
Introduction to high temperature polymer blends
Abstract:
A review of definitions and the overall rationale for the production of high temperature polymer blends is provided. The discussion is divided essentially into two parts: miscible and immiscible blends. It is pointed out that one concern with miscible polymer pairs is that of processing in the miscible state. This phenomenon is dependent on the position of the phase separation temperature relative to the glass transition temperature of the polymer blend. In the case of immiscible blends, the issue of adhesion of the polymers is discussed. Finally, the need for better theoretical models for the prediction of miscibility in polymer blends is highlighted and discussed.
1.1 Introduction
Polymer blends are being used in an increasing number of industrial applications. Sectors ranging from the automotive to the aircraft industry have expressed an interest in developing these materials for specific applications. In fact, for some time, research in blends has been one of the biggest areas of polymer research, in both the industrial and academic world. Recently, requirements for materials in certain areas have become increasingly severe. Temperatures in excess of 200°C for times of hundreds of hours have become stated requirements for some materials. Particularly severe in this regard are requirements from the aircraft industry for engine components where high service temperatures for long periods of time are often normal.
The commercial activity in high temperature blends has been somewhat limited. As already alluded to, one of the major driving forces for higher temperature performance has involved military and aerospace applications. Other emerging application needs have developed, but at a slower rate. Continued needs for high temperature materials will be present in the aerospace as well as in the transportation area where under-the-hood components, friction and wear applications continue to require more demanding performance. Electric and electronic needs will also continue at a high rate of growth requiring higher temperature performance. Finally, high temperature polymer needs will emerge in advanced composites research, which often requires blend technology to optimize the performance of available materials. One approach to meeting these material requirements is to synthesize new polymers. Another method to tailor the properties of materials is through blending of two polymers. In this approach, the goal is to highlight the positive features of both materials while attempting to eliminate the negative features.
This book will address various aspects of high temperature polymers and highlight the advantages of producing such blends. Specifically, this first chapter will introduce the reader to general terminology and definitions and also provide background information on the various classes of high temperature polymer blends. For purposes of these discussions, high temperature polymers are generally defined as materials which have service temperatures of at least 175°C. This definition is arbitrary in nature and is governed by the fact that this is the temperature specified in many aerospace applications. Similarly, high temperature polymer blends are defined by the same use temperature. It should be noted that amorphous polymers that have a service temperature of 175°C have glass transition temperatures in the range of about 200°C, approximately 25°C higher than the actual service temperature. On the other hand, semicrystalline polymers have service temperatures which are largely defined by their melting temperatures. Most high temperature polymers are amorphous, but some are semicrystalline. In the case of these high temperature semicrystalline polymers, those with melting temperature less than 400°C are desirable. Above that temperature, degradation starts to compete with melting.
1.2 General principles of polymer blending
The physical state of mixing which is present in polymer blends can be categorized in three ways: miscible, partially miscible or immiscible. A miscible polymer blend is a homogeneous, single-phase material. In many respects, a miscible blend behaves as if it is a single polymer. If the two components in the blend are miscible only within a certain composition range, the polymers are deemed partially miscible. An immiscible blend contains two distinct phases and is heterogeneous in nature.
The philosophy of blending two polymers is to produce a material which has properties that are tailored to a certain performance level. Since the morphology and resultant physical properties are controlled to a large extent by the miscibility and phase behavior, it is quite easy to understand why there is so much interest in controlling the phase structure in blends. In fact, this topic has evolved into a central area of polymer research during the last 40 years. One of the first ideas, that as a rule polymer blends are immiscible, needs to be reevaluated due to the increasing number of miscible or partially miscible polymer pairs reported in the literature (see, for example, Paul and Newman, 1979).1 Despite this high level of activity, much of the work remains based on art and intuition rather than on science. Most of the work performed on high temperature polymer blends has involved the definition of miscible polymer pairs and their phase separation characteristics.2−6 Not much work has been done to predict miscible pairs. In fact, this is an open area of research in the entire area of polymer blends.
Present theoretical models of polymer blends generally focus on the extreme cases of either random coil mixing or rigid rod mixing. For treating random coil mixtures, the Flory–Huggins formalism7 is usually used to describe the free energy of mixing. In this formalism, miscibility is largely dominated by the enthalpy part of the free energy. On the other hand, Flory8, 9 has treated the mixing of rigid rods with random coils, the concept upon which molecular composites are based. In these systems, the two polymers being mixed have vastly different molecular conformations. Phase separation is easily introduced in such systems. It should be recognized that this separation is based solely on entropic effects.
Two extreme cases have, then, been established in the literature. Random coil/random coil mixing is controlled by enthalpic effects, while the mixing of rigid rods and random coils is dominated by entropic effects. However, since most high temperature polymers contain hetero-cyclic units, they possess restricted rotation in their backbone. In fact, it is precisely this restricted rotation which leads to the high glass transition temperature of these polymers. Thus, these high temperature polymers can be regarded as rigid, or at least semi-rigid, in overall conformation. An adequate theoretical model is still to be developed for the mixing of such polymers.
1.3 Thermodynamics of polymer blends
A miscible polymer blend is a homogeneous, single-phase material. Various experimental methods, many of which will be discussed in Chapter 2, have been developed for defining such a structure in a mixture of polymers. Among these, a single glass transition temperature (Tg) is the most widely accepted criteria for miscibility. It is generally assumed that polymer blends which display a single Tg are mixed at the molecular level. Based on these criteria, various models10, 11 have been reported in the literature for the description of the relationship between Tg and the blend composition.
A necessary condition for two polymers to form a miscible blend is that the free energy of mixing, ΔGmix, should be less than zero, where ΔGmix is given by the following equation:
where ΔHmix is the enthalpy of mixing, T is the temperature and ΔSmix is the entropy of mixing. Obviously, one way for ΔGmix to be negative in Equation [1.1] is for ΔHmix to be negative. This is generally accomplished through specific interactions, such as dipole-dipole or hydrogen bonding.
Simply having a negative free energy of mixing does not guarantee phase stability. The necessary condition for phase stability in a blend composition x at a fixed temperature and pressure is that the second derivative of the free energy with respect to composition is greater than zero, i.e.,
Without satisfaction of this condition, the miscible system is thermodynamically unstable and phase separation will occur.
One of the most common models for treating the free energy of mixing in polymer blends is the Flory–Huggins treatment.7 That model assumes that the free energy of mixing, ΔGmix, is given by:
where Θ1 and Θ2 are the volume fractions of polymers 1 and 2, respectively, N1 and N2 are the degrees of polymerization of polymers 1 and 2, and χ12 is called the ‘chi’ parameter and is used to describe the enthalpic interaction between polymers 1 and 2. This, as originally developed, is strictly applicable only to polymers which have random coil conformations. It is also assumed that the entropic and enthalpy contributions to the free energy of mixing are completely independent of each other.
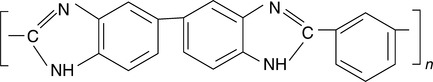
Based on the rigid conformation of many high temperature polymers, it is expected that the criteria for miscibility of such polymers will be even stricter than for random coil polymers. However, several high temperature miscible polymer blends have been discovered and reported in the literature 2−6,12−16 which are based on poly(2,2′ (m-phenylene)-5-5′ bibenzimidazole) (PBI), the chemical structure of which is shown in Fig. 1.1. Chapter 7 discusses these blends in detail but it is to be noted that the fundamental reason for the observation of miscibility in these systems is the occurrence of functional groups in the polymer backbone which can interact with the functional groups present on the backbone in the other polymer in the mixture. Thus, miscibility in these systems is an example of a specific interaction which leads to a negative enthalpy of mixing.
One of the issues with miscible polymer blends in general is the temperature range of their miscibility. If such materials are to be processed in the melt state as miscible blends, there must be a temperature window between the glass transition temperature and the phase separation temperature which is large enough for melt processing. If this processing window is too small, the blends will either phase separate or have too high a viscosity for melt processability.
In the case of the PBI blends discussed above, phase separation takes place a few degrees above the glass transition temperature for most blend compositions.17 This can be seen from the phase diagram for PBI/Ultem poly (ether imide) blends, as shown by Karasz and MacKnight.17 This suggests that the miscibility which is observed is a metastable phenomenon which is controlled by kinetic factors. The observed phase separation also indicates that it will be difficult to melt process the blends in the miscible state, due to the reasons already discussed.
There have been several miscible high temperature polymer pairs defined in the literature. Several of these pairs are miscible from solution but are immiscible when processing is attempted in the melt state. These results indicate that the blends phase separate when heated above their glass transition temperature. This further shows that kinetic factors as well as thermodynamic factors are important in the observed miscibility. Also, the role of the solvent in the observed miscibility needs to be better understood. One of the current technical challenges is to widen the temperature range between the glass transition temperature of the blend and its phase separation temperature, to allow miscible blends to be processed in the melt state.
1.4 Immiscible blends
In this section, three types of immiscible blend will be briefly introduced. The first will be molecular composites, which are generally accepted as partially miscible blends but are not truly miscible at the molecular level. The second type of immiscible blend involves mixtures of two thermoplastic polymers, and a discussion is provided of the features that govern the observed structure. The final type will be mixtures of thermosets with high temperature thermoplastic polymers.
1.4.1 Molecular composites
The concept of molecular composites was first proposed by Flory in the late 1970s. These systems are based on the mixing of a rigid rod polymer and a random coil polymer, vastly different molecular conformations. Flory8,9 predicted that phase separation is easily induced in these systems and is based solely on entropic effects.
Experimental work on molecular composites has focused on attempting to kinetically delay the phase separation with a desirable morphology before the thermodynamics leads to complete immiscibility. Most of this work was initially performed as part of a program sponsored by the United States Air Force. For example, poly (p-phenylene terephthalamide) (PPTA) and poly (p-phenylene benozbisthiazole) (PBT) have been successfully dispersed in a Nylon 66 matrix.18−22 Kyu et al.23 have reported that there are interactions present in a PPTA and Nylon 6 system and that phase separation can be thermally induced in molecular composites based on these two polymers. Thermally induced phase separation has also been observed in the PBT/Nylon 6 system, when the melting temperature of the Nylon component is reached. Finally, Moore and Mathias24 reported a unique method for the preparation of molecular composites using an in situ polymerization process in which the anion of the PPTA was used as the initiator for the anionic polymerization of acrylamide in the formation of a Nylon 3 matrix.
The molecular composites discussed thus far involved processing of the two polymers from a common solvent. One of the major challenges of this approach is the identification of a proper solvent that can be used. In addition, most of the suitable solvents are quite corrosive in nature. That fact, as well as the control of phase separation and, hence, the final properties have largely hindered further widespread industrial development of these materials.
Initial applications of molecular composites are expected to be in the aerospace area. Matrix materials based on molecular composites which are reinforced with carbon fiber are expected to be a key area of interest. The use of a molecular composite as a matrix for advanced composites should provide a significant increase in the modulus and strength over that obtainable with conventional thermoplastics and thermosets. Electronic applications are another area where molecular composite commercialization should continue to emerge.
1.4.2 Immiscible thermoplastic blends based on liquid crystal polymers
It has been established in the literature25 that one of the key parameters which established the morphology and, hence, the physical properties of blends which are based on two immiscible polymers is the viscosity ratio for the two component polymers. The ratio can usually be controlled through parameters such as the molecular weight of the polymers and the melt processing temperature. Through that approach, it is possible to observe a variety of different morphologies with the same two immiscible polymers.26
Nowhere is that fact more apparent than in blends of liquid crystal polymers (LCPs) with other thermoplastic polymers. It is not the intent of the present chapter to completely review all the work done in the area of blends which contain LCPs. Instead, that will be the focus of Chapter 5. The objective of the present discussion is to make general observations and show how those general phenomena may be applicable to other high temperature polymer blends as well.
In these types of blends, the size, shape and distribution of the LCP phase depends on many factors, such as the blend composition, the processing conditions, the viscosity ratio of the component polymers at the shear rate which is being used in the processing, and the rheological characteristics of the thermoplastic matrix polymer. This observation is present in the work of Acierno et al.27 who demonstrated that different morphologies could be observed in the same LCP/polycarbonate blend simply by varying the processing temperature. The observed morphological differences were attributed solely to different viscosity ratios at the different processing temperatures.
Several reports have appeared in the literature which discuss the lowering of the viscosity of a thermoplastic polymer with the addition of a small amount of the LCP component. These studies have been performed on a variety of polymers including polyamide,28 poly (ether imide),29 poly (ether sulfone)30, 31 and polycarbonate.32−34 All of these studies reported a lowering of the viscosity of the traditional thermoplastic polymer with the addition of various small amounts of the LCP component.
This result is interpreted as being due to a lubricating effect of the LCP on the melt. This is because the groups of LCP molecules, called domains, slide past each other resulting in a lubrication of the polymer melt.
This lowering of the melt viscosity allows the LCP to act as a processing aid for the conventional thermoplastic. Cogswell et al.35−37 concluded that, in order for this effect to be realized, the temperature range at which the conventional polymers are melt processed should overlap the temperature range in which the LCP forms an anisotropic melt. In that work, as in the other previously reported studies, the melts had lower viscosities than the pure thermoplastic polymers and, therefore, the processing temperature could be lower. The advantages of the subsequent reduction in the processing temperature include reduced energy consumption and less degradation of polymers that are sensitive to high temperatures. Also, a lowering of the viscosity allows for easier filling of large and complex molds in injection-molding applications.
The other principal effect observed in blends of LCPs with thermoplastic polymers is the utilization of the LCP as a reinforcement for the more flexible polymer. In numerous studies reported in the literature38−53 improved mechanical properties in the blends have been observed. Most of these studies have attempted to explain the changes in the mechanical properties in terms of the morphology of the LCP domains in the blends. The most widely used experimental technique to study the morphology has been the scanning electron microscope (SEM).
The fact that LCPs can act as reinforcing agents in a blend has led some workers to model the mechanical behavior of the blends using theories of composites. Thus, Kohli et al.47 showed that the modulus of highly drawn melts can be treated effectively by a simple rule of mixtures. That is, the modulus of a blend is given by
where υ1 and υ2 are the volume fractions of the two components in the blend and M1 and M2 are the corresponding modulus values of the two polymers. These same workers showed that the moduli values of samples in which the LCP forms spherical particles can be treated using an inverse rule of mixtures.
One of the main drawbacks of using LCPs as a reinforcement is the poor adhesion to the matrix polymer. This lack of adhesion between two immiscible polymers is a general phenomenon and appears to be common to all blends which contain LCPs. An approach to address this problem was proposed by Akkapeddi et al.54 They proposed blending thermotropic oligomers and isotropic polymers in the presence of a particulate material such as talc or silica. The purpose of the particulate is twofold. First, it appears to reduce the phase separation between the thermotropic oligomer and the matrix polymer. Second, its presence helps improve the dispersion of the oligomer in the polymer. The final blend has an increased tensile modulus, tensile strength and abrasion resistance compared to blends in which the particulate was not used.
Summarizing the work on immiscible blends which contain LCPs, two interesting phenomena have been demonstrated in the literature. First, the addition of an LCP to a thermoplastic polymer can be used to lower the melt viscosity, allowing for lower processing temperatures and, perhaps, easier mold filling. The second phenomenon which has been described is the use of LCPs as reinforcing agents. This observation has been made with several thermoplastic polymers, and the processing conditions for obtaining this morphology have been defined.
The biggest technical issue which has prevented more widespread use of these blends is the lack of adhesion between the LCP and the matrix polymer. Additional work needs to be focused on solving this problem before the full potential of these blends can be realized. Effective ways to compatibilize the two polymers need to be further developed. This is a general effect of blends of two immiscible high temperature polymers, and additional work is needed in this area.
1.4.3 Semi-interpenetrating networks
The idea behind semi-interpenetrating networks (semi-IPNs) is to combine the processability of thermoplastic polymers with the high temperature performance of cross-linked thermosetting materials. Such mixtures should possess the desirable features of both types of materials. Most of the semi-IPNs contain continuous phases, and the components are immiscible at the molecular level.
St. Clair et al.55, 56 used the concept of sequential semi-IPNs to produce two unique IPNs which are based on acetylene-terminated imide oligomers and thermoplastic polyimides, particularly a material designated LARC-TPI. Pater and other57−59 used the concept of simultaneous semi-IPNs to make semi-IPNs which are based on PMR-15 mixed with such materials as LARC-TPI, NR150B2 and Thermid 600 polyimides. Other examples of semi-IPNs which fit into the category of high temperature materials include thermoplastic modified bismaleimides (BMI). As one example, BMI has been mixed with condensation polyimides such as PI2028060 or with terminated poly (arylene ether ketone) oligomers.58 The observed miscibility in semi-IPNs which are based on thermoplastic BMIs was found to be further improved when a BMI and thermoplastic both prepared from aromatic diamines were blended.61, 62
1.5 Conclusions
Several common themes have emerged in all of the systems that have been discussed. The first is related to the high temperature involved in processing high temperature blends. This high temperature means that possible degradation of the polymers is always an issue. Such degradation may often compete with this processing of the polymer mixture. A second issue is the control of the phase separation in the blends. Sometimes, neither complete miscibility nor immiscibility is desired in blend systems, but instead some intermediate situation. Thus, complete miscibility may not be the optimum situation, but enough adhesion between the two phases must be present to obtain a desired physical property profile. The mechanism of the transformation from a miscible to immiscible system and vice versa is presently not well understood. The thermodynamics of phase separation may be understood, but the kinetics is still to be developed.
The morphology which results from the phase separation of a binary polymer mixture during a transition across the phase boundary is an area of considerable interest, but is one hardly addressed in the case of high temperature polymer mixtures. The resultant rheology of phase separated polymers will be a function of the morphology. Prediction of the rheological changes which accompany the morphological changes is an area where further research is needed.
In addition, in blends which contain two-phase separated polymers, adhesion of the two phases is of primary importance for utilizing the blend. Additional research needs to be done to understand the features that provide compatibilization in high temperature polymer blends and the optimum structure of compatibilizing agents for such blends. An example of this has been presented in the case of blends which contain liquid crystal polymers, but the same point is true of all blends based on immiscible high temperature polymers.
Better theoretical models and predictive tools need to be developed which can be used to understand the structural features in polymers which lead to miscibility. Presently, most of the information obtained in this regard has been done through trial and error. Then, when a miscible system is found, efforts are expended to rationalize the observed miscibility. A different approach would be to develop theoretical schemes which can predict miscibility beforehand and, then, experimentally produce such systems. Unfortunately, the present computer technology does not yet adequately allow for the incorporation of explicit molecular details in such models. When such developments become available, a major step in the development of high temperature polymer blends will be achieved.