
Chapter 10
Major Anticancer Chemotherapies
10.1 Introduction
Diseases desperate grown
By desperate appliance are relieved,
Or not at all
William Shakespeare, Hamlet, Act IV, Scene 3
Cancer has been treated surgically since antiquity. Following the discovery of X-rays in 1896, physicians and scientists were quick to adapt radiation as an anticancer therapy. With the development of modern systemic chemotherapy in the latter half of the 20th century, the treatment of solid tumors came to follow a “cut-burn-poison” model. In the previous chapter we covered the “poison” of chemotherapy, and we discuss the “burn” of radiation in the next. Here, we follow the previous chapter with additional history and background on some of the particular poisons in common use. The name is apt—white arsenic, a favorite poison of the Middle Ages, was used effectively against chronic leukemia in the late 1800s. Although arsenic subsequently fell out of favor as an anticancer drug, arsenic trioxide was recently revived as a highly effective therapy for promyelocytic leukemia (PML) [39, 53]. Indeed, arsenicals enjoy a colorful history as a therapeutic for a variety conditions that dates to antiquity [53]. Just as appropriately, modern cancer chemotherapy arose from poison gas research that followed the First World War and blossomed during the second.
In early 1942, several investigators at Yale University, in coordination with the wartime Office of Scientific Research and Development, began studying the nitrogen mustards [19], a class of chemical derived from the sulfur mustards, the infamous “mustard gas” that had been used to terrible effect on the battlefields of World War I. By this time, it was well established that both mustard gas and its less reactive cousin, nitrogen mustard, were potently cy-totoxic to rapidly proliferating cells, particularly those of the hematopoietic (blood-forming) system and the gastrointestinal tract. Moreover, the effect of nitrogen mustard on cells appeared similar to that of X-rays (and indeed, both induce similar breaks in the DNA, although by different mechanisms). Motivated by such data, Gilman and colleagues took, at first, a single mouse with a transplanted lymphoid tumor and treated it with nitrogen mustard until the tumor completely disappeared. After some time, the tumor recurred and was treated again, only this time regression was less complete. When it recurred a third time, no further treatment affected it.
Despite this and other studies with similar results, the compound eventually was tested in humans.1 Initially, a patient with terminal lymphosarcoma, moribund and riddled with tumors, was selected and treated to extraordinary effect, his tumors shrinking and disappearing in days. However, mirroring the mouse, the cancer recurred, and a second course gave a diminished response. When the disease began to progress again, further treatment had little effect. The clinical trajectories of these early trials in both mouse and human foreshadowed the clinical experience of chemotherapy in the decades to come, and it remains a common trajectory to this day: the development of treatment resistance has always been the hobgoblin of chemotherapy (see Chapter 9 for more details.)
Because of the secrecy of the chemical gas program, these and other trials were conducted behind closed doors, with patient charts even reporting treatment by “0.1 mg. per kg. compound X given intravenously” [19]. In 1946 wartime secrecy restrictions were lifted, and a series of publications reporting these results ushered in the modern era of cancer chemotherapy. In the next two decades, a host of similar agents with antitumor activity would be developed. Our goal in this chapter is to introduce the biological and medical aspects of some particular chemotherapies in widespread use, as well as some particular mathematical treatments, contra the more general treatment given in the prior chapter.
10.2 Alkylating and alkalating-like agents
The alkylating agents make up the largest and oldest class of chemothera-peutics. They target the DNA, RNA, and proteins, although their interaction with DNA is generally considered to be the dominant mechanism for cytotox-icity. Many alkylating agents are small organic molecules with two reactive functional groups. The first forms a covalent link to a nitrogenous base in the DNA, forming what is called a monofunctional adduct. The second functional group can then react with another base, covalently cross-linking them to form a bifunctional adduct. These cross-links may be either intra-strand or cross-strand, and they interfere with DNA processing machinery (e.g., for replication and (or) transcription) and often distort the helical structure of the DNA.
Alkylating agents are widely used to treat various leukemias, but they have limited selectively for tumor cells, often causing “collateral” damage to non-target cells. What selective cyotoxicity they possess is believed to be primarily due to (1) high proliferation rates of tumor cells, and (2) deficiencies in DNA repair mechanisms in tumor cells. The latter is particularly important in testicular cancer, as defective DNA repair is linked to hypersensitivity to cisplatin in this disease [33].
10.2.1 Nitrogen mustards
The nitrogen mustards were the first anticancer chemotherapies, and are derivatives of the sulfur mustards, which, as mentioned above, are chemical warfare agents. Their antitumor activity was discovered by a group studying such agents during World War II, and they were secretly tested on the first human subjects in 1942. It was not until secrecy restrictions were lifted in 1946 that these investigations were published publicly [19]. They are classified as alkylating agents and produce DNA cross-links by the mechanism outlined above. The first agents studied were tris(β-chloroethyl)amine and methyl-bis(β-chloroethyl)amine (mustine). All other drugs in the nitrogen mustard class are based on mustine, and the basic structure of such drugs is shown in Figure 10.1. Nitrogen mustards covalently bind to guanine residues in the DNA to form adducts and form cross-links between opposite strands of DNA.
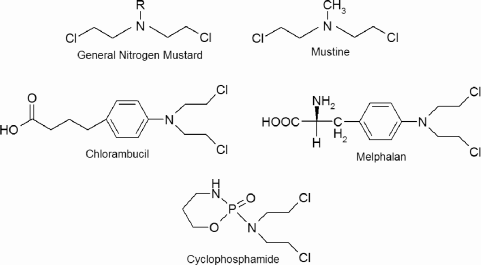
The basic structure of the nitrogen mustards, along with some of the specific drugs in this class.
In the 1950s and 60s two major classes of nitrogen mustard derivatives were developed specifically to target tumor cells. The reactivity of the nitrogen mustard base is also reduced in these drugs, which allows more of the drug to reach the DNA target intact. The aromatic nitrogen mustards replace the methyl group of nitrogen mustard with an (R-Phe) group, stabilizing the nitrogen and reducing drug reactivity. Chlorambucil and melpha-lan are two such drugs. Melphalan (L-phenylalanine mustard) incorporates the amino acid residue L-phenylalanine, allowing it to be taken up by the L-phenylalanine active transport mechanism. Therefore, it may selectively target cells undergoing rapid protein synthesis, such as rapidly proliferating tumor cells. An advantage of the aromatic nitrogen mustards is that they can be given orally.
The oxazaphosphorines are nitrogen mustard pro-drugs—inactive agents that become activated by metabolic mechanisms after introduction to the body. The major drug in this class is cyclophosphamide, an inactive transport form of the drug with a much higher therapeutic range than do agents with direct alkylating ability. Ifosfamide is a derivative of cyclophosphamide that is metabolized more slowly and has a somewhat different therapeutic profile. Moreover, fractionation of ifosfamide dose schedules and delivery by continuous infusions of between 1 and 5 days decreases off-target toxicity, increases the maximum allowable dose, and improves therapeutic efficacy.
The uptake and efflux mechanisms for many of the nitrogen mustards have been well quantified, and many enter the cell by carrier-mediated transport. Mustine enters cells primarily by active transport mediated by the choline carrier; choline is a natural substance similar in structure to mustine [20].
Uptake of melphalan appears to be mediated by two separate active transport mechanisms involved in amino acid transport [4, 21], while efflux likely occurs only by simple diffusion. Goldenberg et al. [21] measured cell-to-interstitium concentration ratio for melphalan to be 11.0 ± 3.1 in physiological conditions, and Begleiter et al. [2] showed that about 80% of intracellular drug was exchangeable with the extracellular compartment. The rate of change in intracellular concentration of the drug can be described as the sum of two Michaelis-Menten terms for uptake and a first-order term for efflux:
where C(t) and [S] are intra- and extracellular drug concentrations, respectively, Vmax1 = 1.1 ± 0.4 × 10−16 mol/cell/min, Km1 = 8.0 ± 3.0 × 10−5 M, Vmax2 = 2.2 ± 2.4 × 10−17 mol/cell/min, Km2 = 1.0 ± 0.7 × 10−5 M [4], and k = 0.13 ± 0.05 [2].
Cyclophosphamide is similarly transported into the cell by two saturable processes [22], and the kinetics of DNA damage by cyclophosphamide have been studied both in vitro [7] and in vivo [9, 50].
Chlorambucil is more water-soluble than other nitrogen mustards [49], and transport across the membrane occurs by passive diffusion, with intracellular concentration reaching equilibrium with the extracellular concentration very rapidly [1]. The kinetics of chlorambucil metabolism and binding to DNA, RNA, and proteins have been studied quantitatively by several groups [1, 27].
For a comprehensive quantitative review of membrane transport of the nitrogen mustards the reader may consult the 1980 review by Goldenberg and Begleiter [20]. To our knowledge, the nitrogen mustards have not been studied using more complicated dynamical models.
10.2.2 Platinum-based drugs
The cytotoxic action of cisplatin (cis-diamminedichloroplatinum(II)) was accidently discovered in 1965. It was first used in humans as an anticancer agent in 1971 and approved by the FDA in 1978 [33]. It remains a frontline treatment for ovarian and testicular cancer and has been the focus of several mathematical models. Metastatic testicular cancer in particular is often hypersentitive to cisplatin, and cisplatin-based chemotherapy regimes have increased 5-year survival rates from 5% to over 80% [33] making it one of the few solid metastatic tumors for which systemic chemotherapy is curative. All other platinum-based drugs are based on cisplatin, which has the chemical structure ; the structures of this and several other platinum drugs are shown in Figure 10.2.
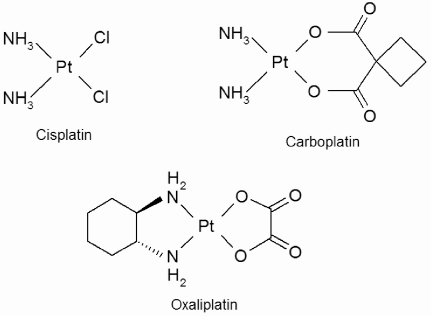
Cisplatin’s major mechanism of action as a cytotoxic agent requires intracellular activation of the molecule by aquation (displacement by water) of one of the two Cl groups. This allows activated cisplatin to bind covalently to purine DNA bases to form a cisplatin-DNA adduct. Aquation of the other Cl group leads it to bind to a second DNA base, thus cross-linking the DNA strand [33]. The formation of such mono- and bifunctional adducts interferes with replication and transcription and ultimately leads to cellular apoptosis by a sequence of events that is not yet fully understood.
Platinum-based drugs are often classified as alkylating agents, and while their mechanism of action is similar to alkylating agents they contain no alkyl group and therefore such a classification is technically incorrect.
Cisplatin can cause severe damage to the kidneys (nephrotoxicity) and gastrointestinal tract. Carboplatin (cis-diammine(l,l,-cyclobutanedicarboxylato)-platinum(II)), a second-generation compound, replaces the chlorine groups with a more stable leaving group, and has vastly reduced nephrotoxicity compared to cisplatin [33, 35]. Myelosuppression (inhibition of bone marrow) is the dose-limiting toxicity for carboplatin. Cisplatin and carboplatin have similar efficacy in the treatment of ovarian cancer [33].
Cisplatin is highly polar, which limits its diffusion across the non-polar cell membrane. Copper transport proteins appear to determine cisplatin uptake and efflux. Copper transporter-1 (CTR1) plays a major role in uptake, and copper efflux proteins remove cisplatin from the cell. Intracellular thiols detoxify cisplatin, and DNA-repair mechanisms can repair cisplatin lesions [33].
There is a great deal of quantitative information on the kinetics of cisplatin-DNA binding and cross-linking as well as binding kinetics between various thiols and cisplatin. Several models of in vitro uptake and cytotoxicity have been developed. The relatively simple mechanism of action and the relative wealth of quantitative data for cisplatin and previous work make it an excellent choice for modeling.
There are several studies on the pharmacokinetics of plasma cisplatin, which can be reasonably described by a one-compartment model. The kinetics of DNA binding and cross-linking by both cisplatin and carboplatin were quantified by Knox et al. [35], and later studies have quantified the kinetics of cisplatin binding to thiols such as glutathione [8, 25] and metallothionein [24]. Based on their experimental data, Sadowitz et al. proposed a simple mathematical model accounting for cisplatin transport across the cell membrane, detoxification by intracellular thiols, and binding to the DNA [44]. El-Kareh and Secomb have developed several dynamical models of the uptake kinetics and cytotoxicity of cisplatin [15, 16].
10.2.3 Nitrosoureas
The nitrosoureas were developed in the 1960s, following the finding by the Cancer Chemotherapy National Service Center screening program that ni-trosoguanidines had weak activity against intraperitoneal leukemias in mice. The nitrosoureas are structurally similar, are very lipophilic, have a low extent of ionization, and do not readily bind plasma proteins. Therefore, they can cross the blood-brain barrier and enter the central nervous system (CNS). Interest in these agents began with the discovery that the drug was partially effective against leukemia cells implanted intracerebrally [45]. At that time, no other agents had activity against CNS tumors, so this finding spurred development of this class of drugs.
The two most successful and commonly used nitrosoureas are 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU, carmustine) and 1-(2-chloroethyl)-3-cyclo-hexyl-1-nitrosourea (CCNU, lomustine). Because of their ability to cross the blood-brain barrier, they are frequently used to treat CNS malignancies, including malignant gliomas (primary brain tumors). Delayed bone-marrow suppression is the dose-limiting toxicity. While there is conflicting data, the evidence overall suggests that the nitrosoureas are equally cytotoxic to non-proliferating and proliferating cells [11, 26].
In the body, BCNU and CCNU break down fairly rapidly into 2-chloro-ethyldiazene hydroxide and isocyanate [3, 49]. The former undergoes further reactions to form a chloroethyl carbonium ion which in turn forms mono-and bifunctional DNA adducts [49]. In solution, the half-lives for BCNU and CCNU have been variously measured as 46 and 53 minutes [52], and 57 and 64 minutes [3], respectively. Both agents appear to cross the cell membrane by passive diffusion, and the isocyanate decomposition product readily enters cells by diffusion. However, the carbonium ion that alkylates the DNA is not taken up [3], and therefore breakdown of the nitrosoureas before they have reached their cellular target may represent a barrier to therapy. This is one aspect of these drugs that could be studied with a theoretical model.
Weikam and Deen [52] developed a quantitative kinetics scheme that described DNA alkylation by the nitrosureas that successfully explained in vitro dose-response curves in 9L rat glioma cells. Kohn [34] reports some quantitative data on the kinetics of DNA crosslinking by nitrosoureas.
10.2.4 Methylating agents
Methylating agents form monofunctional DNA adducts and typically add a methyl group to guanine residues. Important methylating agents are dacar-bazine, procarbazine, and temozolomide.
10.3 Antitumor antibiotics
In general, the term antibiotic can refer to any compound derived from a microbial source—like bacteria of the genus Streptomyces, for example—that kills cells. A number of such compounds effectively kill cancer cells and are hence called antitumor or antineoplastic antiobiotics to distinguish them from their more famous siblings, the antimicrobial antibiotics. A number of such antineoplastic antibiotics are in common use in chemotherapy. Their primary modes of action are to inhibit nucleic acid function in some way.
10.3.1 Anthracyclines
The anthracyclines are a class of antitumor antibiotics derived from Step-tomyces bacteria with a variety of mechanisms of action. They are currently one of the most useful and widely used chemotherapies, with activity against a very broad range of neoplasms. The most commonly used are daunorubicin, doxorubicin, idarubicin, and epirubicin. All are very similar to doxorubicin, which remains the standard and most commonly used anthracycline.
Doxorubicin has received a great deal of attention from theorists, who have proposed many competing mathematical models for treatment using this agent [12, 14, 17, 28, 41, 46]. (For details, see Chapter 9.) Its plasma pharma-cokinetics have been quantified using two- and three-compartment models [13, 23, 42].
10.3.2 Mitomycin-C
One agent of particular interest is mitomycin-C, which produces intra-strand DNA cross-links by a complex mechanism that requires enzymatic reduction for activation [49]. The requirement for bioreduction suggests that mitomycin-C, unlike most other chemotherapeutics, may have significant activity in hypoxic tumors since such environments are reducing. Indeed, Te-icher et al. have consistently found mitomycin to be toxic to hypoxic cells [48, 47], and it is equally effective against proliferating and non-proliferating cells [11].
10.3.3 Bleomycins
The bleomycins are a family of glycopeptides first isolated from Strep-tomyces verticullus in 1966. They are used in combination chemotherapy against several lymphomas, head and neck cancers, and germ-cell tumors [6]. Bleomycin is most successfully used in cisplatin-etoposide-bleomycin combination therapy, which is 80% curative for testicular cancer [6, 33]. The bleomycins are fairly large molecules (MW ~ 1500 Da) with a common core structure and a variable sugar moiety and positively charged tail [6]. The commercial formula is called Blenoxane and is a mixture primarily of bleomycins A2 and B2. The term bleomycin therefore refers to a variety of similar molecules, although “bleomycin” is typically regarded as a single agent in the literature.
Bleomycin targets the DNA by several related pathways that induce single and double stranded DNA breaks. Such damage is very similar to that induced by ionizing radiation (see Chapter 11). Bleomycins are hydrophilic and do not diffuse across the cell membrane. They bind to a surface receptor and enter the cell by receptor-mediation endocytosis [6, 37]. Although membrane transport of bleomycins is inefficient, internalization of only a few hundred molecules is needed to cause cell death [37].
Once inside the cell, bleomycin must be activated by an oxygen species and a reduced transition metal co-factor (nearly always Fe2+). The activation cascade and subsequent mechanism for DNA damage is complex and variable. For a thorough characterization of the process that includes a partial quantification of the kinetics the reader may consult the review by Burger [5]. In brief, reduced iron (Fe2+) and O2 form a complex with bleomycin. The O2-Fe2+-BLM complex equilibrates with . The latter complex is converted to activated bleomycin, Fe3+-BLM-OOH [5]. Several other pathways involving superoxide and hydrogen peroxide can also generate activated bleomycin [5].
In the absence of a DNA target, activated bleomycin is unstable and undergoes irreversible molecular suicide. Otherwise, it associates with DNA in a sequence specific manner. That is, it targets GpC or GpT sequences with a specificity that is also affected by nearby nucleotides [5]. Bleomycin also tends to target DNA linker regions (between nucleosomes) of genes being actively transcribed [6], and “hot spots” for attack appear to exist [5]. Once associated with the DNA, bleomycin always attacks the C4ʹ position of the doexyribose sugar, removing the hydrogen and forming a DNA radical. A series of radical reactions then occur following one of two pathways. If O2 is available a break in the DNA backbone is created. If not, an abasic site in the DNA (monomer location lacking either a purine or pyrimidine) is produced [6, 5].
Following formation of the initial DNA lesion, bleomycin is rapidly reactivated. Since reactivation apparently occurs faster than dissociation of bleomycin from DNA [37], a second lesion is often formed near the first, accounting for the large number of double-stranded DNA breaks induced by bleomycin. It has been estimated that a single bleomycin molecule can induce 8–10 DNA strand breaks [37], and 10–20% of DNA cleavage events are double-stranded breaks [5].
The in vitro dose-response curve for bleomycin toxicity is always concave upward (it plateaus) when plotted on a semilogarithmic plot.
10.4 Antimetabolites
The principle antimetabolites in cancer chemotherapy are the antifolates, which primarily interfere with nitrogenous base synthesis, thereby inhibiting DNA and RNA synthesis. The antifolates are generally considered to be S-phase specific.
Folic acids are part of the biosynthetic pathway that produces thymidylate, or deoxythymidine monophosphate (dTMP), one of the nucleosides required for DNA synthesis. The pathway converts dUMP (deoxyuridine monophos-phate) to dTMP. The key enzyme is thymidylate synthase (TS), which oxidizes N5,N10-methylenetetrahydrofolate (5,10-CH2FH4) to 7,8-dihydrofolate (7,8-FH2), after which tetrahydrofolate must be regenerated. This is accomplished in two steps: the first is catalyzed by dihydrofolate reductase (DHFR) and the second by serine hydroxymethyl transferase.
Antifolates disrupt dTMP synthesis either by directly targeting TS or by inhibiting DHFR and thus depleting the 5,10-CH2FH4 necessary for TS activity. Tetrahydrofolates are necessary for de novo purine synthesis, which is also blocked by DHFR inhibition.
DHFR inhibitors have been in use for over 60 years, beginning with am-inopterin in 1948. It was replaced by methotrexate (MXT) in 1956, which is still commonly used. Another compound, 5-fluorouracil, is a pyrimidine analog that binds to and permanently deactivates TS. As a uracil analogue, it also has antitumor activity through incorporation into RNA.
10.5 Mitotic inhibitors
The mitotic inhibitors primarily interfere with microtubule dynamics, generating a dysfunctional mitotic spindle causing G2/M phase block or death during M phase. Therefore, although they do affect cells during interphase, they are considered M-phase specific drugs. The two major classes of mitotic inhibitors are the vinca alkaloids and taxanes, although other mitotic inhibitors exist. Such drugs have been the (at least indirect) focus of many mathematical models studying the efficacy of cell-cycle specific chemotherapies (see Chapter 9).
10.5.1 Taxanes
The taxanes are a relatively recent development in cytotoxic chemotherapy, only coming into widespread use in the 1990s, although their development spanned several decades. In the 1960s, National Cancer Institute (NCI) screening programs found that the crude extract of Pacific yew tree (Taxus brevifolia) bark had anticancer activity. In 1971, the active ingredient pacli-taxel was identified, and in 1983 clinical trials began. Trials were hampered by hypersentivity reactions, which were eventually tempered with pre-treatment steroids and a switch to long (e.g., 24 hours) infusion times. Finally, in 1992 the FDA approved paclitaxel for advanced ovarian cancer [40].
The two major taxanes in clinical use are paclitaxel (Taxol) and docetaxel (Taxotere). Like the vinca alkaloids (see below), they are considered mitotic spindle inhibitors, although they act by stabilizing, rather than inhibiting, microtubule formation. Both pacli- and docetaxel comprise a complex taxane ring system and an ester side chain. The drugs differ in this side chain [40].
Paclitaxel acts on tubulin, a major building-block of the cytoskeleton. Tubu-lin is a dimer, with subunits α and β, that polymerize to form microtubules. Microtubules are normally in dynamic equilibrium with tubulin dimers, and paclitaxel binds reversibly to tubulin (preferably the β subunit) shifting the equilibrium to favor microtubule formation [40].
Paclitaxel binding greatly increases microtubule stability, but microtubules are inherently dynamic polymers that undergo constant and rapid remodeling. The two remodeling processes are dynamic instability, where micro-tubules transition between lengthening and shortening phases at their ends, and treadmilling, where tubulin is added to one end of the microtubule at the same rate at which it is removed from the other [31]. At low concentrations, paclitaxel inhibits these dynamic processes, and the microtubules that form are dysfunctional. Microtubules are one of the polymers that make up the cellular cytoskeleton, and microtubule dysfunction can affect cell motility, intracellular transport, and response to growth factors. However, their most dramatic role is in forming the mitotic spindle during mitosis, which has a deranged morphology in taxane-treated cells [31]. The taxanes are therefore considered to be M-phase specific, inhibiting cell proliferation primarily by blocking the cell cycle at the G2-M phase transition [43]. Cytotoxicity increases three-fold from G1 to G2 [10]. Taxanes also may retard the cell’s response to growth factors during interphase [43].
Cells treated with paclitaxel develop abnormal asters and bundles of micro-tubules. The asters appear in G2/M phase and have not been associated with toxicity. On the other hand, microtubule bundles form throughout the cell cycle and are associated with toxicity, while decline of these structures correlates with paclitaxel resistance [43]. However, the jury is still out regarding the precise mechanism of cytotoxicity, and more subtle effects than these on microtubule dynamics may be clinically more important [31].
The taxanes are characterized by biphasic plasma pharmacokinetics and are highly protein-bound [40]. Transport across the membrane occurs by passive diffusion, but P-glycoprotein expression associated with multi-drug resistance (MDR) causes active efflux of the drug [40]. At equilibrium, the intracellular drug concentration is several hundred times greater than extracellular concentration, as nearly all intracellular drug binds to cellular components [36]. Kuh et al. [36] developed a model of intracellular pharmacokinetics for paclitaxel. Panetta [38] proposed a mathematical model of paclitaxel treatment of an in vivo tumor consisting of proliferating and quiescent cells (see Chapter 9).
10.5.2 Vinca alkaloids
The vinca alkaloids were isolated from the Madagascar periwinkle plant (Catharanthus roseus, formerly Vinca rosea) in the 1960s. Periwinkle had been used extensively in folk medicine to treat various disorders, including diabetes. This prompted laboratory investigation, and although the anti-diabetic activity failed to be substantiated, the vinca alkaloids were found to suppress the bone marrow and have antileukemic activity [29]. The earliest, best studied, and most widely used vinca alkaloids are vincristine and vinblastine.
At high concentrations, the vinca alkaloids cause concentration-dependent disintegration of microtubule polymers, and this was the first mechanism for cytotoxicity identified. However, at much lower concentrations the vinca alkaloids kinetically stabilize microtubules, causing subtle disorganization of the mitotic spindle [30]. This inhibition of mitotic spindle function causes cells to become arrested at metaphase; this in turn correlates strongly with inhibition of cell proliferation.
Therefore, while the taxanes induce microtubule polymerization and the vinca alkaloids inhibit polymerization, both classes of drugs inhibit micro-tubule dynamics at low concentrations, which may represent a common mechanism for their clinical anticancer activity [31, 32]. Interestingly, the differential effects at different concentrations may be explained (for vinca alkaloids) by the fact that vinblastine has two binding sites. At low concentrations, it binds with high affinity to sites at the microtubule ends (Kd = 1.9 µm, 16-17 binding sites / microtubule), thus inhibiting microtubule remodeling without significantly affecting overall mass. At higher concentrations it binds to low affinity sites on the microtubule surface to cause depolymerization [30].
The vinca alkaloids also appear to have a profound ability to destroy tumoral vasculature. The cytoskeleton of endothelial cells can be very rapidly destabilized by vinca alkaloids, leading to cell death and loss of blood flow to the tumor within minutes. Moreover, endothelial cells of the developing and immature tumor vasculature appear to be selectively targeted [32]. Jordan and Wilson [32] published a nice review of agents that target microtubules.
Like the taxanes, the vinca alkaloids bind extensively to cellular components, and at equilibrium the intracellular concentration is much greater than the extracellular concentration [30].
10.6 Non-cytotoxic and targeted therapies
A number of treatment modalities other than cytotoxic chemotherapies exist, as we briefly discuss here. Prostate and breast cancers are treated hormonally: androgen deprivation by castration or anti-androgens has long been the cornerstone of prostate cancer treatment (see Chapter 5 for a thorough discussion), and anti-estrogens are used to prevent breast cancer recurrence. More recently, monoclonal antibodies targeting either cancer-specific, mutated proteins, or cytokines involved in cancer progression, have also been used. In [18], the authors discussed at length another targeted therapy, the treatment of chronic myelogenous leukemia with the drug, imatinib.
References
[1] Bank BB, Kanganis D, Liebes LF, Silber R: Chlorambucil pharmacoki-netics and DNA binding in chronic lymphocytic leukemia lymphocytes. Cancer Res 1989, 49:554–559.
[2] Begleiter A, Grover J, Goldenberg GJ: Mechanism of efflux of melphalan from L5178Y lymphoblasts in vitro. Cancer Res 1982, 42:987–991.
[3] Begleiter A, Lam HP, Goldenberg GJ: Mechanism of uptake of ni-trosoureas by L5178Y lymphoblasts in vitro. Cancer Res 1977, 37:1022– 1027.
[4] Begleiter A, Lam HY, Grover J, Froese E, Goldenberg GJ: Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res 1979, 39:353–359.
[5] Burger RM: Cleavage of nucleic acids by bleomycin. Chem Rev 1998, 98:1153–1170.
[6] Chen J, Stubbe J: Bleomycins: Towards better therapeutics. Nat Rev Cancer 2005, 5:102–112.
[7] Crook TR, Souhami RL, McLean AE: Cytotoxicity, DNA cross-linking, and single strand breaks induced by activated cyclophosphamide and acrolein in human leukemia cells. Cancer Res 1986, 46:5029–5034.
[8] Dabrowiak JC, Goodisman J, Souid AK: Kinetic study of the reaction of cisplatin with thiols. Drug Metab Dispos 2002, 30:1378–1384.
[9] DeNeve W, Valeriote F, Edelstein M, Everett C, Bischoff M: In vivo DNA cross-linking by cyclophosphamide: Comparison of human chronic lymphatic leukemia cells with mouse L1210 leukemia and normal bone marrow cells. Cancer Res 1989, 49:3452–3456.
[10] Donaldson KL, Goolsby GL, Wahl AF: Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int J Cancer 1994, 57:847–855.
[11] Drewinko B, Patchen M, Yang LY, Barlogie B: Differential killing efficacy of twenty antitumor drugs on proliferating and nonproliferating human tumor cells. Cancer Res 1981, 41:2328–2333.
[12] Eikenberry S: A tumor cord model for doxorubicin delivery and dose optimization in solid tumors. Theor Biol Med Model 2009, 6:16.
[13] Eksborg S, Strandler HS, Edsmyr F, Näslund I, Tahvanainen P: Phar-macokinetic study of i.v. infusions of adriamycin. Eur J Clin Pharmacol 1985 28:205–212.
[14] El-Kareh AW, Secomb TW: A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxoru-bicin to tumor cells. Neoplasia 2000, 2:325–338.
[15] El-Kareh AW, Secomb TW: A mathematical model for cisplatin cellular pharmacodynamics. Neoplasia 2003, 5:161–169.
[16] El-Kareh AW, Secomb TW: A theoretical model for intraperitoneal delivery of cisplatin and the effect of hyperthermia on drug penetration distance. Neoplasia 2004, 6:117–127.
[17] El-Kareh AW, Secomb TW: Two-mechanism peak concentration model for cellular pharmacodynamics of doxorubicin. Neoplasia 2005, 7:705– 713.
[18] Everett RA, Zhao Y, Flores KB, Kuang Y: Data and implication based comparison of two chronic myeloid leukemia models. Math Biosc Eng 2013, 10:1501–1518.
[19] Gilman A: The initial clinical trial of nitrogen mustard. Am J Surg 1963, 105:574–578.
[20] Goldenberg GJ, Begleiter A: Membrane transport of alkylating agents. Pharmacol Ther 1980, 8:237–274.
[21] Goldenberg GJ, Lam HY, Begleiter A: Active carrier-mediated transport of melphalan by two separate amino acid transport systems in LPC-1 plasmacytoma cells in vitro. J Biol Chem 1979, 254:1057–1064.
[22] Goldenberg GJ, Land HB, Cormack DV: Mechanism of cyclophos-phamide transport by L5178Y lymphoblasts in vitro. Cancer Res 1974, 34:3274–3282.
[23] Greene RF, Collins JM, Jenkins JF, Speyer JL, Myers CE: Plasma phar-macokinetics of adriamycin and adriamycinol: Implications for the design of in vitro experiments and treatment protocols. Cancer Res 1983, 43:3417–3421.
[24] Hagrman D, Goodisman J, Dabrowiak JC, Souid AK: Kinetic study on the reaction of cisplatin with metallothionein. Drug Metab Dispos 2003, 31:916–923.
[25] Hagrman D, Goodisman J, Souid AK: Kinetic study on the reactions of platinum drugs with glutathione. J Pharmacol Exp Ther 2004, 308:658– 666.
[26] Hahn GM, Gordon LF, Kurkjian SD: Responses of cycling and non-cycling cells to 1,3-bis(2-chloroethyl)-1-nitrosourea and to bleomycin. Cancer Res 1974, 34:2373–2377.
[27] Hill BT: Studies on the transport and cellular distribution of chloram-bucil in the Yoshida ascites sarcoma. Biochem Pharmacol 1972, 21:495– 502.
[28] Jackson TL: Intracellular accumulation and mechanism of action of dox-orubicin in a spatio-temporal tumor model. J Theor Biol 2003, 220:201– 213.
[29] Johnson IS, Armstrong JG, Gorman M, Burnett JP Jr.: The vinca alkaloids: A new class of oncolytic agents. Cancer Res 1963, 23:1390– 1427.
[30] Jordan MA, Thrower D, Wilson L: Mechanism of inhibition of cell proliferation by vinca alkaloids. Cancer Res 1991, 51:2212–2222.
[31] Jordan MA, Toso RJ, Thrower D, Wilson L: Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA 1993, 90:9552–9556.
[32] Jordan MA, Wilson L: Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004, 4:253–265.
[33] Kelland L: The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007, 7:573–584.
[34] Kohn KW: Interstrand cross-linking of DNA by 1,3-bis(2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res 1977, 37:1450–1454.
[35] Knox RJ, Friedlos F, Lydall DA, Roberts JJ: Mechanism of cytotoxicity of anticancer platinum drugs: Evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA. Cancer Res 1986, 46:1972–1979.
[36] Kuh HJ, Jang SH, Wientjes MG, Au JL: Computational model of intracellular pharmacokinetics of paclitaxel. J Pharmacol Exp Ther 2000, 293:761–770.
[37] Mir LM, Tounekti O, Orlowski S: Bleomycin: Revival of an old drug. Gen Pharmac 1996, 27:745–748.
[38] Panetta JC: A mathematical model of breast and ovarian cancer treated with paclitaxel. Math Biosci 1997, 146:89–113.
[39] Papac RJ: Origins of cancer therapy. Yale J Biol Med 2001, 74:391–398.
[40] Pazdur R, Kudelka AP, Kavanagh JJ, Cohen PR, Raber MN: The tax-oids: Paclitaxel (Taxol) and docetaxel (Taxotere). Cancer Treat Rev 1993, 19:351–386.
[41] Ribba B, Marron K, Agur Z, Alarcón T, Maini PK: A mathematical model of doxorubicin treatment efficacy for non-Hodgkin’s lymphoma: Investigation of the current protocol through theoretical modelling results. Bull Math Biol 2005, 67:79–99.
[42] Robert J, Illiadis A, Hoerni B, Cano JP, Durand M, Lagarde C: Phar-macokinetics of adriamycin in patients with breast cancer: Correlation between pharmacokinetic parameters and clinical short-term response. Eur J Cancer Clin Oncol 1982, 18:739–745.
[43] Rowinsky EK, Donehower RC, Jones RJ, Tucker RW: Microtubule changes and cytotoxicity in leukemic cell lines treated with taxol. Cancer Res 1988, 48:4093–4100.
[44] Sadowitz PD, Hubbard BA, Dabrowiak JC, Goodisman J, Tacka KA, Aktas MK, Cunningham MJ, Dubowy RL, Souid AK: Kinetics of cis-platin binding to cellular DNA and modulations by thiol-blocking agents and thiol drugs. Drug Metab Dispos 2002, 30:183–190.
[45] Schabel FM Jr, Johnston TP, McCaleb GS, Montgomery JA, Laster WR, Skipper HE: Experimental evaluation of potential anticancer agents VIII. Effects of certain nitrosoureas on intracerebral L1210 leukemia. Cancer Res 1963, 23:725–733.
[46] Sinek JP, Sanga S, Zheng X, Frieboes HB, Ferrari M, Cristini V: Predicting drug pharmacokinetics and effect in vascularized tumors using computer simulation. J Math Biol 2009, 58:485–510.
[47] Teicher BA, Holden SA, al-Achi A, Herman TS: Classification of anti-neoplastic treatments by their differential toxicity toward putative oxygenated and hypoxic tumor subpopulations in vivo in the FSaIIC murine fibrosarcoma. Cancer Res 1990, 50:3339–3344.
[48] Teicher BA, Lazo JS, Sartorelli AC: Classification of antineoplastic agents by their selective toxicities toward oxygenated and hypoxic tumor cells. Cancer Res 1981, 41:73–81.
[49] Thurston DE: Chemistry and Pharmacology of Anticancer Drugs. Boca Raton, FL: CRC Press, 2006.
[50] Wang JY, Prorok G, Vaughan WP: Cytotoxicity, DNA cross-linking, and DNA single-strand breaks induced by cyclophosphamide in a rat leukemia in vivo. Cancer Chemother Pharmacol 1993, 31:381–386.
[51] Urano M, Fukuda N, Koike S: The effect of bleomycin on survival and tumor growth in a C3H mouse mammary carcinoma. Cancer Res 1973, 33:2849–2855.
[52] Weinkam RJ, Deen DF: Quantitative dose-response relations for the cytotoxic activity of chloroethylnitrosoureas in cell culture. Cancer Res 1982, 42:1008–1014.
[53] Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H: How acute promye-locytic leukaemia revived arsenic. Nat Rev Cancer 2002, 2:705–713.
1A ubiquitous myth, appearing widely in the literature and even in textbooks, holds that a 1943 wartime incident in Bari, Italy, was the inspiration for chemotherapy. The story goes that sailors and civilians exposed to mustard gas were observed to experience bone marrow suppression, giving physicians the idea of using the gas as chemotherapy. This tale is certainly false, as the first human trials had begun a year before the Bari incident.