whether phase splitting takes place by simply inspecting the shape of the Gibbs free energy: phase splitting occurs only when the Gibbs free energy contains a concave segment because it is then possible to connect two points of the curve with a straight line that lies below the curve. By contrast, in a convex curve, any straight line between two points lies above the curve, producing a two-phase system with higher Gibbs energy (and thus less favorable) than the single-phase liquid. Therefore, the presence of a concave portion is necessary and sufficient condition for phase splitting. The compositions of the coexisting liquids are then identified by drawing a double-tangent line, that is, one that is tangent to the Gibbs free energy at both contact points.
Equilibrium and Stability
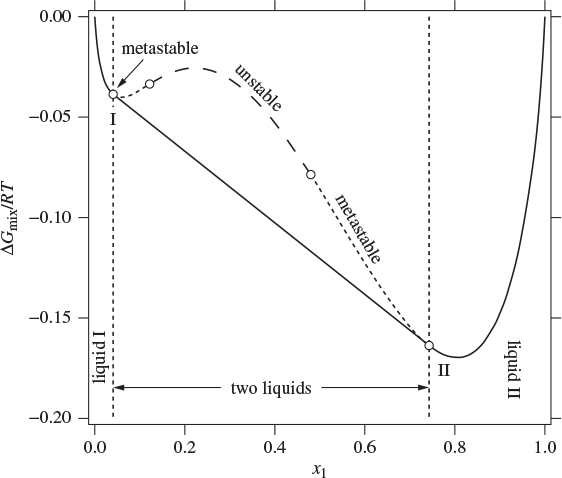
Figure 13-3: Stable (solid lines), metastable (dotted line), and unstable (large dashes) portions of the Gibbs free energy of solution. Concave parts on the Gibbs curve are unstable; convex parts are stable or metastable.
Having identified points B″ and C″ as the miscibility limits, the Gibbs graph must be corrected by removing the portion of the curve that represents nonequilibrium states. To do that, we erase the portion of the curve between points B″ and C″ and connect the two points with a straight tie line. The resulting Gibbs free energy is shown in Figure 13-3 by the solid line with points B″ and C″ now relabeled as I and II, respectively. The system consists of a single liquid in the regions ![]() and
and ![]() ; between
; between ![]() and
and ![]() the system forms two liquid phases and the Gibbs free energy of this two-phase mixture lies on the straight tie-line that connects points I and II. The portion of the Gibbs energy between points I and II contains one concave region near the center (shown by large dashes in Figure 13-3) with two convex portions to each side. The convex parts represent metastable states and may be observed under special experimental conditions.2 States on the concave portion are unstable and cannot exist as a single liquid.
the system forms two liquid phases and the Gibbs free energy of this two-phase mixture lies on the straight tie-line that connects points I and II. The portion of the Gibbs energy between points I and II contains one concave region near the center (shown by large dashes in Figure 13-3) with two convex portions to each side. The convex parts represent metastable states and may be observed under special experimental conditions.2 States on the concave portion are unstable and cannot exist as a single liquid.
2. Metastable states have a strong tendency to revert to the more stable states. To observe them experimentally one must avoid impurities and surface imperfections of the containing vessels, which tend to act as nucleation sites of the more stable phases.
Note
Mathematical Conditions for Stability
The stability criteria can be put in mathematical form. Convexity is indicated by positive values of the second derivative. Therefore, stable and metastable states must satisfy the condition
When the second derivative is zero, we reach the limit of stability:
Beyond this point the second derivative is negative, indicating that the Gibbs energy is a concave function of x1 and thus unstable.
A note on the Wilson equation: This equation results in a convex graph for the Gibbs free energy of mixing for all values of the parameters Λ12 and Λ21; therefore, it is not capable of predicting phase splitting. All other models discussed in Chapter 12 can produce concave shapes and thus may be used to describe partially miscible systems.
Construct the Pxy graph of methanol/disulfide at 10 °C using the NRTL equation with the parameters given in Example 13.1. The saturation pressures of the two components at 10 °C are, ![]() ,
, ![]() bar, where 1 refers to methanol and 2 to carbon disulfide.
bar, where 1 refers to methanol and 2 to carbon disulfide.
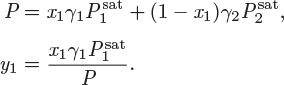
As in usual VLE calculations with a single liquid, the graph is constructed by calculating the bubble pressure P and the corresponding vapor mole fraction at various values of x1. The only difference is that the calculation is performed only within the range of full miscibility, namely, for ![]() and
and ![]() . The activity coefficients are calculated using the NRTL equation with α = 0.2 (see Example 13.1):
. The activity coefficients are calculated using the NRTL equation with α = 0.2 (see Example 13.1):
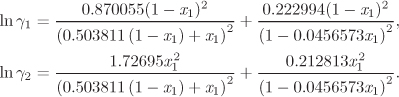
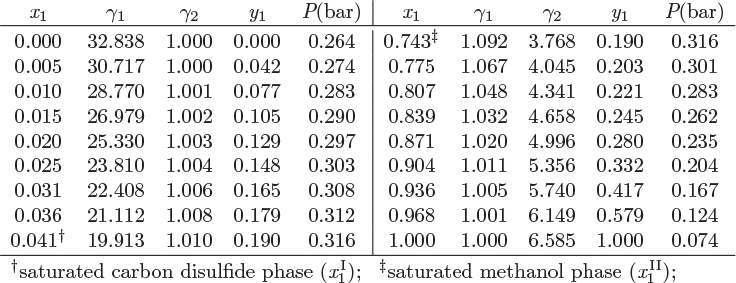
Figure 13-4: Pxy graph of methanol/carbon disulfide at 10 °C (see Example 13.2).
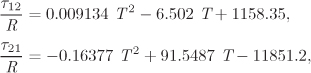
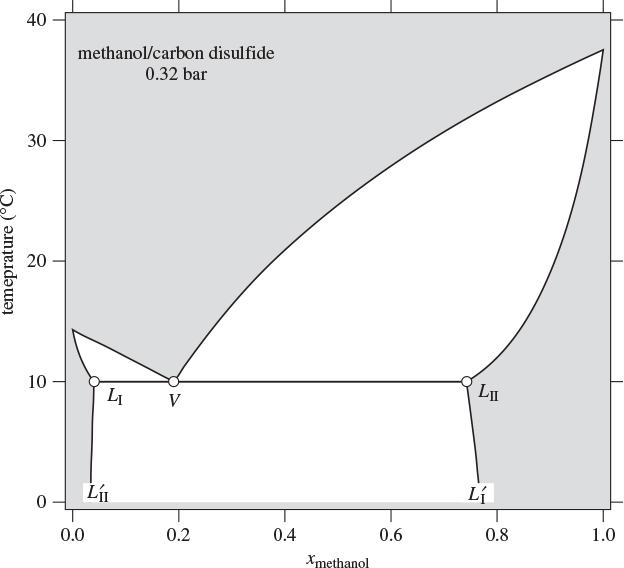
Figure 13-5: Txy graph of methanol/carbon disulfide at 0.32 bar.
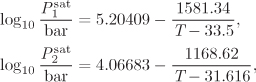
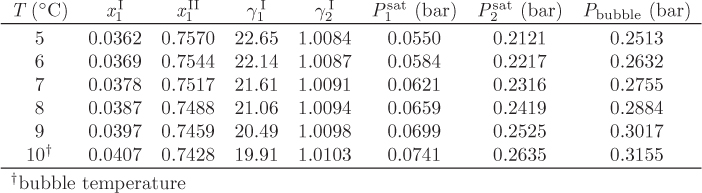
Txy below the bubble temperature: In this part of the calculation we establish the phase boundary between the two liquids provided that the system contains no vapor, i.e., it is below the bubble point. This refers to lines LI − L′I and LII − L′II in Figure 13-5. The calculation is performed as follows: we pick a temperature and we solve eq. (13.1) for ![]() and
and ![]() . Once the compositions of the two liquids are known, we compute the bubble pressure of the two-phase system. If it is below the system pressure of 0.3155 bar, the system is below the bubble point. We then increment the temperature and repeat the calculation, until the bubble point is reached. This calculation produces the liquid-liquid boundary of the phase diagram.
. Once the compositions of the two liquids are known, we compute the bubble pressure of the two-phase system. If it is below the system pressure of 0.3155 bar, the system is below the bubble point. We then increment the temperature and repeat the calculation, until the bubble point is reached. This calculation produces the liquid-liquid boundary of the phase diagram.
Either phase I or II may be used in this calculation. Using phase I and ![]() ,
, ![]() , we find
, we find
P = (0.0361579)(22.6526)(0.0550051) + (0.963842)(1.00843)(0.212145) = 0.25125 bar.
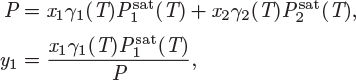
0 ≤ x1 ≤ 0.0407, and 0.7428 ≤ x1≤ 1.
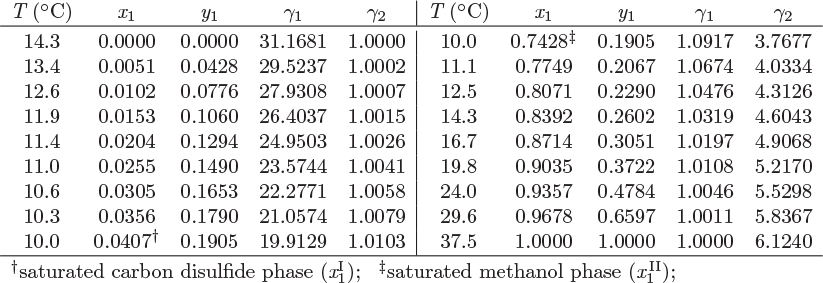
13.3 Liquid Miscibility and Temperature
At fixed pressure the liquid-liquid boundary ceases to exist above the bubble point (in the case of Figure 13-5, 10 °C). If the system pressure is increased, the bubble temperature increases as well, and a similar calculation would reveal that the liquid-liquid boundary extends to even higher temperatures, until the new bubble point is reached. This behavior is seen in Figure 13-6, which shows the Txy graph of methanol/carbon disulfide at three different pressures. For this system, increasing temperature results in increased mutual solubility, as indicated by the convergence of the two liquid branches (dashed line). At the point where the two meet, full miscibility is restored: above the temperature of this point the two liquids are miscible at all compositions. This temperature is called upper consolute temperature (UCT) and is very much analogous to the critical point of a vapor/liquid mixture in the sense that the two phases become indistinguishable at this point. Above the UCT the two components form a homogeneous minimum boiling azeotrope. The relationship between partial miscibility and minimum-temperature azeotropy is not coincidental. Nonideal systems that exhibit positive deviations from ideality indicate unfavorable cross-interaction between components. When mixed they form solutions that have a higher tendency to erase the interaction by forming a vapor at lower temperature than the boiling point of the pure components. If the self-interaction is sufficiently strong, then the system exhibits phase separation, which disrupts the unfavorable interaction. Figure 13-6 demonstrates this gradual transition as a function of temperature.
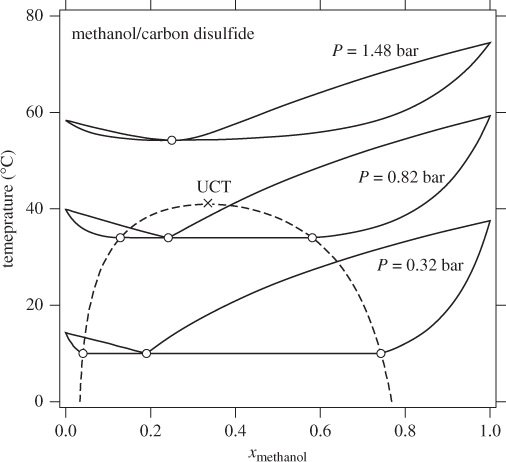
Figure 13-6: Txy graphs of methanol/carbon disulfide at various pressures. Above the upper consolute temperature (UCT) the system exhibits full miscibility (see Example 13.3).
In the system methanol/carbon disulfide, the lower part of the solubility curve is terminated at the freezing boundary (not shown). Many systems exhibit similar behavior with an upper consolute temperature. Other behaviors are encountered as well, as shown in Figure 13-7. Some systems exhibit a lower consolute temperature, with full miscibility at low temperatures and partial miscibility above. It is also possible, though less common, to observe an upper and a lower consolute temperature (case c in Figure 13-7). In some cases, there are two disjoint two-phase regions. Such systems exhibit partial miscibility at low temperature, full miscibility at intermediate temperatures, and partial miscibility again at higher temperatures.
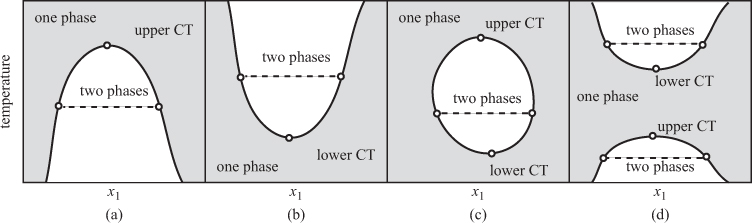
Figure 13-7: Miscibility curves in binary liquids. Dashed lines are tie lines between two liquids. (CT = consolute temperature).
13.4 Completely Immiscible Liquids
When the mutual solubility of the liquids is so low that they may be regarded as completely immiscible, certain simplifications are possible so that we may construct the phase diagrams without the need for activity coefficients. These simplifications arise from the fact that the two-liquid phases each practically consist of the pure components.
Before we show how to calculate these phase diagrams we will examine their features first. Figure 13-8 shows the Txy graph of water(1) / toluene(2) at 1 bar. This system phase-separates and because the mutual solubility is very low, the two phases can be considered pure. In the water-rich phase we have ![]() and in the toluene-rich phase
and in the toluene-rich phase ![]() . As a result, the pure liquid phases have been reduced to a vertical line, one at xw = 0 and one at xw = 1. The region marked W + T indicates the two-liquid region. A point in this region splits into two-liquid phases that are essentially pure water and toluene, respectively. The region marked V + W represents equilibrium between water-rich liquid (which we have assumed to be pure water) and vapor that contains both components. The region marked V + T represents equilibrium between toluene-rich liquid (which we have assumed to be pure toluene) and vapor containing both components. We will demonstrate the calculations of the phase diagram with an example.
. As a result, the pure liquid phases have been reduced to a vertical line, one at xw = 0 and one at xw = 1. The region marked W + T indicates the two-liquid region. A point in this region splits into two-liquid phases that are essentially pure water and toluene, respectively. The region marked V + W represents equilibrium between water-rich liquid (which we have assumed to be pure water) and vapor that contains both components. The region marked V + T represents equilibrium between toluene-rich liquid (which we have assumed to be pure toluene) and vapor containing both components. We will demonstrate the calculations of the phase diagram with an example.
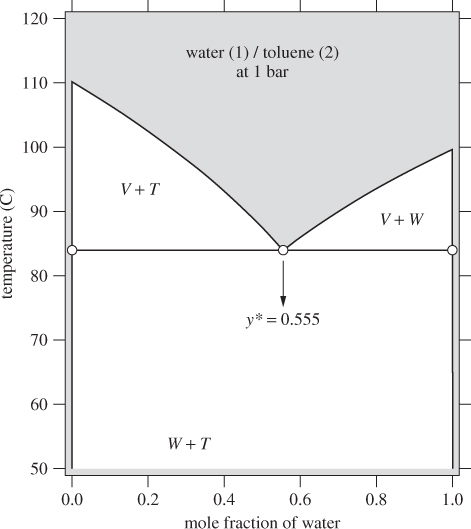
Figure 13-8: Txy graph of water-toluene assuming complete immiscibility.

The mole fraction ![]() is
is
For the construction of the Txy graph we must consider the ranges yw = 0 to ![]() , and
, and ![]() to 1 separately. This is dictated by the fact that a different liquid is present in each of these two ranges.
to 1 separately. This is dictated by the fact that a different liquid is present in each of these two ranges.
Region ![]() . In this region the liquid is pure toluene. No water is contained in the liquid phase. Therefore, the equilibrium condition is written for toluene only:
. In this region the liquid is pure toluene. No water is contained in the liquid phase. Therefore, the equilibrium condition is written for toluene only:
This equation defines the dew line. The simplest way to calculate the dew line is to set the temperature and solve for yt. Notice that the temperature in this range of the graph must be between T* and ![]() . The mole fraction of water is then obtained as yw = 1 − yt. For example, at 90 °C we find
. The mole fraction of water is then obtained as yw = 1 − yt. For example, at 90 °C we find
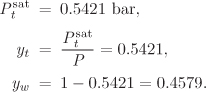
The procedure is continued until the entire range ![]() is covered.
is covered.
Region ![]() . In this region the liquid is pure water and the equilibrium condition is written for water only:
. In this region the liquid is pure water and the equilibrium condition is written for water only:
This equation describes the dew line to the right of point ![]() . The procedure is the same as above. We fix the value of the temperature to a value between T* and
. The procedure is the same as above. We fix the value of the temperature to a value between T* and ![]() and solve for yw. For example, at 90 °C we find
and solve for yw. For example, at 90 °C we find

The complete Txy graph is obtained by repeating the calculation for various temperatures in the range T* to ![]() .
.
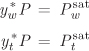
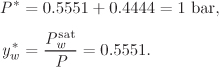
(These values are the same as in the previous example because the temperature given here happens to be the same as the temperature at the triple point at 1 bar.) Having determined the conditions at the triple point we calculate the graph by considering the regions below and above ![]() separately.
separately.
In the region below ![]() the liquid is pure toluene. No water is contained in the liquid phase. Therefore, the equilibrium condition should be written for toluene only:
the liquid is pure toluene. No water is contained in the liquid phase. Therefore, the equilibrium condition should be written for toluene only:

In the region above ![]() the equilibrium condition is written for water only:
the equilibrium condition is written for water only:
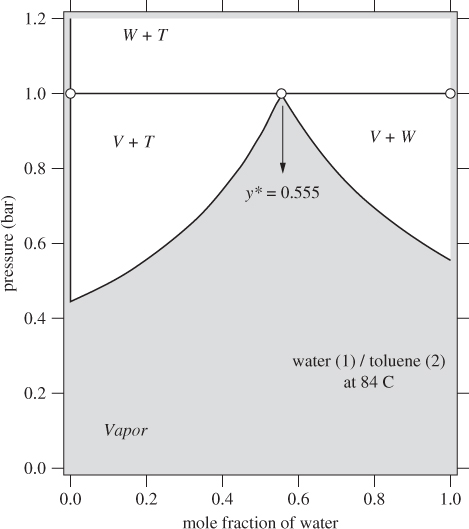
Figure 13-9: Pxy graph of immiscible liquids.
On Complete Immiscibility
It is important to understand the limitations of these simplified calculations. While we have assumed complete immiscibility, in reality there is some amount, however small, of toluene dissolved in water, and of water in toluene. The assumption of complete immiscibility allows us to calculate the bubble and dew points quite accurately. This simplification, however, may not be appropriate for other calculations. For example, the presence of toluene in drinking water is regulated by EPA to a maximum of 1 mg/L, corresponding to a mol fraction of approximately 2 × 10−7. If the purpose of the calculation is to determine compliance with regulations, the assumption of complete immiscibility would be inappropriate. An accurate activity coefficient model is needed for this calculation.
13.5 Solubility of Gases in Liquids
In the examples considered so far, both components are liquids at near ambient conditions. This allows us to calculate the fugacity in solution using the generalized form of the Lewis-Randall equation, eq. (12.16), which requires the fugacity of the pure liquid. Many systems of industrial and biological significance involve a component that at ambient conditions is a gas above its critical temperature. The system water/oxygen is one such example. The solubility of oxygen, an important process for sustaining underwater life, is a case of vapor-liquid equilibrium in which one component (oxygen, Tc = 154.58 K) is a gas above its critical temperature.
The solubility of a gas solute in a solvent is a problem of phase equilibrium. Suppose that we contact oxygen with water at 25 °C. Some oxygen dissolves in water, some water evaporates into the gas phase, and an equilibrium state is established between the two phases. This state is governed by the equality of fugacities: the fugacity of dissolved oxygen is equal to the fugacity of oxygen in the gas, and the fugacity of water in the liquid is equal to that in the vapor. The fugacity of water can be calculated by the general form of the Lewis-Randall equation. For the oxygen, however, we need a different approach, since the component is above its critical temperature. This is done using Henry’s law, a relationship that gives the fugacity of a solute in the limit of infinite dilution:
Here, fi is the fugacity of the solute, xi is the mol fraction of the solute, and ![]() is Henry’s law constant. Henry’s law represents the limiting behavior of fugacity as the concentration of solute approaches zero. This situation is demonstrated in Figure 13-10. As the mol fraction of solute is reduced to zero, fugacity also
is Henry’s law constant. Henry’s law represents the limiting behavior of fugacity as the concentration of solute approaches zero. This situation is demonstrated in Figure 13-10. As the mol fraction of solute is reduced to zero, fugacity also
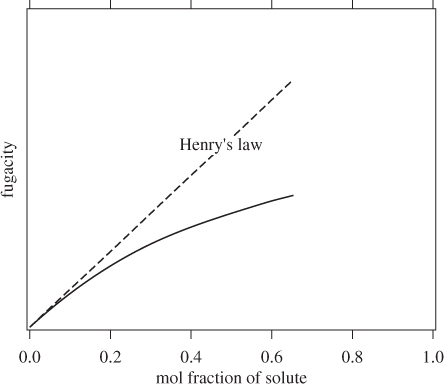
Figure 13-10: Fugacity of solute in the limit of infinite dilution.
approaches zero. Henry’s law constant is the slope of the fugacity at xi = 0. Henry’s law in eq. (13.10) represents a linear extrapolation of fugacity from point xi = 0 into the region xi > 0. Within a small range of compositions, the linear extrapolation is essentially indistinguishable from the actual fugacity. In this region, Henry’s law provides an accurate estimate of the fugacity of solute. At higher concentrations the linear extrapolation and the actual fugacity diverge and Henry’s law breaks down. For gases with very low solubility (for example, most gases in water at near room temperature), eq. (13.10) provides an acceptable approximation.
VLE Using Henry’s Law
Calculations of vapor-liquid equilibrium involving a gas dissolved in a liquid are performed using the Lewis-Randall rule for the fugacity of the liquid component and Henry’s law for the vapor component. Using the subscript s for the solvent, and i for the gas, the equilibrium criterion for the two components is,
where P is the total pressure, and ys, i, xs, i are the mole fractions in each phase (ys + yi = xs + xi = 1). The first equation expresses the equality of fugacities of the solvent in the gas and in the liquid, and the second equation expresses the equality of fugacities of the gas species in the gas phase and in the solution. From eq. (13.12), the solubility of the gas in the liquid is
This result states that the solubility (mol fraction of gas in the liquid) is proportional to the partial pressure of the gas above the liquid and inversely proportional to the value of ![]() . That is, a high value of Henry’s law constant indicates low solubility, and vice versa.
. That is, a high value of Henry’s law constant indicates low solubility, and vice versa.
If Henry’s law constant is known, eqs. (13.11) and (13.12) may be used to calculate the composition of phases at equilibrium. In many cases of practical interest the solubility of the gas in the liquid is so low that the liquid phase is nearly the pure solvent. This allows us to write γs ≈ 1, which eliminates the need for an activity coefficient model and simplifies the VLE equations to
Any of the standard VLE calculations can be performed but the simplest one is the bubble-P calculation. First, we add the above equations to eliminate the gas-phase compositions,
Next we set xs = 1 − xi and solve for the concentration of the solute in the liquid:

This gives the solubility of the gas at total pressure P. The remaining compositions are calculated easily by back substitution.
Other Units for Henry’s Law Constant
Solving eq. (13.12) for ![]() we have
we have
where Pi is the partial pressure of the gas above the saturated liquid and xi is the mol fraction of the gas in the liquid. This equation expresses Henry’s law constant as the ratio of the partial pressure of the gas to the solubility of the gas in the liquid. Henry’s law constant can be measured experimentally by application of this equation: the solvent of interest is brought into contact with the gas of interest and the value of ![]() is obtained as the ratio of the partial pressure of the gas over the solubility. Because solubility can be expressed in different units, Henry’s law constants are often reported in various forms. Common in the chemistry literature is the use of molality for concentration. Molality is the amount of solute in mol divided by the mass of the solvent, in kg. Molality can be converted into mol fraction by simple mass balance equations. The relationship between the two is
is obtained as the ratio of the partial pressure of the gas over the solubility. Because solubility can be expressed in different units, Henry’s law constants are often reported in various forms. Common in the chemistry literature is the use of molality for concentration. Molality is the amount of solute in mol divided by the mass of the solvent, in kg. Molality can be converted into mol fraction by simple mass balance equations. The relationship between the two is
where ci is the concentration in units of molality (mol/kg) and Ms is the molar mass of the solvent (kg/mol). Since Henry’s law is usually applied in the dilute limit (xi ≪ 1), the relationship between molality and mol fraction can be approximated by a simple proportionality,
In molality units, Henry’s law is written as
where ![]() is Henry’s law constant when liquid concentrations are expressed in molality and has units of bar kg/mol. Comparing with eq. (13.10), the relationship between
is Henry’s law constant when liquid concentrations are expressed in molality and has units of bar kg/mol. Comparing with eq. (13.10), the relationship between ![]() and
and ![]() is
is
in which we used xi/ci = Ms, from eq. (13.18).
Note
Alternative (and Potentially Confusing) Forms of Henry’s Law Constant
Henry’s law constant as given in eq. (13.10) is the standard definition in the chemical engineering literature. In other branches of science various alternative definitions are encountered. Such definitions can be reconciled with the definition given here but, in what is a potential source of confusion (and of serious error in the calculation), these other forms are also commonly referred to as “Henry’s law constants.” In the environmental chemistry literature, the definition that seems to be in common use is the reciprocal of the definition given here. Using the symbol kH, this constant is defined as
where ci is the molality of the dissolved gas and Pi is its partial pressure. This is the form of Henry’s law constant that is reported in the NIST WebBook. Another expression is in the form of the ratio
where ![]() and
and ![]() is the concentration of species i in the liquid and in the gas phase, respectively. This has the form of a partition (or distribution) coefficient and is dimensionless. When obtaining values of Henry’s law constant from the literature it is important to be clear about the definition associated with the reported values.
is the concentration of species i in the liquid and in the gas phase, respectively. This has the form of a partition (or distribution) coefficient and is dimensionless. When obtaining values of Henry’s law constant from the literature it is important to be clear about the definition associated with the reported values.
A further observation has to do with the relative magnitudes of the various terms in eq. (13.15). For a solvent of low volatility, that is, at low temperature compared to its boiling point, ![]() is small, both compared to the total pressure P and Henry’s law constant,
is small, both compared to the total pressure P and Henry’s law constant, ![]() . Dropping the saturation pressure from the numerator and denominator of eq. (13.15) we obtain the simplified expression,
. Dropping the saturation pressure from the numerator and denominator of eq. (13.15) we obtain the simplified expression,
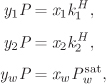
y1 + y2 + yw = 1, x1 + x2 + xw = 1.

By numerical substitution with ![]() bar we find
bar we find
x1 = 4.82 × 10−6, x2 = 8.372 × 10−6.
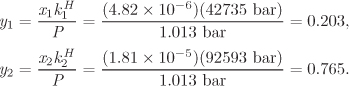
xw = 0.999987, yw = 0.0313421.
Comments If the volatility of the solvent is neglected (this is equivalent to setting ![]() in the above equations), then
in the above equations), then ![]() ,
, ![]() and the solubility of gas component i is
and the solubility of gas component i is
Temperature and Pressure Effects on Henry’s Law Constant
For a given system of solute/solvent, Henry’s law constant is a function of temperature and total pressure. The effect of pressure is small and can be neglected because pressure has generally little effect on properties in the liquid phase. Temperature is a more sensitive variable and must be accounted for. Commonly, literature data on Henry’s law constant are presented in the form of a temperature dependent equation of the form,
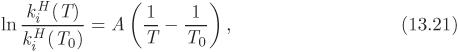
where ![]() is the value of Henry’s law constant at some known temperature T0, often chosen as T0 = 298.15 K. The parameter A (with units of K) is obtained through numerical fitting of data and depends on the nature of the solute and solvent. Occasionally, more complex equations are used to capture more accurately the effect of temperature.3 For many gases at low pressures, increasing temperature increases solubility. This trend is often reversed at higher temperatures.
is the value of Henry’s law constant at some known temperature T0, often chosen as T0 = 298.15 K. The parameter A (with units of K) is obtained through numerical fitting of data and depends on the nature of the solute and solvent. Occasionally, more complex equations are used to capture more accurately the effect of temperature.3 For many gases at low pressures, increasing temperature increases solubility. This trend is often reversed at higher temperatures.
3. See, for example, Sandler [3].
Note
Henry’s Law and Formal Thermodynamics
Henry’s law owes its name to William Henry, the British chemist who reported the linearity between solubility and partial pressure in the early 1800s. The empirical observation of linearity was made independently of thermodynamics and took the force of a “physical law.” It is not a new physical principle, however, and the constant it introduces is fully accounted for by thermodynamics. To establish the relationship between formal quantities introduced earlier and Henry’s law constant, we return to the general expression for the fugacity of a species in a mixture in eq. (10.15). Applying this equation to the infinite dilution limit, we have
where ![]() is the fugacity coefficient of component at the infinite dilution limit (xi → 0). Comparing eq. (13.22) with eq. (13.10), we obtain a relationship between Henry’s law constant and the fugacity coefficient at infinite dilution,
is the fugacity coefficient of component at the infinite dilution limit (xi → 0). Comparing eq. (13.22) with eq. (13.10), we obtain a relationship between Henry’s law constant and the fugacity coefficient at infinite dilution,
Using In ![]() , we further relate
, we further relate ![]() to the residual partial Gibbs energy:
to the residual partial Gibbs energy:
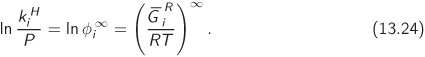
Recall that the fugacity coefficient can be calculated from an equation of state, as discussed in Chapter (10). Therefore, Henry’s law constant can be obtained from the equation of state as well.
The dependence of Henry’s law constant on pressure is obtained from the known dependence of the residual Gibbs energy. The partial derivative with respect to temperature is
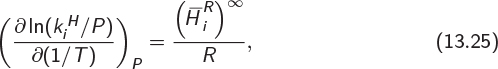
and with respect to pressure is4
4. For a given system of solute/solvent, Henry’s law constant is a function of pressure and temperature but not of composition. This is because it refers to a very specific composition of the solution, the infinite dilution limit.
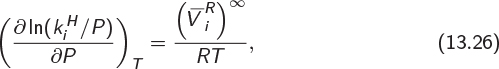
where ![]() is the residual partial molar enthalpy and
is the residual partial molar enthalpy and ![]() the residual partial molar volume, both in the limit of infinite dilution. Since molar (and partial molar) volumes in the liquid phase are small, the right-hand side of eq. (13.26) is a small number and makes a contribution only if pressure varies enormously. Returning to eq. (13.25), we integrate with respect to temperature between temperatures T0 and T. The result is
the residual partial molar volume, both in the limit of infinite dilution. Since molar (and partial molar) volumes in the liquid phase are small, the right-hand side of eq. (13.26) is a small number and makes a contribution only if pressure varies enormously. Returning to eq. (13.25), we integrate with respect to temperature between temperatures T0 and T. The result is

This is the same as eq. (13.21). That is, the empirical fit in eq. (13.21) is of the form expected from the theoretical considerations under the assumption that the residual partial molar enthalpy of solute at infinite dilution is constant. In practice, the factor ![]() is treated as an adjustable parameter and is determined from measurements of Henry’s law constant at two temperatures.
is treated as an adjustable parameter and is determined from measurements of Henry’s law constant at two temperatures.
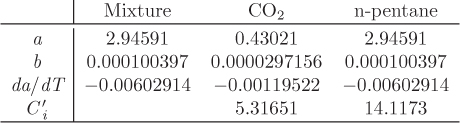
Figure 13-11 shows an SRK calculation of Henry’s law constant as a function of temperature and pressure (see Example 13.9). As we expect, the effect of temperature is much stronger than that of pressure.
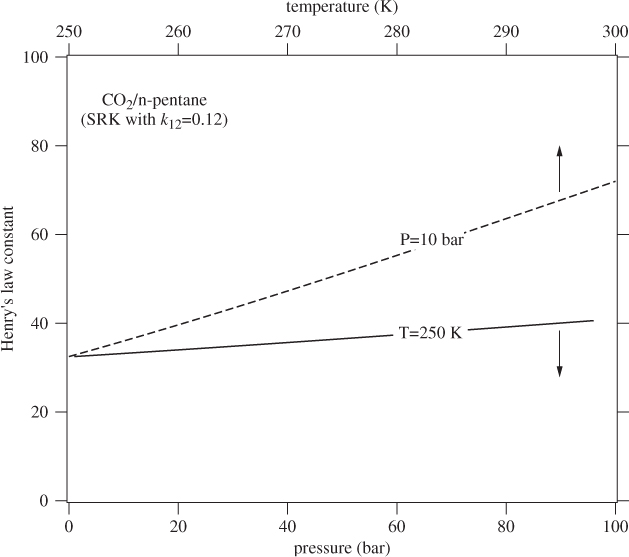
Figure 13-11: SRK calculation of Henry’s law constant for carbon dioxide in n-pentane. The solid line is plotted against pressure (bottom axis) at constant temperature; the dashed line is plotted against temperature (top axis) at constant pressure.
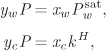
A′ = 0.681898, B′ = 0.0483024.
Z3 − Z2 + 0.631263Z − 0.0329373 = 0,
Z = 0.0570364.
Infinite Dilution and Ideal Solution as Reference States
Henry’s law was introduced as a way of calculating the fugacity of a component in solution when the component is above its critical temperature at the temperature of the solution. Nonetheless, Henry’s law may be used even when the component is below its critical point. There is a certain symmetry between the Lewis-Randall rule, which applies in the limit xi → 1, and Henry’s law, which applies in the limit xi → 0. The relationship is demonstrated in Figure 13-12, which shows the fugacity of carbon dioxide in n-pentane, plotted as a function of the mol fraction of carbon dioxide at constant temperature. In this case carbon dioxide is below its critical temperature (304.2 K) and forms a liquid solution at all compositions between 0 and 1. The Lewis-Randall rule gives the fugacity of component by the linear relationship
This is a straight line that is tangent to the true fugacity at xi = 1. As a tangent line, it provides a good approximation of the true fugacity over some range of compositions near xi = 1. Beyond this range the linear form diverges from the true fugacity and the Lewis-Randall rule is corrected through the use of the activity coefficient:
Henry’s law gives the fugacity of component by the linear relationship
This straight line is tangent to the true fugacity at the opposite corner, at xi = 0. As with the Lewis-Randall rule, it provides a good representation of fugacity within a limited region near xi = 0 but breaks down at higher concentrations. This failure can be corrected using a new activity coefficient, ![]() :
:
Figure 13-12 provides a graphical interpretation of these activity coefficients. When the real fugacity of component i is given by point A, the Lewis-Randall calculation gives point B, and Henry’s law gives point C. The respective activity coefficients are the factors by which we must multiply the fugacity at B and C to bring these points right on A.

Figure 13-12: Fugacity of carbon dioxide in n-pentane (saturated liquid at 250 K).
Both the Lewis-Randall rule and Henry’s law represent limiting behaviors where the fugacity of component may be calculated using simple relationships. A system that behaves according to the Lewis/Randall rule is called “ideal in the Lewis-Randall sense” (or simply, ideal solution), and a system that obeys Henry’s law is called “ideal in the sense of Henry’s law.” And in both cases, activity coefficients are introduced to account from deviations from the “ideal” behavior.
13.6 Solubility of Solids in Liquids
The dissolution of solids in liquids arises often in practice because the liquid phase provides a more homogeneous environment for contact between components as well as for chemical reactions. Solids generally have a finite solubility in a liquid solvent. Exceeding the solubility limit produces a two-phase system, a solid in contact with the solution. This problem of solid liquid equilibrium (SLE) is treated by the same general thermodynamics tools developed so far. When the liquid is saturated in the solid component (solute), the solute satisfies the equilibrium criterion,
Here, ![]() is the fugacity of the component in the solid, and
is the fugacity of the component in the solid, and ![]() its fugacity in the liquid, which can be expressed, using an activity coefficient, in terms of the mole fraction of the dissolved solid and the fugacity of the solute as a pure liquid at the temperature of the solution. Since the solute does not exist as a pure liquid at the temperature of the solution,
its fugacity in the liquid, which can be expressed, using an activity coefficient, in terms of the mole fraction of the dissolved solid and the fugacity of the solute as a pure liquid at the temperature of the solution. Since the solute does not exist as a pure liquid at the temperature of the solution, ![]() refers to a hypothetical liquid and must be calculated by extrapolation from the liquid phase. The situation is demonstrated in Figure 13-13, which shows the pressure-enthalpy graph of the pure solute. The pure solute at temperature T of the solution is shown by point S, which lies in the solid region. The hypothetical liquid at T is shown by point L. This point lies on the dashed line L′L, which represents the hypothetical liquid isotherm at T, that is, the isotherm that is obtained by assuming that the liquid at L′ does not solidify but continues to exist as a liquid. The fugacity of point
refers to a hypothetical liquid and must be calculated by extrapolation from the liquid phase. The situation is demonstrated in Figure 13-13, which shows the pressure-enthalpy graph of the pure solute. The pure solute at temperature T of the solution is shown by point S, which lies in the solid region. The hypothetical liquid at T is shown by point L. This point lies on the dashed line L′L, which represents the hypothetical liquid isotherm at T, that is, the isotherm that is obtained by assuming that the liquid at L′ does not solidify but continues to exist as a liquid. The fugacity of point ![]() and of point L is
and of point L is ![]() . For each of these fugacities we apply eq. (10.4)
. For each of these fugacities we apply eq. (10.4)
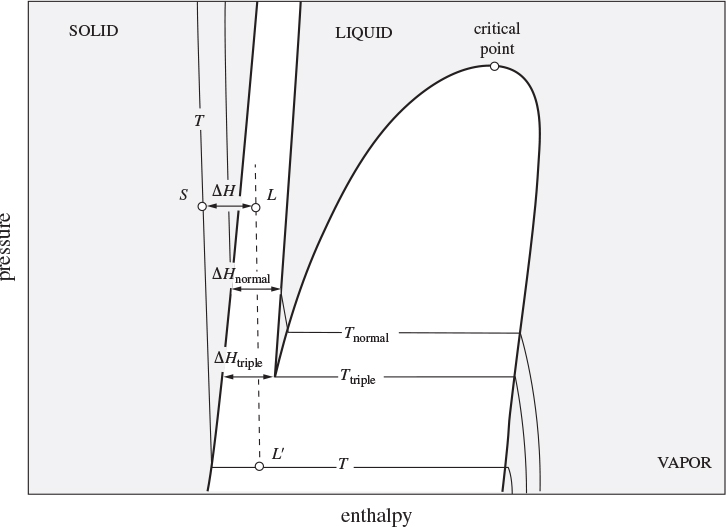
Figure 13-13: Hypothetical liquid state L of pure solute for the calculation of solubility. Dashed line LL′ is a hypothetical liquid isotherm at the same temperature as solid-state S.
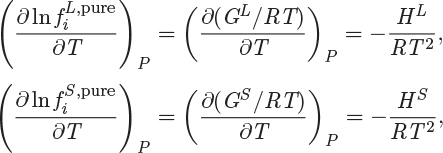
and upon taking the difference we have
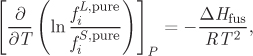
where ΔHfus = HL − HS is the heat of fusion for the hypothetical melting S → L at T. Next we integrate the ratio of fugacities with respect to temperature from the normal melting point (states Sn, Ln) to the hypothetical melting point (states S, L) noting that the fugacities of the solid and liquid at the normal melting point are equal to each other since this is an equilibrium state.5 Finally, we assume the heat of fusion to be constant and equal to its value at the normal melting point. The result of the integration is
5. The fugacities at S and at the hypothetical liquid L are not equal because this is not a pair of coexisting phases.

where Tn is the normal melting point. Combining this result with eq. (13.27) we obtain the final expression for the solubility of the solute:
If solute and solvent are similar in chemical structure and interaction, their solution is approximately ideal (γi ≈ 1). In this case we obtain a simpler relationship for the ideal solubility of solid solute in a solvent at temperature T:
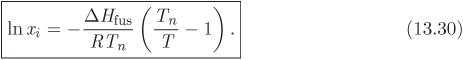
It is interesting to observe that the result depends on the heat of fusion. This alludes to the fact that the dissolution of a solid is akin to melting, a process that disrupts the bonding between molecules in the solid phase. In the case of dissolution this disruption is caused not by temperature but by the presence of the solvent. Even in the absence of specific interaction (γi = 1), the solvent is capable of bringing a certain amount of solute into the solution.
Before we demonstrate the application of this equation, a few comments on the derivation. The use of the normal melting point as a starting point for the integration of the fugacity ratio is done for convenience, since the melting temperature and heat of fusion at 1 atm are usually available from tables. Since the heat of fusion does not change much with temperature, other temperatures where the heat of fusion is known can be used.6 The derivation assumes that the heat of fusion is independent of temperature. More accurate expressions require the heat capacity of the solid and liquid (see, for example, Sandler [3] or Prausnitz [4]). Another assumption that was made but not explicitly stated is that the solid phase is pure solute, that is, no solvent is dissolved into the solid. This is usually an acceptable assumption.
6. In some texts the result is given in terms of the melting temperature and heat of fusion at the triple point. For most solids the temperature of the triple point is fairly close to the normal melting point so that either choice gives essentially the same result.
ΔHN = 19061 J/mol, ΔHB = 9866.3 J/mol,
xN = 0.43566.
xB = 0.918.
Phase Diagram and Freezing Point Depression
The calculation of Example 13.10 can be repeated at other temperatures to obtain the solubility of naphthalene and benzene in each other. Figure 13-14 shows the calculated solubility lines plotted against the mole fraction of naphthalene (the solubility of benzene is plotted against xN = 1 − xB). Point A corresponds to the solubility of naphthalene in benzene at 40 °C and point A′ to the solubility of benzene in naphthalene at 0 °C. Both solubilities increase with increasing temperature, as eq. (13.30) indicates. The solubility lines also indicate the phase boundary of the system. To see why this is so, let us examine point A, which gives the solubility (maximum mole fraction) of naphthalene in benzene at 40 °C. To the left of this point we have a homogenous solution because the mol fraction of naphthalene is less than the solubility limit. To the right, we have a heterogeneous mixture that consists of a liquid that is saturated in naphthalene, along with a solid phase of undissolved naphthalene, since the amount present is above the solubility limit. Therefore, the solubility curve of naphthalene marks the boundary between the homogenous liquid (above) and the heterogeneous mixture of liquid with undissolved naphthalene, below. Similarly, the solubility curve of benzene marks the boundary between the homogeneous solution (above) and a liquid mixed with undissolved benzene, below. The two curves meet at point E, which marks the end of each solubility curve. Line BEN is formed by applying eq. (13.30) for benzene from xN = 0 (xB = 1) to ![]() (eutectic point), and the same equation for naphthalene from
(eutectic point), and the same equation for naphthalene from ![]() to xN = 1. To complete the phase diagram we draw the tie line that passes through point E (see graph on the right pane of Figure 13-14). The graph represents the phase diagram of two completely immiscible solids whose liquid is completely miscible at all compositions. It is very similar to the Txy graph of two completely immiscible liquids (see Figure 13-8) and it is read in a similar manner. The region above line BEN is a homogeneous liquid. All other regions are a mixture of two phases, two solids (below CED), or a liquid and a solid (regions between lines CED and BEN). Line BEN is the freezing line of the mixture: approached from above (liquid), it represents the point where a solid phase forms. Line CED is the melting line: approached from below (mixture of two solids), it represents the temperature where a liquid forms. In metallurgy, the freezing and melting lines are known as solidus and liquidus, respectively.
to xN = 1. To complete the phase diagram we draw the tie line that passes through point E (see graph on the right pane of Figure 13-14). The graph represents the phase diagram of two completely immiscible solids whose liquid is completely miscible at all compositions. It is very similar to the Txy graph of two completely immiscible liquids (see Figure 13-8) and it is read in a similar manner. The region above line BEN is a homogeneous liquid. All other regions are a mixture of two phases, two solids (below CED), or a liquid and a solid (regions between lines CED and BEN). Line BEN is the freezing line of the mixture: approached from above (liquid), it represents the point where a solid phase forms. Line CED is the melting line: approached from below (mixture of two solids), it represents the temperature where a liquid forms. In metallurgy, the freezing and melting lines are known as solidus and liquidus, respectively.
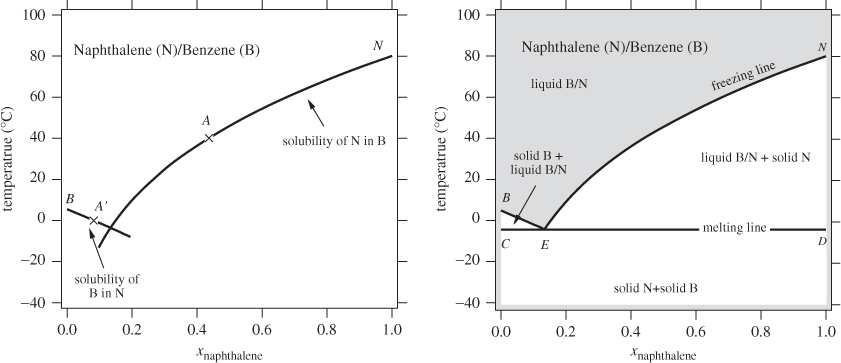
Figure 13-14: Ideal solubility between naphthalene (N) and benzene (B). Left: solubility curves; right: phase diagram. (See Example 13.10.)
A characteristic of such systems is that the freezing line is curved downwards, that is, mixtures of the two components have a lower freezing point than either component. This is known as freezing point depression. A common application of this phenomenon is in deicing. By mixing ice with a nonvolatile solute, the ice-solute mixture becomes liquid even at temperatures where pure water would be solid.

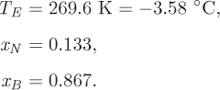
13.7 Osmotic Equilibrium
Osmosis is a process in which a system establishes equilibrium between two regions separated by a partition that is permeable to some but not all components. A typical situation is that of a mixture of two components one of which is small enough to pass through the membrane but the other is too big to fit. Osmosis is an important process in biology, as most living organisms depend on membranes to regulate selective transport through them. It is also the basis of reverse osmosis, a separation process that employs a semipermeable membrane to purify a component from a mixture. The basic osmotic experiment consists of a solution that contains a large solute and which is brought into contact with the pure solvent via a membrane that is permeable to the solvent but not to the solute (Figure 13-15).7 Initially both compartments are at the same temperature and pressure. In the equilibrated system, the pressure is higher in the compartment that contains both components due to the transfer of solvent from compartment A. In reverse osmosis, pressure is applied to compartment B causing the transfer of solvent into compartment A. This process produces pure solvent from a solvent/solute mixture and is used commercially in water purification.
7. In the case demonstrated in Figure 13-15, compartment A contains solvent only. This is not necessary. If both compartments initially contain solvent and solute, but at different concentrations, solvent will be transferred from the compartment with the higher concentration to the one with lower concentration.
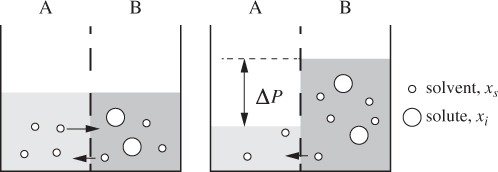
Figure 13-15: The osmotic process: Two regions establish equilibrium via a semipermeable membrane. At equilibrium, pressure is higher in the compartment where both components are present.
In the basic osmotic process, the equilibrium state is one in which pressure and composition in the two compartments are unequal. This is a case of constrained equilibrium: The construction of the membrane prevents the equilibration of pressures; it also prevents the equilibration of the chemical potential of the solute, since it cannot pass through the membrane. On the other hand, it permits the equilibration of the solvent, which is free to pass between both sides, and the equilibration of temperature, since molecular transport, even of one species only, necessarily transports energy between the two sides. The equilibrium conditions for this situation are,
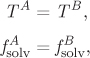
where A and B indicate the compartment (A is pure solvent, B is solvent plus solute). Fugacity ![]() refers to pure solvent at pressure PA; fugacity
refers to pure solvent at pressure PA; fugacity ![]() refers to mixed solvent with mol fraction xs at pressure PB = PA + Π, where Π is the osmotic pressure and is equal to the pressure difference between the two compartments at equilibrium. For the fugacities we write,
refers to mixed solvent with mol fraction xs at pressure PB = PA + Π, where Π is the osmotic pressure and is equal to the pressure difference between the two compartments at equilibrium. For the fugacities we write,

where ![]() and
and ![]() is the mol fraction and activity coefficient of the solvent in the compartment that contains the solute. The fugacities of pure solvent that appear in these two equations are related via the Poynting equation:
is the mol fraction and activity coefficient of the solvent in the compartment that contains the solute. The fugacities of pure solvent that appear in these two equations are related via the Poynting equation:
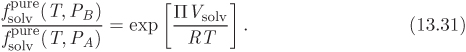
where Vsolv is the molar volume of the solvent. Combining these results and solving for the osmotic pressure we obtain,

As we see, the osmotic pressure of the solution depends on the activity of the solvent, ![]() , in the solution that contains the solute. A simpler expression is obtained in the limit of infinite dilution, where the activity coefficient of the solvent is approximately unity. Using the approximation – ln xsolv ≈ 1 − xsolv = xi, which is valid when xsolv ≈ 1, eq. (13.32) becomes,
, in the solution that contains the solute. A simpler expression is obtained in the limit of infinite dilution, where the activity coefficient of the solvent is approximately unity. Using the approximation – ln xsolv ≈ 1 − xsolv = xi, which is valid when xsolv ≈ 1, eq. (13.32) becomes,

where xi = 1 − xsolv is the mol fraction of the solute. Noting that xi/Vsolv ≈ ci, where ci is the molar concentration of the solute (in mol per unit volume), another form of eq. (13.33) is8
8. To obtain this result, write xi = ni/(ni + ns) and work out the ratio xi/Vsolv:
For low concentrations of solute, the molar volume of the solvent is equal to that of the solution, that is, (ni + ns)Vsolv ≈ Vtot, and the above becomes equal to the molar concentration of the solute.
The general result is eq. (13.32) while eqs. (13.33) and (13.34) are approximations when the concentration of the solute is low. To apply the general form of the equation we must know the activity coefficient of the solvent, which depends on concentration. This dependence is not always known and to circumvent this difficulty, an alternative expression for the osmotic pressure is developed that is easier to accommodate experimental data. In this form, the osmotic pressure is expressed as a series expansion in ci:

This is known as osmotic virial expansion and is analogous to the virial expansion of the compressibility factor. At very low solute concentrations, the linear and higher-order terms in ci on the right-hand side are negligible and eq. (13.35) reverts to (13.34). The coefficients B(T), C(T), and others are the osmotic virial coefficients, and for a given solute/solvent system they are functions of temperature only. These coefficients can be obtained experimentally by fitting experimental values of the osmotic pressure to a polynomial function of concentration. The higher coefficients are difficult to obtain accurately and in practice the series is usually truncated past the second or third term.
Note
Chemical Potential as Driving Force for Diffusion
The osmotic experiment demonstrates that the driving force for diffusion is chemical potential, not concentration. In the initial state, the chemical potential of the solvent is different in the two compartments. It is higher in compartment A (pure solvent), and lower in compartment B (mixed solvent). This is seen by writing
where ![]() is the fugacity of pure solvent, and
is the fugacity of pure solvent, and ![]() is the fugacity of mixed solvent. Since xB < 1, the chemical potential of the solvent in A is higher and causes the transfer of solvent molecules from A to B. The same arguments apply to the solute except that the semipermeable membrane does not allow this component to equilibrate (this analogous to an adiabatic wall that prevents the equilibration of temperature between two regions). The identification of the chemical potential as the driving force for molecular transport is important because it helps us understand the conditions of thermal, mechanical, and chemical equilibrium better. Pressure is the driving force for bulk (convective) motion; mechanical equilibrium requires uniformity of pressure. Temperature is the driving force for the transfer of heat; thermal equilibrium requires uniformity of temperature. Chemical potential is the driving force for molecular transport (diffusion); chemical equilibrium requires uniformity of the chemical potential of any species.
is the fugacity of mixed solvent. Since xB < 1, the chemical potential of the solvent in A is higher and causes the transfer of solvent molecules from A to B. The same arguments apply to the solute except that the semipermeable membrane does not allow this component to equilibrate (this analogous to an adiabatic wall that prevents the equilibration of temperature between two regions). The identification of the chemical potential as the driving force for molecular transport is important because it helps us understand the conditions of thermal, mechanical, and chemical equilibrium better. Pressure is the driving force for bulk (convective) motion; mechanical equilibrium requires uniformity of pressure. Temperature is the driving force for the transfer of heat; thermal equilibrium requires uniformity of temperature. Chemical potential is the driving force for molecular transport (diffusion); chemical equilibrium requires uniformity of the chemical potential of any species.
This discussion brings us to a question that may still be lingering: Why do we call it “chemical potential”? Just as electrons move in response to an electric potential, chemical species move in response to their chemical potential. This motion is from regions of high potential to regions of low potential. The imbalance allows the transport of solvent against a pressure gradient: the chemical potential “pushes” the solvent with stronger force than the force due to pressure. In other words, chemical potential can produce a real mechanical force. Equilibrium with respect to molecular transport is established when the chemical potential is uniform in all regions where a species can wander. In the osmotic experiment, the chemical potential in the side of the pure solvent remains constant. On the solute side it is lower but increases continuously with molecular transport because both the mole fraction of the solvent and pressure increase. At equilibrium, solvent molecules continue to pass freely from one side to the other but there is no net change to either side because the chemical potential is uniform. At this point, the force due to chemical potential balances exactly the mechanical force exerted by the pressure difference.
Reverse Osmosis
In the osmotic process, shown in Figure 13-15, pure solvent is brought into contact with a solvent/solute solution resulting in elevated pressure on the side of the solution. If the solution is subjected to pressure, this will cause a reverse transfer of solvent from the solution to the pure solvent because pressure increases the chemical potential of the solvent in the solution and forces it pass on the other side. This process is called reverse osmosis or hyperfiltration, and may be used to purify a solvent. One of the most prominent applications is in desalination of seawater. In desalination by reverse osmosis, sea water (containing approximately 3.5% wt of salts of mostly Na+ and Cl− ions) is compressed to a value above its osmotic pressure and is brought into contact with a semipermeable membrane that allows water to pass but not any of the ions. The process is shown schematically in Figure 13-16. Effectively, pressure causes water to pass through the membrane, which acts as a filter that keeps out the ions, hence hyperfiltration. However, there are important differences between regular filtration and reverse osmosis. In filtration, the applied pressure is needed to overcome the resistance to the flow of the liquid through the filter; even small pressure will produce a trickle of flow. In reverse osmosis, pressure establishes a gradient of chemical potential from the seawater side to the freshwater side that squeezes freshwater out of the water/salt solution. For this process to work, the pressure on the seawater side must be at least equal to the osmotic pressure of the seawater solution. If it is less, freshwater will pass into the seawater by forward osmosis. In desalination, the goal is to produce a stream of pure solvent (water). The same process, however, can be used to concentrate a dilute solution of a solute. An important advantage of reverse osmosis is that it requires no heat (its energy input is entirely in the form of work), which makes it possible to operate at room temperature. For this reason reverse osmosis finds application in the separation of temperature-sensitive products such as proteins.
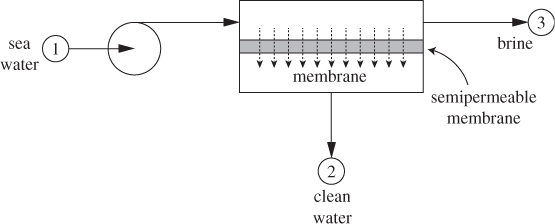
Figure 13-16: Schematic of desalination process by reverse osmosis.

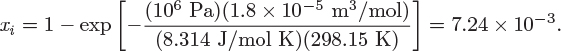
13.8 Summary
The processes discussed in this chapter demonstrate the great variety of phase equilibrium that can arise beyond the basic vapor-liquid problems discussed in most of the previous chapters. Many other systems could be included: The adsorption of gases onto solids (used in the removal of pollutants from air), the distribution of detergents in water/oil systems, the wetting of solid surface by a liquid, the formation of an electrochemical cell when two metals make contact are all examples of multiphase/multicomponent equilibrium. They all share one important common element: their equilibrium state is determined by the requirement that the chemical potential of any species must be the same in any phase where the species can be found. These problems are beyond the scope of this book. The important point is this: The mathematical development of equilibrium (Chapter 10) is extremely powerful and encompasses any system whose behavior is dominated by equilibrium.
13.9 Problems
Problem 13.1: Determine the solubility of hexane in methanol and decane at 25 °C based on the available activity coefficients at infinite dilution:
a) Hexane in methanol: ![]() ,
, ![]()
b) Hexane in decane: ![]() ,
, ![]() .
.
Hint: Assume that the Margules equation is valid for these systems.
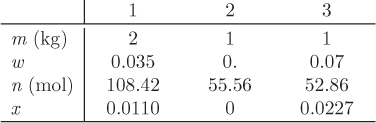
Problem 13.2: A tank contains a mixture of water and carbon dioxide. The mol fraction of CO2 in the liquid is 5.0 × 10−4 and the temperature is 70 °C.
a) Calculate the total pressure as well as the composition of the vapor phase.
b) To what temperature should you bring the system in order to increase the mol fraction of CO2 in the liquid by a factor of 5, if the total pressure is to remain constant?
c) Calculate the composition of the vapor phase for the conditions of part (b). Henry’s constant of CO2 is
with T in kelvin. Notice that Henry’s law constant is expressed in units of molality.
Problem 13.3: The fugacity coefficient of CO2 in liquid n-pentane at 344.15 K, 2.93 bar approaches the value ϕ = 39.2 as the mol fraction of CO2 approaches zero. Use this information to calculate Henry’s law constant for this system.
Problem 13.4: A chemical process produces a waste stream that is mostly air but contains dimethlyamine (DMA) at a mol fraction of 0.01. To satisfy emission standards, the gas stream must be purified to contain no more than 100 ppm of DMA. To achieve this concentration, the gas is passed through a liquid scrubber in which water is sprayed from the top of the tank while the gas rises. During the contact between the drops and the gas, some DMA is transferred to the liquid phase. The inlet water contains no DMA and the exiting streams may be assumed to be in equilibrium with each other. The process is operated at 25 °C, 1 bar.
a) Calculate the required flow rate of water to achieved the desired purification. Report the result in kg of water per kg of air.
b) If the water flow rate must remain less than 50 kg water/kg of air, what is the required pressure?
c) Critique the assumption that the exit streams are at equilibrium.
Additional data: Henry’s law constant for DMA at 25 °C is 1.84 bar.
Problem 13.5: The VLE data in the table below were obtained using the SRK equation. The system under consideration is carbon dioxide (1) and cyclohexane (2). Using only data given in this problem, answer the following questions:
a) What is the saturation pressure of cyclohexane at 250 K?
b) Calculate the solubility (mole fraction) of CO2 in cyclohexane at 250 K under total pressure of 1 bar.
c) A vapor mixture of carbon dioxide/cyclohexane is to be separated by partial condensation in a vapor-liquid separator at 250 K so that carbon dioxide is received at a purity of 80%. Determine the pressure and the mole fraction of CO2 in the liquid.
d) If the feed stream in the previous question contains 30% CO2 by mole, calculate the recovery of carbon dioxide (i.e., moles of CO2 in the purified stream as a percentage of moles CO2 in the feed).
e) Clearly state and justify your assumptions.

Problem 13.6: Pure water (stream A) and H2S (stream B) are brought into contact in a bubbler where they reach equilibrium at 1 bar, 50 °C. The gas stream that leaves the bubbler is then compressed to 5 bar and subsequently passes through a heat exchanger which cools the compressed stream to 25 °C.
a) Determine the composition of streams D and C.
b) Determine whether stream G contains any liquid and if so, calculate the fraction of the liquid.
Additional data: The saturation pressure of water (Pw) and Henry’s law constant for H2S in water (kH) are given below as a function of T:
ln Pw = −37.224 + 0.16686 × T − 0.00017985 × T2,
ln kH = −14.13 + 0.11365 × T − 0.00015146 × T2.
In the above, T is in kelvin while both ![]() and KH are in bar.
and KH are in bar.
Problem 13.7: An air stream that contains 8% ammonia (by mol) is treated in absorption tower that removes 95% of the ammonia. In this unit, the gas stream is brought into contact with freshwater at 1 bar, 20 °C. In a simplified treatment of the process, assume that the liquid that exits the tower is in equilibrium with the exiting air stream. Determine the required flow rate of water per mol of the air stream entering the unit. The following equilibrium data are available:

Data from Wark, Warner, and Davis, Air Pollution: Its Origin and Control, 3rd ed., (Boston: Addison-Wesley, 1998), p. 329.
Problem 13.8: Water and normal heptane are essentially immiscible.
a) What is the bubble temperature of a liquid mixture that contains 50% by mol normal heptane at 1 bar?
b) What is the dew temperature of a vapor mixture with 50% normal heptane at 1 bar?
c) What is the dew pressure of an equimolar vapor mixture at 50 °C?
Problem 13.9: Water and normal octane are practically immiscible in each other.
a) Calculate the dew temperature at 2 bar of a vapor mixture that contains 65% water and 35% octane (by mol). What is the composition of the first liquid to condense?
b) A mixture that contains 75% water and 25% octane (by mol) is brought to 115 °C, 2 bar. Which phases are present?
c) 100 mol of vapor mixture that contains 75 mol water and 25 mol octane is cooled at constant P = 2 bar until 10 mol of liquid octane have been collected. How many moles of water have condensed at that point?
The saturation pressures of the two components are given by the equations below (P in bar, T in C):
PW = e11.6832−3816.44/(T+227.02), PO = e9.3222−3120.29/(T+209.52)
where W stands for water and O stands for octane.
Problem 13.10: Water and normal octane are essentially immiscible in each other. Consider a solution of the two components that contains 32% water by mol. In all of the following the temperature is 50 °C.
a) At what pressure does the liquid begin to boil?
b) What is the composition of the first bubble?
c) Which phase boils off first?
d) What is the pressure when the first liquid phase boils off?
e) At what pressure does the second liquid phase disappear?
f) What is the composition of the vapor at that point?
Additional data: You may use Antoine equations given in Problem 13.9.
Problem 13.11: Water and normal octane are essentially immiscible in each other. Consider a solution of water and normal octane containing 80% water by mol. In all of the following the pressure is 1 bar.
a) At what temperature does the liquid begin to boil?
b) What is the composition of the first bubble?
c) Which phase boils off first?
d) What is the temperature when the first liquid phase boils off?
e) At what temperature does the second liquid phase disappear?
f) What is the composition of the vapor at that point? Antoine parameters are given in the Problem 13.9.
Problem 13.12: At 100 °C water (w) and nitrobenzene (n) are only partially miscible. At this temperature, the solubility of nitrobenzene in water is 0.147 mol %, while the solubility of water in nitrobenzene is 8.3 mol %.
a) Assuming that each liquid phase behaves ideally with respect to the concentrated species (that is, γw = 1, in the water-rich phase and γn = 1 in the nitrobenzene-rich phase), calculate the activity coefficient of each component at infinite dilution.
b) Show that if boiling occurs under constant pressure, the boiling temperature must remain constant until one of the two-liquid phases completely evaporates.
c) 100 mol of the water-rich phase are mixed with 100 mol of the nitrobenzene-rich phase at 100 °C and the pressure is adjusted until boiling starts. What is the pressure?
d) Calculate the composition of the vapor phase in the previous part.
e) If boiling continues indefinitely, which liquid phase will disappear first?
f) Draw a qualitative Pxy graph for this system at 100 °C. Show all the important features on the graph. The saturation pressure of nitrobenzene at 100 °C is 21 Torr.
Problem 13.13: The activity coefficients for the system hexane/ethanol at 85 °C are given by
a) Construct the Pxy graph for this system at 85 °C.
b) Determine the bubble pressure of a mixture with the overall composition xhex = 0.5 and report the composition of all the phases present.
Additional data: The saturation pressures of the pure components are
Problem 13.14: Benzene and water are essentially immiscible in each other. Consider a liquid produced by mixing 25 moles of benzene with 75 moles of water at 1 bar, 25 °C:
a) At what temperature does the liquid begin to boil?
b) What is the composition of the first bubble?
c) Which phase boils off first?
d) What is the temperature when the first liquid boils off?
e) At what temperature does the second liquid phase disappear?
f) What is the composition of the vapor at that point?
Additional data: The Antoine equations for the two components are given below (temperature in K, pressure in Torr):
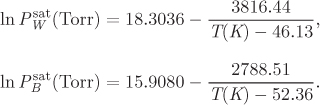
Problem 13.15: Butanol and water are partially miscible liquids. At 1.013 bar, the bubble point of the two-phase system is at 93 °C, and the mol fraction of butanol in the two liquids is 4% and 40%, respectively. Determine the activity coefficients of the two components in the two liquids at 93 °C. Make any suitable assumptions. Additional data: The saturation pressures of butanol and water at 93 °C are 0.387 bar, 0.7849 bar, respectively.
Problem 13.16: A desalination process such as the one in Figure 13-16 is used to produce freshwater from seawater that contains 3.25% wt salts. If the operating pressure is 42 bar, determine the amount (kg) of freshwater that is produced per kg of seawater. Assume seawater to be a solution of NaCl and take its density to be that of freshwater.