
Thermodynamics of high temperature polymer blends
Abstract:
The performance and subsequent properties of polymer blends are highly dependent on the blend’s phase structure. For example, a miscible mixture of two polymers will have different features to an immiscible mixture of the same two polymers. Additionally, the manner in which a transformation from a miscible blend to an immiscible blend occurs will affect the ultimate properties. These features can be categorized under the topic of thermodynamics of polymer blends. This chapter discusses some features of the thermodynamics of blends that contain high temperature polymers, highlighting those which are most important in defining the blend phase structure. A comparison is made with other polymer blends, and important differences are noted.
4.1 Introduction
The performance of polymer blends depends on the properties of the polymeric components, as well as on how they are arranged in space. The spatial arrangement is controlled to a large extent by the thermodynamics of the system. The term ‘thermodynamics’ invariably brings to mind ‘miscibility’. The aim of this chapter is to discuss the thermodynamic features of high temperature polymer blends and highlight studies that have focused on a determination of the features that lead to miscibility in such systems.
A miscible polymer blend is a homogeneous, single phase material. Several experimental methods, many of which have already been discussed in Chapter 2, have been developed for defining such a structure in a mixture of polymers. Among these methods, the occurrence of a single glass transition temperature, or Tg, is the most widely accepted criteria for miscibility. It is generally assumed that polymer blends that display a single Tg are molecularly mixed. Based on those criteria, various models1,2 have been developed in the polymer literature for a description of the relation of a blend Tg and the blend composition.
A necessary condition for two polymers to form a miscible mixture is that the free energy of mixing, ∆Gmix, should be less than zero. In general, ∆Gmix is given by the following equation:
where ∆Hmix is the enthalpy of mixing, T is the temperature and ∆Smix is the entropy of mixing. Obviously, one way for ∆Gmix to be negative is for ∆Hmix to be negative. This is generally accomplished through specific interactions between the two polymers in the mixture, such as dipole–dipole interactions or hydrogen bonding. However, simply having a negative free energy of mixing does not guarantee phase stability. The necessary condition for phase stability in a blend composition of x at a fixed temperature, T, and pressure, P, is that the second derivative of the free energy with respect to composition is greater than zero, i.e.
Without the satisfaction of this condition, the miscible system is thermodynamically unstable and phase separation will occur.
One of the most common models for dealing with the free energy of mixing between two polymers is the Flory–Huggins model.3 That model assumes that the free energy of mixing, ∆Gmix, for the two polymers is given by:
where Θ1 and Θ2 are the volume fractions, respectively, of polymers 1 and 2, N1 and N2 are the degrees of polymerization of polymers 1 and 2 and χ is the so-called ‘chi’ parameter used to describe the enthalpic interaction between polymers 1 and 2. This model, as originally developed, is strictly applicable only to polymers that have random coil configurations. It is also an assumption that the entropic and enthalpic contributions to the free energy are completely independent of each other.
Flory4,5 first proposed the concept of molecular composites which are systems based on the mixing of a rigid-rod polymer and a random coil polymer, vastly different molecular conformations. The theoretical prediction was made that phase separation is easily induced in such systems. The phase separation in such blend systems is based solely on entropic effects. This is an important difference with random coil mixtures in which the driving force for phase separation is usually non-favorable enthalpic interactions.
Two extreme cases of behavior have, then, been theoretically examined in the literature. Mixing of two random coil polymers is controlled by enthalpic effects while the mixing of rigid-rod polymers with random coils is dominated by entropic effects. Since most high temperature polymers contain some form of heterocyclic units, they have restricted rotation in their backbone. It is exactly that restricted motion that often leads to the high glass transition temperature observed in high temperature polymers. Thus, most high temperature polymers can be treated as rigid, or at least semi-rigid, in overall conformation. An adequate theoretical model is still to be developed for the mixing of such polymers that have a molecular conformation between random coils and rigid rods.
It should be noted that there have been attempts to produce molecular modeling results and simulations of high temperature polymer blends. The most extensive of these efforts was published by Jacobson et al.6 They used a simple short chain molecular model that incorporates both inter and intra-molecular interactions. Using that model, estimates of the net interaction energies for a series of high temperature polymer blends were calculated and used to predict miscibility. The results were in general agreement with experiments and could be used to focus the direction of additional experimental work.
4.2 Blending miscible high temperature polymers
There have been several miscible high temperature polymer pairs that have been identified in both patent and open literature. Several of these polymer pairs are miscible when solutions are formed and films are cast. However, when processing is attempted in the melt state, immiscibility results. These results suggest that the blends phase separate when heated above their glass transition temperature. Also, kinetic factors along with thermodynamic factors seem to be affecting the observed miscibility.
The role of the solvent itself also needs to be better understood in the overall scheme of the miscibility observed. It may be that the presence of the solvent produces a kinetically favorable situation for miscibility but, upon attainment of thermodynamic equilibrium, that situation no longer exists. One of the challenges for many of the systems that will be discussed is to broaden the temperature range between the glass transition temperature of the initially miscible blend and its phase separation temperature. Such an effect will allow miscible blends to be processed in the melt state, thus eliminating the need for processing and handling of solvents in the fabrication of products from the blends.
4.3 Poly (2,2' (m-phenylene)-5-5' bibenzimidazole) (PBI) blends
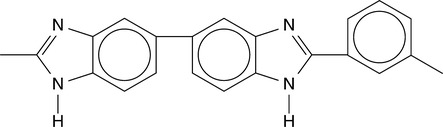
One of the most highly studied high temperature polymers in miscible blends is poly (2,2´ (m-phenylene)-5-5´ bibenzimidazole) or PBI, the chemical structure of which is shown in Fig. 4.1. The fundamental reason for the observation of miscibility in PBI-based systems is the presence of the N-H functional group that can interact with the functional groups which are present on the backbone of other polymers. Thus, miscibility in these types of systems is an example of a specific interaction that leads to a negative enthalpy of mixing.
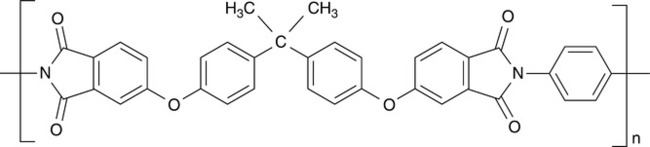
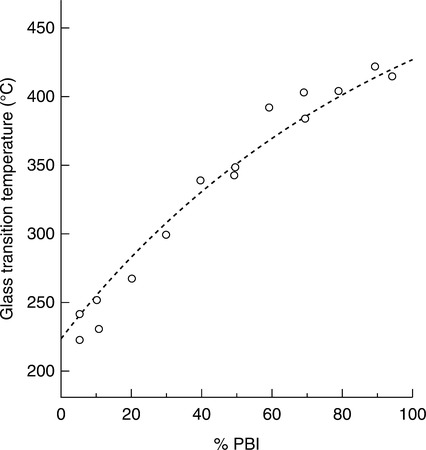
The most extensively studied PBI blend system is PBI blended with a polyetherimide, originally available from GE, Ultem 1000.7 The chemical structure of Ultem is shown in Fig. 4.2. The early work on that blend led to the phase separation diagram shown in Fig. 4.3. This figure shows that a processing window exists for blends that contain in excess of 75% PBI and also for blends that contain less than about 20% PBI. In this case, the processing window is defined as those temperatures between Tg and the phase separation temperature. For example, for an 85/15 PBI/Ultem blend ratio, the glass transition temperature is 400 °C and the processing window is 25 °C.
Infrared studies of these blends showed that hydrogen bonding exists between the N-H groups of PBI and the carbonyl group of Ultem8 and that the hydrogen bonding relaxes during the thermal treatment phase separation.9,10 In subsequent work,11 a direct relationship was found between the strength of the hydrogen bonding of the component polymers, the glass transition temperature of the blends and the solvent diffusion rate of both water and 1,2,4 trichlorobenzene. These latter studies conclude that there is a partial miscibility between the two polymers and that phase inversion occurs between 0.4 and 0.6 Ultem content.
The phase behavior observed with the PBI/Ultem blends is to be contrasted with the phase separation observed in blends of PBI with another polyimide, XU218 from Ciba Geigy.12 That system showed phase separation only above 400 °C. The actual temperatures are determined by the blend composition. Thus, the phase behavior observed in PBI/Polyimide blends is dependent both on the type of polyimide and the thermal history of the blends.
In further investigations of PBI blends, Jaffe et al.13 showed that PBI is miscible with certain polyimides that contain the hexafluoroisopropylidene or 6 F moiety, i.e. CF3–C–CF3 . The miscibility and phase behavior in these blend systems is dependent on the overall structure of the polyimide. In addition, the amount of 6 F chemical group affects the Tg of the polyimide itself and, hence, the Tg of any miscible blends.
Those studies of blends of PBI with various polyimides were subsequently extended to include other polymers. For example, it was shown that PBI and polysulfone form immiscible mixtures.14 However, it was later shown15 that the introduction of functional groups, such as sulfonate groups, into the polysulfone polymer chain resulted in the formation of miscible blends with PBI. It was shown that the sulfonation degree as well as the blend compo-sition controls the miscibility behavior. FT-IR analysis conf rmed that the observed miscibility is due to specif c interactions between the PBI N-H groups and the sulfonate groups on the polysulfone.
4.4 Polyimide blends
There are other high temperature polymers that have also been shown to form miscible mixtures. Polyimides (PIs) have already been discussed in blends with PBI. Other miscible PI blends have been reported with poly (ether-ether ketone) (PEEK),16 polyethersulone (PES)17 and sulfonated PEEK.18 Although the mechanism for miscibility in PBI/PIs was demonstrated to be related to hydrogen bonding between the chains of the two components, the mechanism of miscibility in some of these other blend systems was not as clear.
In order to better understand the miscibility in polyimide-based systems, Sun et al.19 prepared pairs of polyimide blends with different molecular structures in two ways: mixing of the polyamic acid precursors with subsequent imidization and direct solution blending of the two polyimides. Dynamic mechanical analysis (DMA) techniques showed that all of the blends prepared in the two different ways are miscible, as evidenced by the existence of only one Tg for all the blends. It was proposed that the miscibility of these polyimide/polyimide blends is a result of the strong inter-molecular charge-transfer interaction between the two chains of the blend components.
Overall, then, the high temperature polymer blends that have been discussed thus far display some type of well-defined specific interaction that leads to the observed miscibility. In the case of PBI blends, it is hydrogen bonding that occurs through the N-H group present on the PBI backbone. For PI blends, there appears to be a charge-transfer interaction that leads to miscibility in mixtures. In both cases, it is clearly the enthalpic part of the free energy of mixing that leads to miscible blends.
4.5 Liquid crystal polymer blends
Such is not the case in the blends of two liquid crystal polymers (LCPs) that were first extensively studied by DeMeuse and Jaffe. In their initial study,20 they examined blends of LCPs that contain copolymers of p-hyrdoxybenzoic acid (HBA) and 6-hydroxynapthoic acid (HNA) of different copolymer ratios. It was surmised that the miscibility observed in the melt state depended on the difference in copolymer ratios between the two component polymers. However, miscibility in the solid state seemed to be present for all the blends examined. This is an interesting contrast to the systems mentioned above because no obvious enthalpic interaction occurs between the two blend components.
In further studies21,22 those preliminary efforts were extended to include LCPs that contained other monomer units such as terephthalic acid (TA) and hydroquinone (HQ). In some cases, miscible systems were defined21 and sometimes immiscible blends were observed.22 For all cases, however, there was not a well-defined specific interaction that could be attached to the observed miscibility. It seems that the entropy of mixing is important in defining the miscibility in these types of systems.
An extensive study of a number of blends that contain only thermotropic liquid crystalline polymers as components was undertaken by Hsieh et al.23 A wide selection of commercial or semi-commercial materials was chosen for that study. Both thermotropic LCPs (TLCPs) that are regarded as rigid and semi-rigid in molecular conformation were studied. The study revealed that TLCPs with few common comonomers and/or with different rigidities are generally found to be immiscible. Polymeric aspects such as molecular chemistry and molecular packing are decisive aspects. For miscibility to occur, both components should be of similar molecular rigidity, rigid or semiflexible, and preferably contain some common monomer or monomers.
Thus, we have two different types of behavior in blends of high temperature polymers. The first situation is exemplified by the PBI blends in which specific interactions through the N-H group of the PBI are responsible for the observed miscibility. The other situation is displayed by the mixtures of two LCPs in which entropic effects are important for the miscibility. These two types of behavior would seem to be the extreme cases. This also suggests that there are blend systems of high temperature polymers in which both enthalpic and entropic effects should be important for miscibility.
This concept was explored by Lee and DiBenedetto24 who introduced a second LCP as a compatibilizing agent in order to improve the adhesion and dispersion between the components of incompatible LCP/thermoplastic blends. The primary reason that the LCP and thermoplastic polymer are immiscible is due to molecular conformation differences or entropy effects, as originally discussed by Flory in his work on molecular composites. The concept for using a second LCP in such blends is that the two LCPs will be miscible due to entropy effects and the second LCP and the thermoplastic polymer will adhere due to specific interactions. The LCP coupling phase used in this work was a copolymer of PET and HBA known as PHB60. Blends of an LCP with both polycarbonate (PC) and PET were prepared with and without the addition of the second LCP coupling phase. Morphological evidence indicated that the LCP reinforcing phase in the ternary systems exhibited improved adhesion and dispersion on a finer scale than in the binary blends that were prepared using the same processing conditions.
This approach was further explored by Hakemi25 who prepared blends that contain both a wholly aromatic and an aromatic-aliphatic LCP that are miscible with each other. The ultimate goal of this approach was to develop multi-component miscible blends that have components of thermoplastics. The miscible blends could be useful as reinforcing agents for the thermoplastic matrix polymer, and because LCPs contain some of the components of the thermoplastic polymer, improved adhesion between the LCP portion and the matrix portion of the mixture is expected. This is another example of an attempt to balance the phase separation inherent in high temperature polymer blends due to molecular conformation differences by enforcing the enthalpic interactions between the two polymers.
Before leaving the topic of LCP/LCP blends, one additional point needs to be stressed. Low molecular weight liquid crystals (LMWLCs) of the same mesophasic class often show miscibility.26 On the other hand, TLCPs that form the same mesophase, usually nematic, do not necessarily exhibit complete miscibility, Thus, in order to implement the ideas of the last several paragraphs, care must be used in choosing the two LCPs to form the miscible blend.
4.6 Molecular composites
As already discussed in the Introduction (Section 4.1), molecular composites, as first envisioned by Flory, show phase separation based strictly on entropic effects. Those entropic effects arise from the differences in molecular conformation between the rigid rod and the random coil component in the mixture. Thus, in order to interrupt the phase separation, one possible approach is to enhance the potential for specific interactions through the incorporation of interacting chemical groups into the polymeric components of the mixture.
Initially, ionic blends were utilized to produce miscibility in molecular composites.27–30 Ionic bonds are stronger and more thermally stable than hydrogen bonds and, thus, are deemed to be more effective at promoting miscibility. Those initial studies focused primarily on molecular composites that were cast from solution. After those initial studies, work was also focused on the development of melt-processable molecular composites. Such mixtures were produced by dispersing rigid-rod molecules, such as ionic versions of Kevlar, poly (p-phenylene terephthalamide) (PPTA), in a matrix of a flexible polymer, such as poly (4-vinylphenol) (PVP).31–33 Relatively low additions of a PPTA anion, which contains the ionic groups directly attached to the back-bone chains, in the PVP matrix, led to miscibility and to a good dispersion of the rigid rods in the matrix. These miscibility effects were attributed to the presence of ionic interactions between the ionic groups of the modified PPTA and the polar groups of the vinyl pyridine units.
This work represents another attempt at overcoming the phase separation inherent in mixtures of polymers of vastly different molecular conformations through the use of specific molecular interactions. That seems to be an effective approach for interrupting the phase separation. The general application of this idea to the development of specific high temperature polymer blends is still to be realized, however. Also, it needs to be better understood how the chemical modification of the rigid-rod component to increase the molecular interactions alters its conformation, if at all, toward more of a semi-rigid polymer. This could also contribute to the enhanced miscibility with other high temperature polymers.
4.7 Conclusions
In general, the production of miscible blends of high temperature polymers is driven by the need to have specific molecular interactions that lead to favorable values of the enthalpy of mixing portion of the free energy of mixing. While this is often also the case with mixtures of flexible coil polymers, the miscibility criteria for mixtures of high temperature polymers seem to be more stringent. As one moves away from random coil/random coil mixing, entropic effects, other than simply molecular weight, seem to play an increasingly important role. This can be seen clearly from the fact that mixtures of true rigid rods with random coils are immiscible based solely on entropic effects that arise from differences in molecular conformations.
Practically speaking, however, very few polymers can be categorized as rigid rod in conformation, but instead fall into the category of semi-rigid in molecular conformation. In fact, most high temperature polymers that have been blended can best be classified as semi-rigid. Therefore, the theories that have been developed to define the thermodynamics of mixtures of polymers do not apply to these polymers.
This is an area for which additional work is recommended. Establishment of a theory or model that explicitly captures the conformation of the two polymers being mixed is highly desired. Presently, extreme cases have been defined, and the transition from one extreme to the other needs to be better understood. Also, the ability of a polymer to change its molecular conformation to maximize possible favorable enthalpic interactions such as hydrogen bonding should be considered in the theoretical development.
From an experimental perspective, it becomes clear that miscible blends offer certain advantages. Further, an obvious way to enhance the possibility of miscibility in mixtures that involve high temperature polymers is through the development of specific interactions. That approach should be further explored with high temperature polymers through the incorporation of chemical groups that can promote such interactions. A caution about that general approach is that the appropriate chemical modification does not cause the polymer to completely lose its high temperature features.
4.8 Sources of further information and advice
There do not appear to be any books devoted solely to high temperature polymer blends, let alone the thermodynamics of such mixtures. A reasonable review article of some of the factors that affect the phase behavior of polymer blends for high temperature applications was provided by Jaffe et al.34