
Chapter 10
Potentiometric Sensors
10.1 Introduction
Analytical determination of inorganic and organic ions is a matter of great interest to various areas such as biomedical sciences, environment monitoring and industry. Ion sensors respond to the demand of continuous ion monitoring, field analysis and in vivo ion determination.
Ion sensors rely on the general property of ions of bearing an electric charge. It is therefore not surprising that ion sensors were first developed based on electrochemical principles in the framework of potentiometry, that is, electrochemistry under equilibrium conditions. However, practical potentiometric ion sensors are not generally based on electrochemical oxidation or reduction processes because such processes display poor selectivity. Rather, potentiometric ion sensors are based on ion-transfer processes occurring at the interface of two immiscible phases. One phase is the test solution and the second phase is constructed so as to interact specifically with the analyte ion. The second phase, usually called an ion-sensitive membrane, can be in the solid state form or a liquid supported on an inert material. Due to the analyte interaction with the membrane, a nonuniform ion distribution appears at the membrane/solution interface. This results in a nonuniform electric charge distribution that gives rise to an electric potential difference. It is this potential difference that produces the response signal of potentiometric sensors. Such ion sensors are by tradition termed ion-selective electrodes (ISEs), although this name is not rigorously in agreement with the underlying physicochemical principles. That is why, the term potentiometric ion sensor will be used throughout this chapter to denote this kind of sensor.
Besides the determination of ions, potentiometric ion sensors can be used in an extensive series of applications involving the determination of nonionic compounds. Such applications have been developed by integrating an ion sensor with a component that reacts with the analyte to produce ions. In these devices, the ion sensor functions as a transducer that generates the response signal. Following this principle, sensors for gases that interact with aqueous solutions to form ions have been developed. Furthermore, if the sensing element contains an enzyme or living cells, biocatalytic potentiometric sensors can be developed providing that the biocatalytic reaction produces detectable ions. Enzymatic potentiometric sensors can be converted readily into immunosensors by including an immunoreactive component. Therefore, potentiometric ion sensors form the basis in the development of a broad range of chemical sensors.
One more point of interest regarding potentiometric ion sensors arises from the fact that sensing material and reagents developed in the framework of this area are also useful in other types of ion sensors such as optical ion sensors (Section 19.2).
Good introductions to the field of ion-selective electrodes are available in various advanced analytical chemistry textbooks. Comprehensive presentations of this topic can be found in a series of monographs and book chapters [1–4]. Practical applications of ion-selective electrodes are amply discussed in refs. [5–7]. Specialized applications in the field of organic analysis [8, 9], biology [10], and medicine [11] have also been comprehensively surveyed. IUPAC nomenclature recommendations for ion-selective electrodes can be found in ref. [12]. Recent advances in this field are presented in a series of review papers (see, e.g., refs. [13, 14]). An historical perspective of ion-selective electrode development is available in ref. [15].
10.2 The Galvanic Cell at Equilibrium
Electrochemical processes involve electron- and ion-transfer processes. An electron transfer between two species produces modification in the oxidation state of the reactants in what is called a redox reaction in the case in which the involved reactants are ionic or molecular species in the solution. When the donor or acceptor species are macroscopic conductor materials, the electron-transfer process is termed an electrochemical reaction. Electrochemical reactions take place in electrochemical cells, that, in the simplest form, consist of two different electronic conductors (electrodes) immersed in an electrolyte solution. Each electrode may, in principle, exchange electrons with species in solutions either spontaneously or under the effect of an applied voltage. A cell in which the electron transfer at electrodes occurs spontaneously is termed a galvanic cell. It is important to keep in mind that the electrochemical cells used in potentiometric sensor applications are galvanic cell.
Before proceeding to the presentation of the galvanic cell characteristics, it is useful to give a short overview of the thermodynamics of electrolyte solutions.
10.2.1 Thermodynamics of Electrolyte Solutions
The thermodynamic state of any chemical species is defined by its chemical potential,![]() , which indicates how much the free energy G of an open system changes in response to an infinitesimal variation in species amount (number of moles, (
, which indicates how much the free energy G of an open system changes in response to an infinitesimal variation in species amount (number of moles, (![]() )), when the temperature (T), the pressure (P) and the number of moles of other species (
)), when the temperature (T), the pressure (P) and the number of moles of other species (![]() ) remain constant:
) remain constant:
(10.1) ![]()
The chemical potential (in J mol−1) depends on the thermodynamic activity (a) as follows:
where ![]() is the chemical potential in the standard state. In turn, the activity is proportional to the concentration c, the proportionality coefficient
is the chemical potential in the standard state. In turn, the activity is proportional to the concentration c, the proportionality coefficient ![]() being termed the activity coefficient (
being termed the activity coefficient (![]() ):
):
In very dilute solutions, when ![]() is close to unity the activity is approximately equal to the concentration.
is close to unity the activity is approximately equal to the concentration.
Only the mean activity coefficient of an ion can be determined experimentally. For a binary electrolyte, the mean activity coefficient ![]() is, by definition:
is, by definition:
(10.4) ![]()
where ![]() and
and ![]() are the individual activity coefficients of the cation and the anion, respectively.
are the individual activity coefficients of the cation and the anion, respectively.
In electrolyte solutions, each ion exerts and, simultaneously, experiences electrostatic forces due to interaction with other ions. That is why the activity coefficient of an ion depends on the ionic strength, I, of the solution, which is determined by the activity (![]() ) and the electric charge (
) and the electric charge (![]() ) of each ion present. So, the ionic strength is defined as:
) of each ion present. So, the ionic strength is defined as:
where the summation involves the activity and the charge of each ion type in the system. At very low values of ionic strength, the mean activity coefficient can be calculated by the following equation:
(10.6) ![]()
Here, A is parameter depending on the dielectric constant of water and the temperature. At a higher ionic strength ![]() can be calculated by means of semiempirical formulas.
can be calculated by means of semiempirical formulas.
The chemical potential of a dissolved neutral species is determined mostly by the solvation process and short-range interactions. Conversely, in the case of an ion any change in the ion concentration brings about a variation in the electric charge density with important effects on the potential energy of the system. This effect can be accounted for by means of the concept of electric potential.
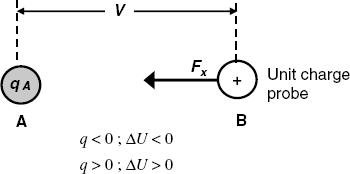
By definition, the electric potential difference between two points A and B (Figure 10.1) represents the change in the potential energy of the system when a unit positive charge arises from B to A in vacuum. The change in the potential energy equates the work ![]() associated with the charge movement in the electric field induced by the charge qA. The charge probe experiences an electrostatic force Fx which is attractive if
associated with the charge movement in the electric field induced by the charge qA. The charge probe experiences an electrostatic force Fx which is attractive if ![]() (as in Figure 10.1) or repulsive when
(as in Figure 10.1) or repulsive when ![]() . Accordingly, the potential energy decreases by the term
. Accordingly, the potential energy decreases by the term ![]() in the first case, as it occurs when an extended spring relaxes. Conversely, it increases by
in the first case, as it occurs when an extended spring relaxes. Conversely, it increases by ![]() in the second case, similar to the case of a spring compression. By definition, the electric potential difference
in the second case, similar to the case of a spring compression. By definition, the electric potential difference ![]() between the two points is the quotient of the work and the local charge at the point A:
between the two points is the quotient of the work and the local charge at the point A:
(10.7) ![]()
Figure 10.1 Definition of the electric potential difference between the points A and B. The unit charge at B moves to the point A where the charge qA is localized. The electrostatic force Fx could be repulsive or attractive according to sign of the charge at the point A. ![]() is the change in the potential energy.
is the change in the potential energy.
It is sometimes convenient to operate with the absolute electric potential that corresponds to the hypothetical situation in which the unit probe arrives at A from an infinite distance in vacuum. By means of this concept, the potential difference (or voltage) between A and B in volts (V) is:
(10.8) ![]()
where ![]() and
and ![]() represents the absolute potential at the point A and B, respectively.
represents the absolute potential at the point A and B, respectively.
If the unit charge probe is replaced by a certain charge q, then the change in the potential energy due to the shift of this charge from infinity to the point A is:
(10.9) ![]()
This is an important conclusion when assessing the effect of inserting an ion at a given point A. Letting z be the ion charge and F the Faraday constant (that is, the charge of a mole of a singly charged ion), it results that the change in potential energy produced by the transfer of one mole of ions is:
The state of an ion can now be described by taking into account both the chemical and electrostatic effects that are included in the electrochemical potential![]() of the ion. The change of the Gibbs free energy (in J mol−1) when a mole of ions with charge z appears in the considered system is obtained by adding the chemical potential, as defined in Equation (10.2) and the electrical term given by Equation (10.10):
of the ion. The change of the Gibbs free energy (in J mol−1) when a mole of ions with charge z appears in the considered system is obtained by adding the chemical potential, as defined in Equation (10.2) and the electrical term given by Equation (10.10):
Here, ![]() stands for the inner potential (also termed the Galvani potential) of a phase and the ion charge z should be included in this equation with the pertinent sign. The inner potential is the sum of two terms, the outer potential (
stands for the inner potential (also termed the Galvani potential) of a phase and the ion charge z should be included in this equation with the pertinent sign. The inner potential is the sum of two terms, the outer potential (![]() ) and the surface potential (
) and the surface potential (![]() ):
):
(10.12) ![]()
The surface potential is associated with the electrical charge localized at the surface of the considered phase. This charge is produced by oriented dipoles or ions accumulated at the surface. The outer potential is the potential difference between vacuum and a position close to the exterior of the surface where the electrostatic effect of the surface charge is absent.
10.2.2 Thermodynamics of the Galvanic Cell
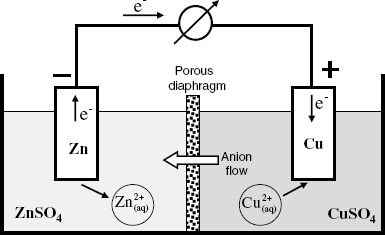
A typical galvanic cell is the Daniel cell (Figure 10.2) which consists of copper and zinc electrodes immersed in solutions of their respective metal sulfates. The two solutions are separated by a porous diaphragm that prevents solutions from mixing but allows ions to migrate across it. Spontaneously, the zinc metal undergoes oxidation and zinc ions are released to the solution, whereas electrons are released to the electrode and that develops a negative charge. At the same time, copper ions in solution are reduced to copper metal by accepting electrons from the copper electrode that thus assumes a positive charge. Consequently, an electron flow from the zinc to the copper electrode takes place when the electrodes are connected via an external circuit. At the same time, anions from the right-hand part flow to the left in order to preserve the electric charge balance. In this way, an electric current is established across the systems, with electrons as charge carriers in the external circuit and ions as charge carriers in the solution. Transition from electronic to ionic conduction is secured by the electrochemical reactions occurring at each metal/solution interface. Schematically, this cell is represented as follows:
Figure 10.2 Schematic representation of a galvanic cell (the Daniel cell).
(10.13) ![]()
Vertical bars in the above scheme represent phase interfaces; as a rule the activity or concentration of each species in the solution is also included in the scheme.
Potentiometry deals with electrochemical systems at equilibrium and it is hence of interest to discuss the characteristics of the electrochemical cell in this state. At equilibrium, the overall reaction rate is zero and no current flows through the system. The cell reaction involving n = 2 electrons is, in this case:
The free energy change in this reaction represents the energy required to transfer nF electrons across a potential difference between the electrodes, which represents the electromotive force (EMF):
The cell reaction (10.14) can be obtained by adding the two half-reactions that are zinc oxidation and copper ion reduction. A system composed of a metal electrode and the surrounding solution is termed a half-cell and the whole cell can be regarded as an assembly of two half-cells, each of them characterized by the pertinent half-reaction. In analytical applications, we are interested in only one of the half-cells, the second one acting as a reference half-cell. The standard half-cell consists of a platinum electrode immersed in a 1 M hydrogen ion activity solution saturated with hydrogen under a hydrogen gas pressure of 1 atm. Such a half-cell is termed the standard hydrogen electrode (SHE). Let us assume that a cell consists of a standard half-cell coupled with a half-cell containing a platinum electrode immersed in a solution containing a redox couple Ox and Red, where Ox is the oxidized form and Red is the reduced form of this couple. This cell is represented as follows:
where ![]() and
and ![]() , and represent the activity of the oxidized and reduced form, respectively.
, and represent the activity of the oxidized and reduced form, respectively. ![]() is the activity of the hydrogen ion in the solution and
is the activity of the hydrogen ion in the solution and ![]() is the partial pressure of hydrogen in the gas phase. The half-reaction at the left-hand half-cell is:
is the partial pressure of hydrogen in the gas phase. The half-reaction at the left-hand half-cell is:
(10.17) ![]()
where ![]() and
and ![]() are the stoichiometric coefficients of Ox and Red, respectively. The electrons needed in this reaction arise from the oxidation of the hydrogen molecule to hydrogen ions in the right hand half-cell. So the cell reaction is:
are the stoichiometric coefficients of Ox and Red, respectively. The electrons needed in this reaction arise from the oxidation of the hydrogen molecule to hydrogen ions in the right hand half-cell. So the cell reaction is:
(10.18) ![]()
According to the general relationship between the Gibbs free energy and the equilibrium constant (K), we have:
(10.19) ![]()
Here ![]() is the standard Gibbs free energy change in reaction (10:18). Since both hydrogen ion activity
is the standard Gibbs free energy change in reaction (10:18). Since both hydrogen ion activity ![]() and hydrogen partial pressure
and hydrogen partial pressure ![]() are equal to unity, they vanish from the above equations. The EMF of the cell in Scheme (10.16) will be denoted by E and, in view of Equation (10.15), is:
are equal to unity, they vanish from the above equations. The EMF of the cell in Scheme (10.16) will be denoted by E and, in view of Equation (10.15), is:
The EMF of a cell like that in Equation (10.16) represents the electrode potential of the left-hand half-cell with respect to the standard hydrogen electrode. This implies that the electrode potential of the standard hydrogen electrode is taken by convention as being zero at any temperature and its value is the origin of the electrode potential scale. The constant term ![]() is the standard electrode potential of the considered half-cell; it depends on
is the standard electrode potential of the considered half-cell; it depends on ![]() .
.
Equation (10.20) is the Nernst equation that relates the electrode potential of a particular redox couple to the activities of the reduced and oxidized forms. However, analytical chemistry operates with concentrations and not with thermodynamic activities. The Nernst equation in terms of concentrations is obtained by substituting each activity term by the ![]() product according to Equation (10.3). One obtains thus the following form:
product according to Equation (10.3). One obtains thus the following form:
where the term ![]() stands for the formal potential. In contrast to the standard potential, which is a thermodynamic constant, the formal potential depends on the activity coefficients and hence, on the solution properties.
stands for the formal potential. In contrast to the standard potential, which is a thermodynamic constant, the formal potential depends on the activity coefficients and hence, on the solution properties.
The electrode potential of a selected half-cell can be measured by assembling it with a reference half-cell as shown in Scheme (10.16). However, preparation and manipulation of the standard hydrogen electrode is rather cumbersome and, as a rule, electrode potentials are measured against a secondary reference electrode. A common reference electrode of this type is the silver/silver chloride half-cell consisting of a silver wire immersed in an AgCl-saturated KCl solution. The half-cell reaction is in this case:
For this half-cell, the Nernst equation gives:
where ![]() is the activity of the chloride ion and
is the activity of the chloride ion and ![]() is a constant. The activities of solid compounds do not appear in this equation because the activity of a pure solid is 1. If the chloride ion activity is kept constant, the electrode potential of the silver/silver chloride potential remains constant and serves as the origin of this electrode potential scale. As demonstrated by reaction (10.22), the metal Ag functions as a source of electrons, while Ag+ in AgCl acts as a sink for electrons. Therefore, the Ag/AgCl system behaves as an electron buffer and any trend to potential shift is counteracted by a shift in the equilibrium (10.22).
is a constant. The activities of solid compounds do not appear in this equation because the activity of a pure solid is 1. If the chloride ion activity is kept constant, the electrode potential of the silver/silver chloride potential remains constant and serves as the origin of this electrode potential scale. As demonstrated by reaction (10.22), the metal Ag functions as a source of electrons, while Ag+ in AgCl acts as a sink for electrons. Therefore, the Ag/AgCl system behaves as an electron buffer and any trend to potential shift is counteracted by a shift in the equilibrium (10.22).
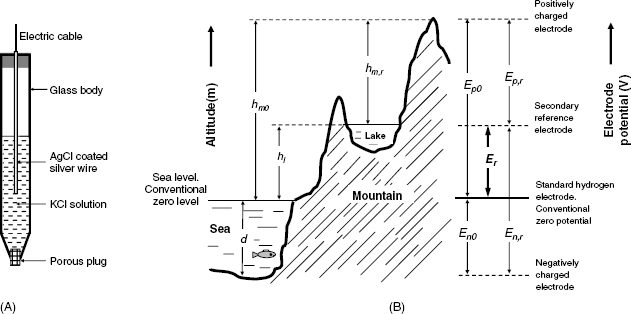
A practical form of this reference electrode is shown in Figure 10.3A. It consists of a glass body containing a KCl solution and a silver wire coated with solid AgCl. At the lower end, it is closed by a porous plug that prevents the inner solution from leaking into the test solution but allows for ion transfer in order to set up electrical contact with the solution. According to Equation (10.23), the electrode potential depends on the KCl concentration and this concentration should be reported when mentioning the reference electrode (e.g., Ag|AgCl (KCl 3.5 M)).
Figure 10.3 (A) A secondary reference electrode: the Ag/AgCl reference electrode; (B) An analogy between altitude measurement and the measurement of the electrode potential. The right-hand diagram shows the rules for electrode potential conversion from the secondary reference electrode scale to the standard hydrogen electrode scale.
Similarly functions the calomel electrode (Hg|Hg2Cl2 (KCl)). A common version of this electrode is the saturated calomel electrode (SCE) in which the internal solution is a saturated KCl solution.
Chloride ions can diffuse from the reference electrode into the test solution. If chloride ions interfere, the Ag/AgCl electrode can be coupled to the solution via an electrolyte bridge filled with an inert electrolyte solution (e.g., KNO3). This combination is termed a double-junction reference electrode because the contact with the test solution is achieved via two liquid–liquid junctions. Alternatively, one can make use of the saturated mercury(I)/mercury sulfate electrode (Hg|Hg2SO4|K2SO4(saturated)). For the purpose of microanalysis or in vivo applications, miniaturized reference electrodes are needed. Such devices can be obtained by microfabrication technology [16].
It is often necessary to convert electrode potentials measured with respect to a secondary reference to corresponding values on the standard hydrogen electrode scale. Conversion can be effected according to the diagram in Figure 10.3B. As an aid to clarity, this figure shows an analogy with the rules for altitude determination with respect to sea level, which is conventionally set as the zero level. More details on reference electrodes can be found in ref. [17]
By convention, the galvanic cell should be represented with the reference electrode at the left and the indicator electrode at the right as in Scheme (10.16), and the EMF is calculated as:
(10.24) ![]()
where ![]() and
and ![]() are the pertinent half-cell potentials.
are the pertinent half-cell potentials.
10.3 Ion Distribution at the Interface of Two Electrolyte Solutions
10.3.1 Charge Distribution at the Junction of Two Electrolyte Solutions. The Diffusion Potential
Whenever different electrolyte solutions are put in contact, a nonuniform ion distribution develops at their interface. Two limiting situations can be envisaged: (a) a permeable interface is formed, which allows for unrestricted transfer of any ion, and (b) a semipermeable interface is formed, which restricts the transfer of certain types of ion. The first case is addressed in this section, while the second case is the subject of the next section.
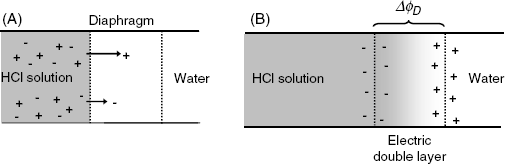
The case of a permeable interface is illustrated by the contact of an HCl solution with pure water (Figure 10.4A). Practically, such a system can be obtained by interposing a porous diaphragm between the two solutions. Due to the difference in concentrations, both H+ and Cl− do diffuse from the left to the right. However, the H+ ion is much faster than Cl−. As a result, a region rich in H+ develops in front (Figure 10.4B), whereas a negatively charged region lags behind. Thus, a double electric layer develops and gives rise to an electric potential difference, ![]() . Under the effect of
. Under the effect of ![]() , the movement of H+ is slowed down while the movement of Cl− is accelerated. When the steady-state is attained, the electrical and diffusional driving forces balance each other and ion distribution across the interface remains invariable.
, the movement of H+ is slowed down while the movement of Cl− is accelerated. When the steady-state is attained, the electrical and diffusional driving forces balance each other and ion distribution across the interface remains invariable. ![]() is termed the diffusion potential or the liquid junction potential.
is termed the diffusion potential or the liquid junction potential.
Figure 10.4 Development of the liquid junction potential at the junction of an HCl solution with water. (A) Initial state (no charge separation). Arrow lengths are proportional to ion mobility. (B) Steady-state conditions: a front of fast cations (H+) precedes the front of slower anions (Cl−) giving rise to a double electric layer. Ions in the intermediate region are not shown.
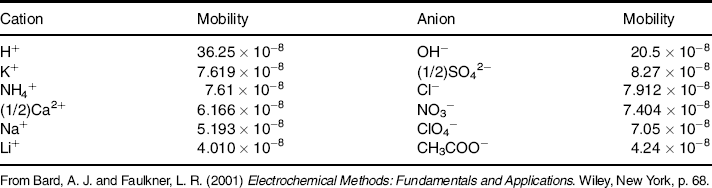
In order to calculate the junction potential, we have to consider the mass transport of ions across the interface. As already mentioned, ion transport occurs under the influence of both the activity gradient across the interface (diffusion) and the electric potential gradient (migration). The characteristic migration velocity of an ion is termed ion mobility and is defined as the increase in the ion velocity when crossing the distance between two points at 1 m distance from each other and subject to a potential difference of 1 V. The mobility unit is therefore the quotient of the velocity unit (m s−1) and the electric field gradient unit (V m−1) that is, m2 s−1 V−1. The diffusion coefficient (D) of an ion is related to the ion mobility (u) as ![]() , where R is the ideal gas constant and T is the absolute temperature. Selected ion mobility values are given in Table 10.1.
, where R is the ideal gas constant and T is the absolute temperature. Selected ion mobility values are given in Table 10.1.
Table 10.1 Ion mobility in water at 25 °C in m2 s−1 V−1
The general case is represented by the junction of two electrolyte solutions, each of them containing various ions. Under the simplifying assumption of a constant activity gradient across the double-layer zone, the diffusion potential is given by the Henderson formula [4]:
In this equation, ![]() and
and ![]() are the ion charge and mobility, respectively,
are the ion charge and mobility, respectively, ![]() and
and ![]() are ion activities at the left hand side and the right hand side of the double layer, respectively
are ion activities at the left hand side and the right hand side of the double layer, respectively ![]() .
. ![]() and
and ![]() could be different from the bulk solution activities. The summation is done over all the ions in the system.
could be different from the bulk solution activities. The summation is done over all the ions in the system.
Diffusion potential values predicted by the Henderson formula (Table 10.2) are in fair accord with the experimental ones. Data in this table can be interpreted by taking into account the ion mobility in Table 10.1. Thus, as K+ and Cl− have quite similar mobilities, the term in front of logarithm becomes very small. Therefore, a KCl solution on one side leads to a low junction potential and this potential decreases as the KCl concentration increases. That is why concentrated KCl solutions are customarily used to fill reference electrodes in order to secure as low a junction potential as possible. The large difference in mobility between H+ and Cl− produces a large junction potential when an HCl solution is put in contact with a diluted KCl solution, but this potential becomes much lower when the KCl solution is 3.5 M, that is, near saturation.
Table 10.2 Liquid junction potentials at 25 °C estimated by Henderson equation.
| Junction | Potential/mV |
| 0.1 M KCl || 0.1 M NaCl | 4.4 |
| 3.5 M KCl || 0.1 M NaCl | −0.2 |
| 3.5 M KCl || 1.0 M NaCl | 1.9 |
| 0.1 M KCl || 0.1 M HCl | −26.8 |
| 3.5 M KCl || 0.1 M HCl | −4.2 |
| From ref. [1], p 72. | |
A junction potential develops always at the contact of the reference electrode with the test solution and adds to the EMF as an unknown and, possibly, randomly variable term. Therefore, although the liquid junction potential is, as a rule, very small, its effect cannot be neglected. Thus, for a potentiometric sensor with a sensitivity of 59 mV, an error of 3 mV in the cell voltage brings about an error of 0.05 pX units, that is, a 12% concentration error. The effect of the junction potential can be minimized by using a double-junction reference electrode with the electrolyte bridge filled with a solution whose ions have similar mobilities. As the two junction potentials have opposite signs, the overall diffusion potential will be very small.
10.3.2 Ion Distribution at an Aqueous/Semipermeable Membrane Interface
The functioning of certain types of ion sensors is based on semipermeable membranes that restrict the mobility of some ions, as is the case, for example, with solid ion exchangers. A solid ion exchanger is a microporous material containing fixed ionic sites that prevent permeation by ions of an opposite charge. Similar behavior is shown by liquid ion-exchanger membranes that include lipophilic charged molecules. The lipophilic ions cannot move into an aqueous solution and, at the same time, prevent the permeation of solution ions of opposite charge.
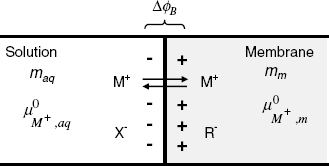
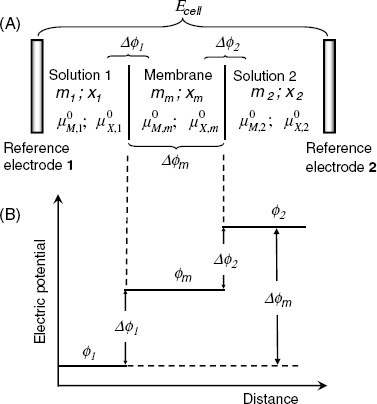
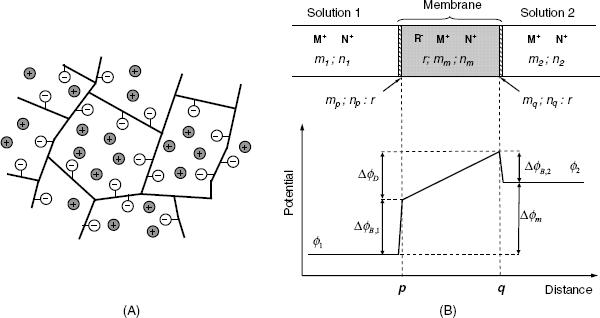
Figure 10.5 displays the situation at the contact of the solution 1 containing the MX salt with a cation-selective membrane. The membrane contains anionic sites R− that cannot move into the aqueous solution. The anion X− is confined to solution 1 due to electrostatic repulsion by the negative sites in the membrane. Conversely, the cations M+ are free to diffuse between the two phases. If the cation activity in solution 1 (maq) is higher than that in the membrane (mm), the cation diffusion to the right gives rise to a charge imbalance that counters cation diffusion (Figure 10.5). When the diffusional and electric driving forces balance each other, the potential difference between the two phases (![]() ), called the phase-boundary potential difference, assumes a constant value. This quantity will be further denoted by the short-hand term boundary potential [1].
), called the phase-boundary potential difference, assumes a constant value. This quantity will be further denoted by the short-hand term boundary potential [1].
Figure 10.5 Ion distribution at a semipermeable phase boundary and development of the boundary potential. The left-hand phase is an aqueous solution, the right-hand one is a semipermeable phase.
In order to derive an equation for the boundary potential, the partition of the cation between the two phases should be considered. At equilibrium, this process is characterized by the partition coefficient![]() defined as follows:
defined as follows:
where maq and mm are the activities of the free metal ions in the solution and in the membrane, respectively. Free ions are present if there are no species able to bind the ion in the solution or in the membrane phase. As the partition coefficient is an equilibrium constant, it is correlated with the standard free energy change of transfer ![]() , and with the chemical potentials of the ion in each phase as follows:
, and with the chemical potentials of the ion in each phase as follows:
Ion transfer from phase 1 to phase 2 is favored by large ![]() values, which implies a high partition coefficient.
values, which implies a high partition coefficient.
The boundary potential equation can be derived from the equilibrium condition for the ion transfer between the two phases, which implies equality of electrochemical potentials (Equation 10.11):
(10.28) ![]()
Taking into account Equation (10.27), one obtains:
where ![]() represents the standard boundary potential. Equation (10.29) is formally analogous to the Nernst equation for the electrode potential.
represents the standard boundary potential. Equation (10.29) is formally analogous to the Nernst equation for the electrode potential.
If the metal ion can bind to a ligand to form a complex either in the solution or in the membrane phase, the free ion activity can be obtained from the stability constant of the complex.
10.4 Potentiometric Ion Sensors – General
10.4.1 Sensor Configuration and the Response Function
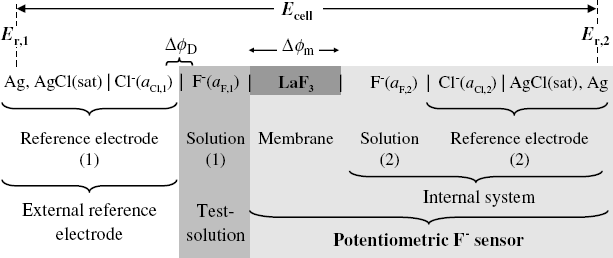
The Nernst equation indicates that the electrode potential can be used to determine the concentration of redox species in solution. However, this approach is not selective at all, as any redox couple in solution will affect the electrode potential. A true potentiometric ion sensor is obtained by inserting an ion-selective membrane between two solutions containing the analyte ion as shown in Figure 10.6 for the particular case of a fluoride ion sensor. The galvanic cell is completed with a second reference electrode.
Figure 10.6 The structure of a galvanic cell for the determination of fluoride ion by means of a fluoride-selective membrane.
The membrane should interact in a specific way with the analyte ion. Thus, a lanthanum fluoride membrane presents selectivity to the fluoride ion. This is due to the fact that, in contact with water, the salt at the surface undergoes partial dissociation accompanied by the transfer of fluoride ions to the solution:
Thus, a certain number of fluoride anions leave the surface and instead remain positive vacancies. If the fluoride ion is present in the solution, the equilibrium (10.30) will be shifted to the left, thus decreasing the density of positive charges at the membrane surface. Therefore, the positive charge density at the membrane surface depends on the analyte concentration in the solution. The same situation arises at the opposite side, where the membrane is in contact with the internal solution. However, the fluoride ion concentration in this solution is kept constant and the charge density at the interface remains constant as well.
It follows that the difference in electric charge density between the two sides of the membrane is dependent on the analyte concentration. Measuring charge densities on such a device is, however, impossible. Instead, one can resort to potential difference measurements. To this end, a reference electrode is immersed in each solution to act as probes that perform transition from ionic conductivity in solution and membrane, to electronic conductivity. A voltmeter connected to the two reference electrodes allows measuring the cell voltage that includes the potential difference across the membrane, termed the membrane potential (![]() ).
).
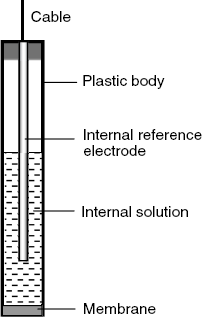
For practical applications, the internal system and the membrane are assembled to form the actual sensor, as shown in Figure 10.7. In order to perform ion determinations, the sensor is dipped in the test solution along with the external reference electrode.
Figure 10.7 Typical configuration of a potentiometric ion sensor.
The response signal is the cell voltage that can be formulated using the symbols in Figure 10.6:
The term ![]() is the liquid junction potential, which develops at the limit where the external reference electrode meets the test solution.
is the liquid junction potential, which develops at the limit where the external reference electrode meets the test solution.
The key term in Equation (10.31) is the membrane potential that depends on the analyte activities as:
(10.32) ![]()
where ![]() and
and ![]() are analyte activities in the internal solution and the test solution, respectively; the charge term is positive for cations and negative for anions. This equation holds only under the assumption that no other ion, except the analyte, affects the membrane potential. As the analyte activity in the internal system is constant, Equation (10.31) can be formulated as:
are analyte activities in the internal solution and the test solution, respectively; the charge term is positive for cations and negative for anions. This equation holds only under the assumption that no other ion, except the analyte, affects the membrane potential. As the analyte activity in the internal system is constant, Equation (10.31) can be formulated as:
The constant term ![]() in this equation includes the two reference electrode potentials as well as the junction potential that is assumed to be constant. Formally,
in this equation includes the two reference electrode potentials as well as the junction potential that is assumed to be constant. Formally, ![]() represents the cell voltage at unit analyte activity; it should be not confused with the standard electrode potential.
represents the cell voltage at unit analyte activity; it should be not confused with the standard electrode potential.
In practical applications, the decimal logarithm is used instead of the natural one and the response function assumes the following form:
By analogy with the pH symbol, the pX symbol, defined in Equation (10.35), is often used instead of the log term (X stands for the symbol of the chemical element).
It follows therefore that the response of a potentiometric ion sensor is a linear function of the logarithm of the analyte activity with a theoretical sensitivity of ![]() , that is, 0.0591/z V per log unit at 25 °C. This ideal response is termed the Nernstian response. In practice, the sensitivity can be slightly smaller than the theoretical value. The
, that is, 0.0591/z V per log unit at 25 °C. This ideal response is termed the Nernstian response. In practice, the sensitivity can be slightly smaller than the theoretical value. The ![]() response increases with the activity in the case of cations, but decreases in the case of anions. Note also that the sensitivity for divalent ions is only half of that for singly charged ions.
response increases with the activity in the case of cations, but decreases in the case of anions. Note also that the sensitivity for divalent ions is only half of that for singly charged ions.
When the cell is coupled to the voltmeter, a current in the femtoampere range flows through the system. This tiny current does not disturb the equilibria. However, it is essential that the membrane elicits some minimal electrical conductivity. To this end, the LaF3 membrane is doped with minute amounts of EuF2. The conductivity issue should be kept in mind when designing any type of ion-sensing membrane.
A common problem arises from the fact that potentiometric sensors respond to ion activity and not to concentration (c), which is the required parameter in analytical applications. However, the activity is proportional to the concentration (Equation (10.3), and, further, the proportionality coefficient ![]() depends on the solution ionic strength (Equation (10.5)). Therefore, the activity term in Equation (10.34) can be substituted by concentration providing that the sample and the standard solutions have similar ionic strengths. The ionic strength can be adjusted by adding to each solution an excess of background electrolyte that does not interact with the membrane or the analyte (e.g., KNO3). Thus, the contribution of sample ions to the total ionic strength becomes negligible and small variations in the analyte concentration have no effect on the activity coefficient. Under these conditions,
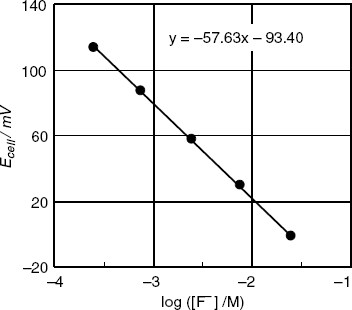
depends on the solution ionic strength (Equation (10.5)). Therefore, the activity term in Equation (10.34) can be substituted by concentration providing that the sample and the standard solutions have similar ionic strengths. The ionic strength can be adjusted by adding to each solution an excess of background electrolyte that does not interact with the membrane or the analyte (e.g., KNO3). Thus, the contribution of sample ions to the total ionic strength becomes negligible and small variations in the analyte concentration have no effect on the activity coefficient. Under these conditions, ![]() is a linear function of log c. As an example, Figure 10.8 shows the calibration graph in terms of log c obtained with a series of fluoride solutions at constant ionic strength. The linear dependence of the signal on the logarithm of the analyte concentration is clear. The sensor sensitivity is given by the slope of the line; in this particular case, it is −57.6 V, that is, slightly lower than the theoretical value (−58.1 mV at 20 °C). This kind of less-than Nernstian response occurs often in practice.
is a linear function of log c. As an example, Figure 10.8 shows the calibration graph in terms of log c obtained with a series of fluoride solutions at constant ionic strength. The linear dependence of the signal on the logarithm of the analyte concentration is clear. The sensor sensitivity is given by the slope of the line; in this particular case, it is −57.6 V, that is, slightly lower than the theoretical value (−58.1 mV at 20 °C). This kind of less-than Nernstian response occurs often in practice.
Figure 10.8 The response of a fluoride potentiometric sensor to the fluoride concentration at constant ionic strength. Temperature: 20 °C.
10.4.2 Selectivity of Potentiometric Ion Sensors
The selectivity of the ion-recognition process can be achieved by complementarity of the analyte and receptor site as far as size, charge and chemical interactions are concerned. However, perfect selectivity cannot be achieved and ions related to the analyte can, in principle, also interact with the receptor. Thus, in the case of the fluoride sensor, interference is produced by hydroxyl ions whose size is quite similar to that of the fluoride ion. Fortunately, in this case, the concentration of the hydroxide ion can be made very low by adjusting the pH to around 5.
If interference by a secondary ion is possible, it can be accounted for by including an additional term in Equation (10.34) to obtain what is known as the Nikolskii–Eisenman equation:
where ![]() is the potentiometric selectivity coefficient and
is the potentiometric selectivity coefficient and ![]() represent the concentrations of the analyte and the interfering ion, respectively, in the test solution. Clearly, good selectivity is obtained when the selectivity coefficient is sufficiently low in order to render the
represent the concentrations of the analyte and the interfering ion, respectively, in the test solution. Clearly, good selectivity is obtained when the selectivity coefficient is sufficiently low in order to render the ![]() term negligible with respect to
term negligible with respect to ![]() . Simultaneous interference by a series of ions bearing the same charge as the analyte is accounted for by the generalized form of the Nikolskii–Eisenman equation:
. Simultaneous interference by a series of ions bearing the same charge as the analyte is accounted for by the generalized form of the Nikolskii–Eisenman equation:
In order to account for the interference of an ion with a different charge, the following equation has been proposed:
where zA and zB represent the charges of the analyte and the interfering ion, respectively. However, due to the different units of the selectivity coefficients in Equations (10.37) and (10.38), this approach does not allow comparison of the effect of two interfering ions with different charges.
Equation (10.36) is based on the following assumptions: (a) both the analyte and the interfering ions have the same charge, and (b) the sensor response to each of the above ions is a Nernstian one. Each of the above assumptions can be inaccurate in many particular cases, which led to more careful approaches being made to the selectivity modeling problem [18, 19].
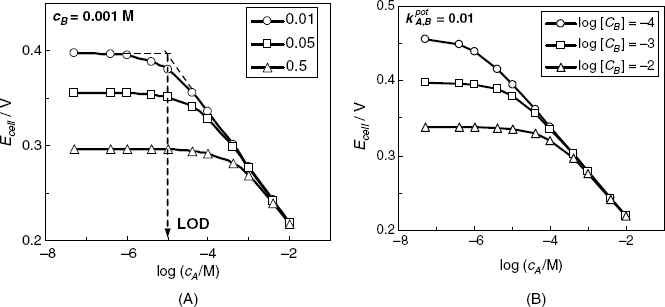
There are several methods for the experimental determination of the selectivity coefficient based on Equation (10.36) [20, 21]. Thus, in the separate solution method, the response to each ion is recorded in the absence of the second ion and Equation (10.36) is used to calculate the selectivity coefficient by means if ion activities producing a similar response to each ion. Other methods belong to the class of mixed-solution methods that rely on measurements in solutions containing both ions of interest. Thus, the fixed interference method is based on the determination of the response to increasing analyte concentration in the presence of a fixed interfering ion concentration, as shown in Figure 10.9A. According to Equation (10.36), the extended linear parts of the response line cross each other at a point where ![]() . Hence, the ratio
. Hence, the ratio ![]() at the intersection point is equal to the selectivity coefficient. The fixed primary ion method is based on the determination of the potential change in response to the addition of increasing interferent concentrations in a solution containing a fixed analyte concentration. The selectivity coefficient is calculated as in the previous method. The three methods above assume that the slope of the response line is the same for both the primary and secondary ion.
at the intersection point is equal to the selectivity coefficient. The fixed primary ion method is based on the determination of the potential change in response to the addition of increasing interferent concentrations in a solution containing a fixed analyte concentration. The selectivity coefficient is calculated as in the previous method. The three methods above assume that the slope of the response line is the same for both the primary and secondary ion.
Figure 10.9 The effect of selectivity on an anion (A−) sensor response. (A) Effect of the selectivity coefficient (inset values) at a constant concentration of the interferent B− (0.001 M). LOD is the lower limit of detection. (B) Effect of the interferent concentration (inset) at a constant selectivity coefficient of 0.01.
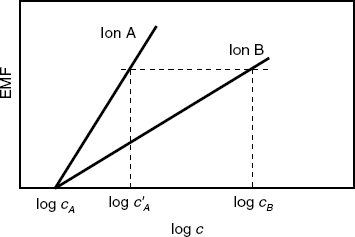
A method independent of Equation (10.36) is the matched potential method that overcomes the difficulty of obtaining accurate results when ions of unequal charge are involved [22]. The selectivity coefficient is defined as the ![]() ratio where
ratio where ![]() and
and ![]() are concentration changes producing the same EMF change in a reference solution.
are concentration changes producing the same EMF change in a reference solution.
Accordingly, the sensor response to ions A and B is recorded independently in the reference solution (Figure 10.10). In the case of ion B, the solution should contain an initial background concentration of ion A. The ion concentrations yielding a similar response (![]() and
and ![]() ) are recorded and the selectivity coefficient is determined using the following equation (See Figure 10.10 for symbol meanings):
) are recorded and the selectivity coefficient is determined using the following equation (See Figure 10.10 for symbol meanings):
(10.39) ![]()
Figure 10.10 Determination of the selectivity coefficient by the matching potential method.
The selectivity coefficient obtained by this method should agree with that obtained by methods based on Equation (10.36) if the ions have similar charge and the response to each of them is Nernstian. Otherwise, there could be large differences between the selectivity coefficients obtained using different methods.
Comprehensive compilations of potentiometric selectivity coefficients are available in refs. [21, 23, 24], whereas criteria and protocols for preparation and evaluation of potentiometric ion sensors can be found in ref. [25].
10.4.3 The Response Range of Potentiometric Ion Sensors
The response range of an ion sensor is the concentration interval within which the sensor response to the analyte activity is in accord with Equation (10.34). As demonstrated below, the limits of the dynamic range are determined by the interaction of other sample components that may interact with the analyte in the solution or with receptor sites in the membrane.
The effect of the selectivity coefficient on the sensor response at a constant interferent concentration is represented in Figure 10.9A. The response is linear only as long as the analyte concentration is sufficiently high and the ![]() term is negligible. At lower analyte concentrations, each ion contributes to some extent and the response function assumes a curved shape. Further, if
term is negligible. At lower analyte concentrations, each ion contributes to some extent and the response function assumes a curved shape. Further, if ![]() , the contribution of the analyte to the total response becomes negligible and the response, which is now determined only by the interferent, assumes a constant value. The intersection of the two linear branches of the response curve defines the lower limit of detection of the ion A− sensor. Curves in Figure 10.9A demonstrate that a low selectivity coefficient results in a lower limit of detection and a broader linear response range. Hence, a potentiometric ion sensor can be used even in the presence of an interferent, but both the limit of detection and the working range depend on interferent concentration. This conclusion is illustrated by the graph in Figure 10.9B which represents the response curve of a sensor with a selectivity coefficient of 0.01 in the presence of an interferent at various concentrations. This graph demonstrates clearly that both the limit of detection and the extent of the working rage depend on the concentration of the interferent.
, the contribution of the analyte to the total response becomes negligible and the response, which is now determined only by the interferent, assumes a constant value. The intersection of the two linear branches of the response curve defines the lower limit of detection of the ion A− sensor. Curves in Figure 10.9A demonstrate that a low selectivity coefficient results in a lower limit of detection and a broader linear response range. Hence, a potentiometric ion sensor can be used even in the presence of an interferent, but both the limit of detection and the working range depend on interferent concentration. This conclusion is illustrated by the graph in Figure 10.9B which represents the response curve of a sensor with a selectivity coefficient of 0.01 in the presence of an interferent at various concentrations. This graph demonstrates clearly that both the limit of detection and the extent of the working rage depend on the concentration of the interferent.
It is clear from the above discussion that the lower detection limit is determined by ions bearing the same charge as the analyte itself. In turn, the upper limit of the response range is a consequence of the fact that ions of opposite charge penetrate into the membrane and produce a marked bending of the response line.
10.4.4 Interferences by Chemical Reactions Occurring in the Sample
As shown in the previous section, interference in the ion-recognition process can be avoided by designing ion-responsive membranes with very low potentiometric selectivity coefficients. Another source of interferences is connected to the chemistry of the analyte ion in the sample. This aspect can be illustrated for the particular case of the fluoride ion. A common feature of all potentiometric ion sensors is that they respond only to the analyte ion in the free ion form. Therefore, if the analyte can react with other sample components, it would be converted into a nondetectable form. Thus, in the case of the fluoride sensor, the fluoride ion is converted to HF and ![]() in acidic solutions. Also, metal ions in the sample (such as Fe3+ or Al3+) form complexes with the fluoride ion. Both protonation and complex formation lead to the masking of the analyte. That is why the pH should be adjusted to about 5 by a buffer system in order to prevent the interference of both H+ and OH− ions. On the other hand, a strong complexing agent should be present in order to mask the metal ions. As the response depends on the ionic strength, an inert electrolyte at constant concentration should also be present in each sample and standard solution. A solution containing all these components is termed a total ionic strength adjustment buffer (TISAB). Thus, the data in Figure 10.8 have been obtained in the presence of a TISAB system that consists of a pH 5 acetate buffer, sodium chloride, and EDTA as a metal ion masking reagent.
in acidic solutions. Also, metal ions in the sample (such as Fe3+ or Al3+) form complexes with the fluoride ion. Both protonation and complex formation lead to the masking of the analyte. That is why the pH should be adjusted to about 5 by a buffer system in order to prevent the interference of both H+ and OH− ions. On the other hand, a strong complexing agent should be present in order to mask the metal ions. As the response depends on the ionic strength, an inert electrolyte at constant concentration should also be present in each sample and standard solution. A solution containing all these components is termed a total ionic strength adjustment buffer (TISAB). Thus, the data in Figure 10.8 have been obtained in the presence of a TISAB system that consists of a pH 5 acetate buffer, sodium chloride, and EDTA as a metal ion masking reagent.
So, in general, two kinds of interference can be expected when using a potentiometric ion sensor. These are (a) interaction of another ion with the receptor sites, and (b) analyte reactions in the solution leading to ion blocking. The first kind of interference is mostly decided by the characteristics of the receptor itself, that is, its selectivity. The second type of interference depends on the sample composition and can be dealt with by means of suitable chemical reactions.
10.4.5 The Response Time of Potentiometric Ion Sensors
The response time depends on the dynamics of all processes involved in the analyte–membrane interaction. Generally, it is defined as the time lapse between the moment of a sudden change in analyte concentration and the instant at which the response change (![]() ) reaches its steady-state value within 1 mV or has reached 90% of the final value.
) reaches its steady-state value within 1 mV or has reached 90% of the final value.
In the case of liquid membranes, the slowest step in the overall process is often the rate of analyte diffusion to the membrane surface and that is why is it is often recommended to provide gentle stirring during the measurement. With solid membranes, redistribution of ions within the membrane in response to the change in the membrane potential often determines the response time. The rate of ion redistribution depends on the membrane thickness (which determines the strength of the electric field) and the membrane conductivity.
10.4.6 Outlook
The key problem in developing a potentiometric ion sensor is the choice of preparation of the sensitive membrane that interacts in a characteristic way with the analyte in order to develop the membrane potential. The first potentiometric ion sensors (such as the pH electrode) were based on glass membranes that form a thin surface layer in contact with aqueous solutions that functions as a cation exchanger. Further, solid crystalline membranes formed of sparingly soluble salts (such as AgCl or metal sulfides) have been developed. Determination of ions that do not form sparingly soluble salts can be performed by means of ion exchangers dissolved in a water-immiscible solvent. The most advanced ion sensors are based on neutral receptors incorporated in liquid membranes. Each of the above alternatives will be reviewed in the next sections.
10.5 Sparingly Soluble Solid Salts as Membrane Materials
10.5.1 Membrane Composition
A well-known crystalline membrane is the single-crystal lanthanum fluoride membrane discussed in Section 10.4.1. However, much more widely used are polycrystalline membranes composed of silver halides or sparingly soluble metal sulfides. Polycrystalline membranes can be obtained as pressed pellets or by incorporation of the salt in silicone rubber or in thermoplastic polymers. As already mentioned, the sensing mechanism of such membranes relies on the effect of the analyte on the dissociation equilibrium of the membrane salt. Therefore, such membranes would be expected to respond to both the composing cation and anion. However, due to the extremely low solubility of the membrane compound, only one of these ions can be present in solution at a significant concentration.
A simple example of this type is the silver chloride membrane that is responsive to both Ag+ and Cl−. However, it displays a series of drawbacks and, consequently, it is used as a 1 : 1 molar ratio mixture with silver sulfide. Silver sulfide membranes are sensitive to both sulfide and silver ions. Mixtures of silver sulfide with a divalent ion sulfide respond to the divalent ion activity and form the basis for the development of potentiometric sensors for cations such as Cu2+, Pb2+, and Cd2+. Determination of anions such as cyanide, iodide and thiocyanate are based on their reactions with the membrane cation to form sparingly soluble salts or stable metal complexes, as shown in next Section.
10.5.2 Response Function and Selectivity
In the following discussion it will be assumed that the membrane consists of a single salt, MX, which is in contact at both faces with solutions containing the ions M+ and X− (Figure 10.11). The behavior of such a membrane is determined by the dissolution of the solid compound MX into the aqueous phase:
Figure 10.11 Variation of the electric potential across a membrane/solutions system in the case if a solid salt crystalline membrane. (A) Cell configuration; (B) potential variation across the system.
This equilibrium is characterized by the solubility constant Ks that depends on the ion activities in the saturated MX solution:
where m and x represent the activity of the cation and anion, respectively. If either M+ or X− is present as an analyte, the activity of the second ion is related to that of the first by Equation (10.41). Figure 10.11 also shows the variation of the electric potential across the system. Note that the potential is constant across the membrane because the ions in the crystal lattice occupy fixed positions and no internal diffusion potential can develop.
The membrane potential difference ![]() can be derived from the equilibrium conditions for reaction (10.40), that, in the case of the first interface, is formulated as follows (the activity of the pure solid MX is taken as equal to unity):
can be derived from the equilibrium conditions for reaction (10.40), that, in the case of the first interface, is formulated as follows (the activity of the pure solid MX is taken as equal to unity):
Equation (10.42) allows the potential difference at the first interface (![]() ) to be calculated, as a function of the cation activity in solution 1:
) to be calculated, as a function of the cation activity in solution 1:
(10.44) ![]()
Similarly, one obtains the following equation for the potential difference at the second interface:
(10.45) ![]()
From the above equations, the membrane potential equation is obtained in the following form:
Using the equilibrium conditions with respect to the anion (Equation (10.43)) and an analogous equation for the second interface, one obtains the membrane potential as a function of the anion activities (x1; x2):
It can be demonstrated in a similar way that a silver sulfide (Ag2S) membrane responds to the sulfide ion (S2−) with the prelogarithm factor of −RT/2F.
Ion activity in the solution 1 can be determined by keeping constant this ion activity in the reference solution 2. Under these conditions, Equation (10.46) gives:
An analogous equation can be derived for the anion.
In order to assess the selectivity, it will be assumed that an X− sensor is in contact with a solution containing both X− and the an interfering anion, Y−. Each anion is involved in a dissociation equilibrium and the activity of each anion is determined by the solubility constants of the pertinent salts:
(10.49) ![]()
x1 and y1 denotes activities of pertinent ions. From the above equations one obtains the activity of the second anion:
As long as y1 is lower than the value given by Equation (10.50), no solid MY forms in the solution and the membrane responds to the X− activity only, according to Equation (10.47). Conversely, if y1 exceeds the limit imposed by the Equation (10.21), solid MY does form in the solution and Equation (10.47) should be adapted by substituting the x1 expression derived from Equation (10.50) into Equation (10.47). It results:
(10.51) ![]()
Hence, if x2 is kept constant, the above equation can be reduced to:
(10.52) ![]()
The above considerations demonstrate that Y− can interfere in the X− determination when ![]() . In the opposite case, the MX membrane turns into a Y− -sensitive membrane. For example, an AgCl membrane responds to the iodide concentration as the solubility of AgI (logKs, AgI = −16.8) is much less than that of AgCl (log Ks, AgCl =− 9.75). However, the limit of detection for the Y− ion depends on the X− concentration in the sample.
. In the opposite case, the MX membrane turns into a Y− -sensitive membrane. For example, an AgCl membrane responds to the iodide concentration as the solubility of AgI (logKs, AgI = −16.8) is much less than that of AgCl (log Ks, AgCl =− 9.75). However, the limit of detection for the Y− ion depends on the X− concentration in the sample.
Quite a similar situation occurs if the membrane cation reacts with a solution component Z to form a soluble metal complex:
(10.53) ![]()
In such a situation, the sensor produces a mixed response to both X− and Z species. However, if the X− activity is negligible with respect to that of Z, the sensor responds in a Nernstian way to the Z activity. Thus, an AgCl membrane shows a Nernstian response to cyanide ion activity if this activity is much higher than that of the chloride ion.
A number of potentiometric ion sensors are based on mixed membranes made of a mixture of silver sulfide (M2S) and a divalent cation sulfide (NS). This is because divalent cation sulfides are generally insulators and silver sulfide is needed in order to impart electric conductivity to the membrane. The dissolution of each sulfide is controlled by the pertinent solubility constant:
where s, m and n are the activities of S2−, M2+ and N2+ ions, respectively, in the test solution. The membrane responds to the M2+ activity, but this, in turn, is determined by the activity of the N2+ ion, as is clear from Equations (10.54) which gives ![]() . Upon inserting this expression into Equation (10.48) one obtains:
. Upon inserting this expression into Equation (10.48) one obtains:
(10.55) ![]()
The above principles have been used to develop potentiometric sensors for Cu2+, Pb2+, and Cd2+.
Solid salt membrane sensors are robust devices characterized by a very long lifetime. The main drawbacks are the poor selectivity and a relatively high detection limit (in the 10−6 M region).
10.6 Glass Membrane Ion Sensors
10.6.1 Membrane Structure and Properties
In the case of membranes composed of sparingly soluble salts, both salt ions can undergo transfer between the solution and the membrane surface. A different kind of membrane is represented by solid ion exchangers in which one of the ions is covalently attached to a solid network. Typical materials of this kind are amorphous silicates (oxide glasses) such as the glass membrane forming the sensing element of the pH glass electrode. In contact with an aqueous solution, a layer of 50 to 100 nm thickness at the glass surface is impregnated with water and assumes a gel structure (Figure 10.12A). This layer displays cation-exchanger properties as it includes negative Si-O− groups attached to a random network which is typical of amorphous solids. Positive ions are free to cross the interface with a solution, but anion permeation is hindered by the fixed negative sites that exert electrostatic repulsion. The surface gel acts therefore as a semipermeable membrane and performs the actual sensing process in glass membranes.
Figure 10.12 (A) Structure of a solid cation exchanger. The gel structure is impregnated with water. Thick lines represent the molecular network; empty circles are immobilized anions; full circles are mobile solvated cations in the embedded solution. (B) Configuration and potential distribution for a solid ion-exchanger membrane. R− indicates immobilized anions within the membrane; M+ and N+ are mobile cations. Lower case symbols indicates the activities of ionic species indicated by pertinent upper case symbols.
Due to the cation-exchange process, the negative charge density within the sensing part can vary in response to the analyte ion activity in the test solution. The same happens at the opposite side of the glass membrane, which is in contact with the internal system of the sensor. The cell voltage of a glass electrode–reference electrode combination is a function of the hydrogen ion activity according to Equation (10.33).
The best-known application of the glass membrane is the potentiometric pH sensor commonly termed the pH glass electrode [26, 27]. The membrane in this case is a sodium and calcium silicate glass. It displays an outstanding selectivity for the hydrogen ion as the single known interference arises from sodium ion at pH > 12. Nevertheless, the introduction of aluminum oxide into the glass imparts certain selectivity to alkali metal and ammonium ions and allows the development of sensors for these ions. However, such sensors should be operated at a high pH in order to avoid interference by the H+ ion. Sufficient electrical conductivity within the compact part of the glass membrane is secured by mobile Na+ ions.
10.6.2 Response Function and Selectivity
In order to derive the response function of a solid ion-exchanger membrane, it will be assumed that the whole membrane consists of a material like that in Figure 10.12A. In other words, it consists of a porous, solid ion-exchanger material containing fixed anionic sites R− at the pore surface where a double electric layer develops by cation attraction. Complete exclusion of the anion X− is achieved when the pore size is close to the double-layer thickness. This allows the repeling action of the R− sites to be effective over the whole volume of the filling solution. If the pore size is slightly greater, moderate anion permeation occurs, whereas when the pore size is very large with respect to the double-layer thickness, the penetration of the anion is unhindered.
A solid ion-exchanger membrane can be approached by the three regions concept (also known as the Teorell, Meyer and Sievers model, more concisely as the TMS model). As the membrane is cation selective, a boundary potential (![]() and
and ![]() ) develops at both faces of the membrane). At the same time, the difference in ion activity between the two sides of the membrane leads to the formation of an internal diffusion potential
) develops at both faces of the membrane). At the same time, the difference in ion activity between the two sides of the membrane leads to the formation of an internal diffusion potential![]() across the membrane (Section 10.3.1). Therefore, a sequence of three distinct regions form across the membrane, as shown in Figure 10.12B. According to this figure, the membrane potential can be obtained as follows:
across the membrane (Section 10.3.1). Therefore, a sequence of three distinct regions form across the membrane, as shown in Figure 10.12B. According to this figure, the membrane potential can be obtained as follows:
If two cations M+ and N+ (e.g., H+ and Na+, respectively) are present in the solution, a competition between these cations will take place in the form of ion exchange between the solutions (s) and the surface layers of the membrane (p and q). At the left side of the membrane, the ion-exchange process is:
(10.57) ![]()
An analogous equilibrium occurs at the right side of the membrane. The equilibrium constant of such a process is termed the ion-exchange constant![]() and this can be formulated as follows using the activity symbols shown in Figure 10.12B
and this can be formulated as follows using the activity symbols shown in Figure 10.12B
In order to derive the internal diffusion potential be means of Equation (10.25) the activity differences (![]() and
and ![]() ) between p and q are needed. These differences can be found from the electrical neutrality conditions: (
) between p and q are needed. These differences can be found from the electrical neutrality conditions: (![]() and
and ![]() ) that yield
) that yield ![]() . As the negative sites in the membrane are fixed, their mobility is zero (uR = 0) the diffusion potential results from the Henderson equation (10.25) as:
. As the negative sites in the membrane are fixed, their mobility is zero (uR = 0) the diffusion potential results from the Henderson equation (10.25) as:
(10.59) ![]()
On the other hand, the boundary potential formulas are, according to Equation (10.29):
(10.60) ![]()
The membrane potential can now be derived by substituting the above ![]() terms in Equation (10.56). One obtains thus:
terms in Equation (10.56). One obtains thus:
(10.61) ![]()
By taking into account the ion-exchange constant equation (10.58) and assuming that n2 = 0 (hence nq = 0), one obtains finally:
This is the Nikolskii–Eisenman equation for a solid ion-exchanger membrane. Assuming that m2 is a constant, Equation (10.62) can be converted into a form similar to Equation (10.38), with the potentiometric selectivity coefficient defined as:
(10.63) ![]()
This equations accounts for the effect of both cations and demonstrates that the response to the M+ activity is linear on a logarithm scale as long if ![]() . The ion-exchange constant, which is included in the
. The ion-exchange constant, which is included in the ![]() , indicates in fact the difference between the ions M+ and N+ affinities for the membrane. The affinity depends on the ion size and the strength of its interaction with the negative sites within the membrane. Thus, the very small hydrogen ion interacts much more strongly than any other monopositive cation in which the charge is spread over a larger volume. The very high mobility of the H+ ion (uM
, indicates in fact the difference between the ions M+ and N+ affinities for the membrane. The affinity depends on the ion size and the strength of its interaction with the negative sites within the membrane. Thus, the very small hydrogen ion interacts much more strongly than any other monopositive cation in which the charge is spread over a larger volume. The very high mobility of the H+ ion (uM ![]() uN) also imparts selectivity for this ion.
uN) also imparts selectivity for this ion.
It was assumed in the approach above that the membrane behaves as a homogeneous ion exchanger. However, in a real glass membrane, ion-exchanger layers form only at the membrane surface, whereas the bulk of the membrane preserves a compact glass structure that hydrogen ions cannot permeate. Consequently, the application of the Henderson equation is a crude approximation. Nevertheless, Equation (10.62) provides a fair description of both sensor response and the effect of interferences by other ions. More advanced theoretical approaches to the glass membrane sensors can be found elsewhere [1, 3].
10.6.3 Chalcogenide Glass Membranes
Chalcogenide glasses contain a large proportion of chalcogen elements, that is, sulfur, selenium and tellurium [28]. They are a kind of amorphous semiconductor with bandgap energies of 1–3 eV. There are marked differences between chalcogenide and oxide glasses due to the character of the metal–chalcogen bond. In oxide glasses, the oxygen–metal bond is purely ionic, whereas in chalcogenide glasses, the metal–chalcogen bond has a pronounced covalent character.
Chalcogenide glass materials have been used for preparing membranes that are selective for various cations and anions [29–31]. Chalcogenide glasses can be prepared from pure elements in evacuated quartz ampoules by heating at a defined temperature (up to 800–1200 K) for 6–20 h. The resulting amorphous material has to be quenched rapidly to room temperature. The solid material is cut into membranes 2–3 mm thick and 5–10 mm in diameter, which are then polished and sealed with epoxy resin into a plastic tube. A solid inner contact is obtained by metal deposition on the inner side of the membrane and attaching a metal wire by means of a conducting adhesive.
The material should include the analyte-element and, in addition, a group 13 (Ga), 14 (Ge) or 15 (As or Sb) element (Table 10.3).
Table 10.3 Chalcogenide glasses as recognition materials in potentiometric ion sensors [30]

The applications of chalcogenide glass sensors partially overlap those of solid salt membranes. However, better detection limit and selectivity have been reported for certain chalcogenide glass sensors. In addition, chalcogenide glasses allows the fabrication of solid membranes for ions such as Na+, Fe3+ and Cr(VI), that cannot be obtained by the solid salt technology. Another advantage emerges from the easy fabrication of thin glass films by pulsed laser deposition [30].
Before being used, chalcogenide glass sensors should be conditioned in a solution containing the analyte ion. During the conditioning, a modified surface layer forms at the surface of the glass. This layer functions as a semipermeable membrane analogous to that in oxide glasses. The conditioning and response mechanisms can vary in accordance with the glass structure and the nature of the analyte ion [29]. Due to the semiconductor character of chalcogenide glasses, the development of the inner diffusion potential involves either electrons (in n-type glasses) or positive vacancies (holes) (in p-type glasses).
10.7 Ion Sensors Based on Molecular Receptors. General Aspects
Solid membranes used in potentiometric ion sensors are characterized by the presence of ion-binding sites that interact with ions in solutions. The selectivity of such membranes is mostly determined by their crystal structure and the possibility of adjusting the selectivity is quite limited. Moreover, determination of a series of important ions, such as Ca2+, Mg2+ as well as organic ions cannot be approached by means of solid membranes. An alternative to solid membranes is based on molecular ion receptors that can interact selectively with the ion of interest. In order to obtain an ion-selective membrane, molecular receptors are embedded in a matrix that allows for ion transfer. Proper tailoring of the receptor structure, conformation and physical properties offers in principle an endless number of opportunities for obtaining receptors with good selectivity for particular ions.
Two different types of molecular ion receptors can be distinguished: ionic receptors (commonly known as liquid ion exchangers) and neutral receptors (also termed ionophores, that is, ion carriers). In the first case, the analyte–receptor interaction is based on electrostatic attraction, whereas in the case of neutral receptors a great variety of interactions can come into play. Although certain natural ionophores are available, numerous synthetic neutral ion receptors have been developed in the framework of supramolecular chemistry [32].
In the initial stage of development, molecular receptors were used in the form of solutions in water-immiscible solvents. Such solutions can be supported in a porous plastic disc. Hence, the name liquid membrane sensor has been assigned to the resulting device. This approach is now obsolete as much better performances have been obtained by incorporating the receptors in a thin film of polymer material to form an ion-sensitive polymeric membrane. As a rule, a polymeric membrane behaves as a typical liquid membrane.
The outstanding importance of molecular receptors resides in their application in both potentiometric and optical ion sensors. The topic of molecular ion receptors has been amply reviewed (see, e.g., refs. [19, 33, 34]).
10.8 Liquid Ion Exchangers as Ion Receptors
10.8.1 Ion Recognition by Liquid Ion Exchangers
Customarily, charged ion receptors are known as ion exchangers because they allow ion exchange between two immiscible phases: an aqueous solution and a nonaqueous solvent in which the ion exchanger is dissolved. In a hydrophobic phase (e.g., a liquid membrane) the receptor is accompanied by a counterion that balance the receptor charge.
Several ionic receptors commonly used in potentiometric sensors are shown in Figure 10.13. The calcium receptor in A is a long-chain dialkyl phosphoric acid ester. Due to the difference in partition coefficients, a membrane based on this receptor discriminates between Ca2+ and Mg2+. Another common ionic receptor for metal cations is dinonylnaphthylsulfonic acid whose anion is shown in Figure 10.13B.
Figure 10.13 Liquid ion exchangers used as ion receptors in potentiometric ion sensors. (A) A dialkyl phosphoric acid ester as Ca2+ receptor; (B) the dinonylnaphthylsulfonic acid anion; a cation receptor; (C) a nickel complex used as nitrate ion receptor; (D) Quaternary ammonium and phosphonium ionic receptors; (E) a porphyrin derivative perchlorate receptor (Ph is a phenyl group); (F) a guanidinium derivative receptor for the ![]() anion.
anion.
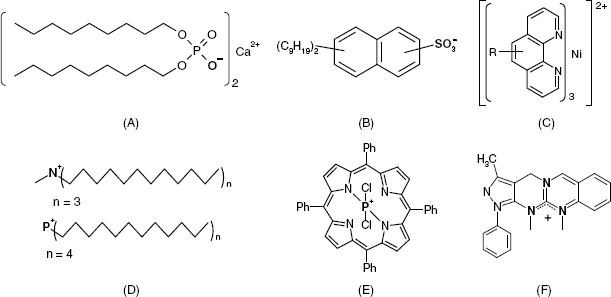
Positively charge receptors are, of course, used in anion sensors. Thus, the nickel complex with o-phenanthroline in Figure 10.13C is suitable for use as a nitrate ion receptor. Fair selectivity arises mostly from the rather high lipophilicity of this complex ion but other anions in the sample can interfere and their removal is recommended prior to the analysis. Figure 10.13D shows a quaternary ammonium receptor for Cl− and a phosphonium cation which is suitable as a receptor for either Cl− or ![]() ion. Quaternary ammonium ions are also suitable receptors for anionic surfactant [35]. A cationic receptor for the
ion. Quaternary ammonium ions are also suitable receptors for anionic surfactant [35]. A cationic receptor for the ![]() is shown in Figure 10.13E. In this case, steric factors, in addition to the partition coefficient, contribute to the selectivity. In contrast to the previous receptors, in which the charge is localized at a single atom, the receptor in Figure 10.13F which is a guanidinium derivative, displays a positive charge distributed over three nitrogen atoms, and this compound has proved useful for sensing the oxygenated
is shown in Figure 10.13E. In this case, steric factors, in addition to the partition coefficient, contribute to the selectivity. In contrast to the previous receptors, in which the charge is localized at a single atom, the receptor in Figure 10.13F which is a guanidinium derivative, displays a positive charge distributed over three nitrogen atoms, and this compound has proved useful for sensing the oxygenated ![]() anion.
anion.
Ionic receptors are in general inexpensive and ready available. However, the selectivity of ionic receptors is rather poor. In addition, certain cation receptors can be extracted into the aqueous solution in the protonated form. For this reason, low pH values should be avoided when using this kind of receptor.
10.8.2 Charged Receptor Membranes
One form of polymeric ion sensing membrane can be fabricated in a very simple way, by dissolving poly(vinyl chloride) PVC (≈33%), a plasticizer (≈66%) and the ion receptor (≈1%) in a suitable solvent (e.g., tetrahydrofuran) and letting the solvent evaporate slowly from a thin solution layer on a glass plate [33, 36]. The membrane is then cut and attached to a PVC tube holder. Other polymers, such as derivatized PVC, silicone rubber, polyurethanes, and polystyrene have also been tested as receptor matrices. However, in some applications, the use of a solvent causes problems. For this reason, photocurable, solvent-free polymers, such as methacrylates have been proposed as matrices.
The plasticizer (Figure 10.14) imparts plasticity to the membrane, but what is more important is that it functions as a solvent for the receptor and other membrane components. That is why the properties of the plasticizer determine to a large extent the performance of the ion sensor. Thus, in the case of an ionic receptor, which has no binding selectivity, the sensor selectivity arises from the partition of the analyte ion into the plasticizer solvent. A large number of plasticizers have been tested with a view to improving lipophilicity, solubility, and selectivity, and a strategy for the design of plasticizers has been advanced in ref. [37].
Figure 10.14 Two common plasticizers used in ion-selective liquid membranes: A nonpolar (A) and a polar (B) plasticizer.
The useful lifetime of a polymeric ion-sensitive membrane is limited by the slow leakage of the membrane components into the aqueous solution.
10.8.3 Response Function and Selectivity
The functioning of a charged receptor-based membrane is based on the ion-exchange process involving an ion M+ in solution (aq) and an ion N+ in the membrane (m) which also contains the ionic receptor R−:
The symbols under the above equation indicate cation activities. This equilibrium is characterized by the ion-exchange constant:
(10.65) ![]()
where Kp,M and Kp,N are the partition coefficients of M+ and N+, respectively (see Equation (10.26)). The extraction into the hydrophobic phase involves preliminary dehydration followed by solvation within the membrane. The hydration energy is much larger than the solvation energy, and, therefore, the partition coefficient depends mainly on the hydration energy of the ion. As a result, the partition coefficient increases with decreasing the degree of hydration. The degrees of hydration decrease with increasing ion size and decreasing ion charge. Thus, the partition coefficient for cation extraction into an anionic receptor-containing non-aqueous solvent increases in the following sequence (the Hofmeister series) [11]:
(10.66) ![]()
where ![]() is a lipophilic organic cation. In the case of organic ions, hydrophilicity, which is imparted by the organic substituents, leads to strong solvation within the membrane phase, which compensates for the hydration energy. For anion extraction the partition coefficient increases as follows:
is a lipophilic organic cation. In the case of organic ions, hydrophilicity, which is imparted by the organic substituents, leads to strong solvation within the membrane phase, which compensates for the hydration energy. For anion extraction the partition coefficient increases as follows:
(10.67) ![]()
where AcO− is the acetate ion and ![]() is a lipophilic organic anion.
is a lipophilic organic anion.
In order to derive the response function of a liquid membrane sensor, consider a semipermeable liquid membrane (which contains the ionic receptor R−) in contact with two solutions 1 and 2 containing the ions M+ and N+ with activities m1 and n1, respectively, in solution 1 and m2 in solution 2 (Figure 10.15). At each side of the membrane, a boundary potential develops according to Equation (10.29), whereas the ion activity gradient across the membrane gives rise to a diffusion potential that can be expressed by the Henderson equation (10.25). Therefore, the potential difference between the two solutions is:
Figure 10.15 Variation of the electric potential across a liquid ion-exchanger membrane inserted between two solutions.
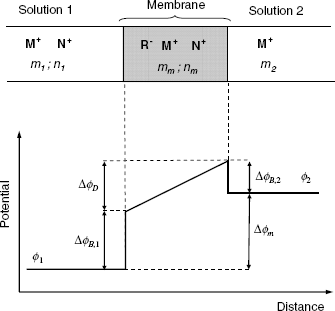
By making appropriate substitutions, the membrane potential equation is obtained in the following form which is valid when there is negligible ion association in the membrane [2]:
(10.69) ![]()
where uM and uN are the ion mobilities within the membrane. Hence, the potentiometric selectivity coefficient is:
(10.70) ![]()
As a rule, there are no major differences between the mobilities of similar ions in the membrane phase and the potentiometric selectivity coefficient can be approximated by the ion-exchange constant, that is, the ratio of the partition coefficients.
10.8.4 Outlook
Because the selectivity of the liquid ion-exchanger membranes depends mostly on the ion hydration energy, no exceptional performances can be expected from this point of view. Despite this limitation, sensors based on liquid ion-exchanger of membrane have proved useful in a series of applications involving both inorganic and organic ions.
10.9 Neutral Ion Receptors (Ionophores)
10.9.1 General Principles
The first neutral receptors used in ion sensors were natural ionophores. Ionophores are lipophilic compounds that can bind ions and transport them across the hydrophobic lipid bilayer of cell membrane. Among ionophores, one can distinguish both cyclic and noncyclic molecule compounds (Figure 10.16). Thus, valinomycin, a cyclic polypeptide (Figure 10.16A) binds potassium ions with an exceptional selectivity over other alkali-metal ions. Ion binding is performed by multiple ion–dipole interactions involving the polar ߝC=O groups in which the oxygen atom bears a partial negative charge. The selectivity for the K+ ion is due to the fact that the ion diameter matches the internal diameter of the macrocycle, which implies that the ion can interact with all available polar oxygen atoms. Smaller ions (Li+, Na+) interact with a smaller number of oxygen atoms, whereas bulkier ions (e.g., Cs+) do not fit inside the macrocycle.
Figure 10.16 Natural ionophores. Dotted lines in (A) indicate the limits of each of the three component peptides in valinomycin.
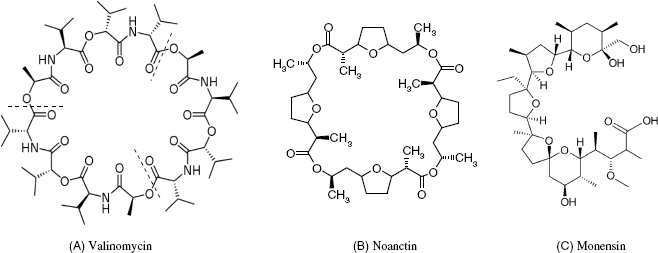
Nonactin (Figure 10.16B) shows a slight preference for ![]() over K+ due to the formation of hydrogen bonds with the oxygen atoms directed towards the interior of the macrocycle.
over K+ due to the formation of hydrogen bonds with the oxygen atoms directed towards the interior of the macrocycle.
An example of a noncyclic ionophore is monensin (Figure 10.16C), which binds various monovalent cations, particularly Na+. As in the case of valinomycin, the cation is bound by ion–dipole interactions, but, due to the noncyclic conformation, ion wrapping is achieved by the receptor molecule folding around the ion.
The examples above indicate the main factors that determine the selectivity of neutral receptors, namely:
The effect of the bonding multiplicity is obvious as it determines to a large extent the strength of the analyte–receptor interaction. The interaction strength depends also on the nature of the chemical bonds involved. Finally, the shape and size fit is crucial for securing as strong as possible analyte–receptor bonding. This depends on the chemical structure and the conformation of the receptor molecule.
So, in contrast to liquid ion exchangers that bind the analyte by strong ion–ion interactions, neutral receptors form relatively weak bonds such as ion–dipole, dipole–dipole and hydrogen bonds. Bond multiplicity, however, enhances the stability of the analyte–receptor complex. In aqueous solutions, the stability of the complex is lessened by ion hydration, but ion solvation is absent in the lipophilic membrane environment, and this enhances the stability of the complex.
The effects of the chemical bonding nature and steric factors on ion recognition by neutral receptors are discussed in the next sections.
10.9.2 Chemistry of Ion Recognition by Neutral Receptors
The feasibility of ion recognition by molecular receptors depends on the type of ion–receptor interaction that takes place. The simplest type of interaction is that of an electrostatic nature which consists of electrostatic attraction between particles of opposite electric charge. Thus, an electrostatic interaction occurs when the receptor is a liquid ion exchanger, that is, a molecular ion. However, electrostatic interactions are also met in the case of neutral receptors provided that the receptor molecule includes strongly polarized chemical bonds. Thus, the carbon–oxygen bond in an organic compound is strongly polarized because of the large difference in electronegativity and the oxygen atom carries a fractional negative charge. A cation experiences attraction by the fractional charge on the oxygen in what is known as an ion–dipole interaction. This kind of interaction is weaker than an ion–ion interaction, but if the ion interacts with multiple dipoles at the receptor molecule, the strength of the ion-receptor bond can be quite large. This kind of interaction is a characteristic of alkali-metal ions.
Less-electronegative nonmetals substituents, such as nitrogen or sulfur, are less polarizable. However, they can donate a lone pairs of electrons to an empty valence orbital in transition-metal ions to form a coordinate bond.
Hydrogen bonding can be involved in the recognition of hydrated cations by means of water bridges between the ion and the negatively polarized atoms in the receptor molecule. In this case, the receptor size must be sufficiently large to incorporate the hydrated ion, which can be much larger than the nonhydrated form.
![]() -Electrons in aromatic rings can also take part in ion-recognition processes. Thus, transition-metal cations such as Fe2+/3+ and Co2+/3+ are known to form coordination compounds with aromatic hydrocarbons by accepting
-Electrons in aromatic rings can also take part in ion-recognition processes. Thus, transition-metal cations such as Fe2+/3+ and Co2+/3+ are known to form coordination compounds with aromatic hydrocarbons by accepting ![]() electrons into an empty orbital. Aromatic molecules form weak bonds by
electrons into an empty orbital. Aromatic molecules form weak bonds by ![]() stacking, which is due to van der Waals force interactions between face-to face oriented aromatic rings.
stacking, which is due to van der Waals force interactions between face-to face oriented aromatic rings. ![]() Stacking is important in organic ion recognition where aromatic substituents in the receptor impart selectivity for aromatic over aliphatic analytes.
Stacking is important in organic ion recognition where aromatic substituents in the receptor impart selectivity for aromatic over aliphatic analytes.
The above types of interaction can also operate in the case of anion receptors. In addition, coordination bonds involving anions can be formed when the receptor contains transition-metal sites.
Metal–ligand interactions have been rationalized within the framework of the hard and soft acids and bases concept (HSAB theory [38, 39]) that refers to the interaction of Lewis acids and bases. A Lewis acid is a molecular entity that is an electron-pair acceptor, whereas a Lewis base is an electron-pair donor. A Lewis acid and a Lewis base are therefore able to form a Lewis adduct by sharing the electron pair provided by the Lewis base. In this context hard implies small and nonpolarizable entities and soft indicates larger entities that are more polarizable. Low polarizability is associated with small size and strong electronegative or electropositive character, whereas high polarizability occurs with bulky entities bearing a small charge. Borderline cases that show intermediate properties can also be identified. Several Lewis acids and bases are shown in Table 10.4.
Table 10.4 Lewis acids and bases classified according to the soft/hard character.
The strongest Lewis acid–base interaction occurs when both reactants are either hard or soft, while the interaction of a hard and a soft reactant is much weaker. Thus, oxygen in ethers (R1–O–R2) is a hard base that interacts strongly with hard Lewis acids such as alkali-metal ions. Conversely, sulfur in thioethers (R1–S–R2) is a soft base and interacts preferentially with soft Lewis acids such as Ag+ or Hg2+. The limiting cases in Lewis acid–base interactions are the ionic bonding (hard–hard character) and the coordinate–covalent bonding (soft–soft character).
It is clear from the above discussion that the selectivity of a neutral ion receptor is determined largely by the hard/soft character of the binding groups. This suggests a broad range of possibilities for tuning receptor selectivity in conjunction with structural and conformational factors.
10.9.3 Effect of Bonding Multiplicity, Steric, and Conformational Factors
As already mentioned, the stability of the metal complex is enhanced if the receptor provides multiple binding sites to the analyte ion; such a molecule is called a multidentate ligand. But, in this case, steric factors also come into play. The conformation of the receptor molecule should be such as to allow the ion to make contact with a maximum number of binding sites at the receptor molecule.
The above principles are illustrated in Figure 10.17 that shows the effect of the receptor (host) shape on the stability of the resulting metal complex. The simplest receptor is a multidentate linear ligand (podand) that is able to interact with the ion by means of each binding site and that folds around the ion forming a chelate complex. Multiple bonding has a synergic effect as the strength of the resulting complex is greater than the sum of each ion-site interaction, an effect known as the chelate effect. Complex stability is impaired by any tension in the ligand molecule that would prompt it to unfold.
Figure 10.17 The chelate, macrocyclic, and macrobicyclic effect. Each triangle represents an ion binding site in the receptor molecule. Adapted with permission from [32]. Copyright 2009 John Wiley & Sons, Ltd.
More stable complexes result when the receptor is a macrocyclic compound shaped such as to be able to wrap round the ion (corand). There is no cause for the ligand to unfold in this case, and this imparts greater stability to the complex. The chelate effect also operates in this case, but, in addition, the macrocyclic effect due to the stable cyclic conformation of the receptor comes into play.
Even greater stability is noticed when the receptor molecule has a three-dimensional configuration with more than one cyclic moiety (that is, a cryptand). Enhanced complex stability arises from the rigidity of the ligand molecule. While in the case of a corand the molecule can suffer slight deformation that reduces the stability of the complex, this is hardly possible with a cryptand.
In conclusion, the stability of the analyte–receptor complex increases with the degree of organization of the receptor molecule. Highly organized receptors display rigid conformation and only an ion matching the receptor size and shape can be accommodated.
Remembering that the receptor is intended to extract the ion from an aqueous solution into a lipophilic membrane, it is clear that the receptor should be a hydrophobic compound. Hydrophobicity is imparted by aromatic or long-chain aliphatic moieties.
10.9.4 Neutral Receptor Ion-Selective Membranes: Composition, Selectivity and Response Function
The fabrication of neutral receptor membranes is similar to that emphasized for the fabrication of liquid exchanger membranes [33]. The preparation consists of a mixture of matrix polymer (PVC), a plasticizer and the receptor. However, a neutral receptor membrane should contain, in addition, a lipophilic ionic additive with an electric charge opposite to that of the analyte. In practice, alkali salts of tetraphenylborate derivatives (Figure 10.18A) are used in cation-selective membranes and tetralkylammonium salts (Figure 10.18B) in anion-selective membranes. The role of the ionic additive is to prevent coextraction of solution counterions as ion pairs with the analyte.
Figure 10.18 Ionic additives for neutral receptor liquid membranes. (A) Potassium tetrakis-[3,5,-bis(trifluoromethyl)phenyl]borate; (B) tridodecylmethyl ammonium chloride.
Ion interaction with the membrane involves ion partitioning into the membrane followed by interaction with the neutral receptor R to yield a complex [MR]+.
In a water/lipophilic solvent system, the partition equilibrium can be expressed as follows:
(10.71) ![]()
A partition coefficient (Kp,M) can be defined with respect to the activities of the free ion in the solution and membrane phase, (ms and mm, respectively):
Formation of the ion complex with the receptor R is represented by the following reaction in which the activity symbol for each species is indicated below the species symbol:
(10.73) ![]()
The driving force in the ion transfer from the solution to the membrane is the change in the free energy that accompanies the complex formation and is correlated with the stability constant of the ion–receptor complex given in the following equation:
The free metal ion activity in the membrane results from the partition coefficient Equation (10.72). On substituting this activity into Equation (10.74) one obtains the activity of the complex as a function of the ion activity in the solution, maq:
Assuming that an interfering cation N+ is also present in the system, it follows that the analyte interaction with the membrane can be represented by the following reaction at equilibrium:
(10.76) ![]()
(aq) and (m) denote the aqueous an membrane phase, respectively; X− is the ionic additive in the membrane. As each cation forms a complex with the receptor, the selectivity coefficient of the recognition process can be expressed as ![]() . By using Equation (10.75) and a similar equation for cN, and assuming that the receptor is in high excess and its concentration changes to a negligible extent, one obtains:
. By using Equation (10.75) and a similar equation for cN, and assuming that the receptor is in high excess and its concentration changes to a negligible extent, one obtains:
As in the case of the liquid exchanger membrane, the membrane potential can be expressed by an equation similar to Equation (10.68). However, the mobilities of the [MR]+ and [NR]+ complexes are similar, being determined by the properties of the receptor. Therefore, no diffusion potential forms across the neutral receptor membrane and the membrane potential is given by the difference in the boundary potentials. If the membrane is inserted between two solutions containing the ion M+ with activities m1 and m2, respectively, the membrane potential is:
(10.78) ![]()
The effect of the interfering ion N+ is accounted for as follows [2]:
(10.79) ![]()
where the potentiometric selectivity coefficient is ![]() . Therefore, the potentiometric selectivity coefficient is mostly determined by the stabilities of ion–receptor complexes involved. The partition coefficients ratio in Equation (10.77) is close to unity and has little effect on selectivity.
. Therefore, the potentiometric selectivity coefficient is mostly determined by the stabilities of ion–receptor complexes involved. The partition coefficients ratio in Equation (10.77) is close to unity and has little effect on selectivity.
The ionic additive in the membrane plays an essential role in determining the limit of detection of the sensor. At a high anion concentration in the solution, the solution anion can be extracted into the membrane as an ion pair with a solution cation. Under these conditions, the sensor becomes anion responsive, which limits the cation response range. However, if an anionic additive is present, the permeation of the solution anions is prevented and the cation response range expands considerably.
The polarity of the plasticizer molecule can also exert a notable influence on the selectivity of neutral receptor-based membranes. When a more polar plasticizer is present, divalent ions are preferred over monovalent ions.
Although the above discussion has focused on cation sensors, similar conclusions apply to anion sensors as well.
In Section 10.9.1 several neutral ion receptors have been introduced. Such compounds are rather expensive and their applications are restricted to certain alkali-metal ions and ammonium. Synthetic chemistry has developed a large number of neutral receptors tailored so as to display selectivity for various cations and anions. By extension, the term ionophore is also used to designate synthetic neutral receptors. The following sections review a series of neutral receptors for both cations and anions.
10.9.5 Neutral Noncyclic Ion Receptors
Several podand-type cation receptors are shown in Figure 10.19. The molecule in (A) contains six soft base groups that interact preferentially with the Zn2+ ion forming a chelate complex. Other transition-metal ions (Cu2+, Pb2+ Hg2+) interfere significantly, but alkali ions are well discriminated against. As the protonation of the nitrogen sites is possible, a good response to Zn2+ is obtained at pH 3.5–6.5. The receptor in Figure 10.19B displays selectivity to the Na+ ion as it forms multiple ion–dipole bonds and folds optimally around this ion. A Ca2+ receptor is shown in Figure 10.19C. It contains six binding sites (ether oxygens) and has no protonable groups that would cause interference by the hydrogen ion, as occurs with ionic receptors.
Figure 10.19 Noncyclic cation receptors.
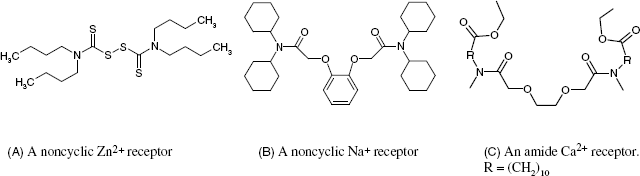
Noncyclic, neutral anion receptors have also be designed [40–42]. Several examples are shown in Figure 10.20. Preorganized bis-thiourea derivatives provide anion recognition by hydrogen bonding. Thus, the compound in Figure 10.20A is selective for the sulfate ion, whereas a similar compound with thiourea moieties separated by a slightly longer distance demonstrates selectivity for Cl−. Anion recognition can also be achieved by analyte coordination to metal centers in the receptor molecule as is the case in Figure 10.20B. The compound in (B) is an organometallic tin-fluorine derivative that can interact with the fluoride ion via coordinate bonding. A binding metal site can also be introduced into the receptor molecule by coordination, as shown in Figure 10.20C for a hydrogenphospahate receptor. In this case, the hydrogenphospahate ion can bind coordinatively to the uranyl center and also to the methoxy groups by hydrogen bonding.
Figure 10.20 Noncyclic neutral anion receptors.
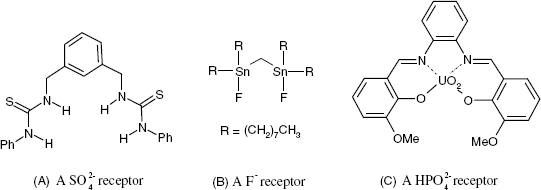
It is of interest to note that neutral receptors for the hydrogen ion have also been developed and tested successfully. Such receptor molecules include a double-bonded tertiary nitrogen atom (ߝC=Nߝ) which becomes positive by protonation. A liquid membrane pH sensor has been validated for pH determination in diary products [43]. An important advantage of the liquid membrane pH sensors arises from the facile miniaturization and the possibility of integration in sensor arrays.
Calixarenes provide a versatile scaffold for assembling binding sites, either at the upper or lower rim, in order to form podand-type receptors. Thus, exceptional Na+ selectivity has been achieved with simple ester substituents such as ![]() . Anion recognition can be performed by calixarenes substituted with coordinated metal ions and amidic or urea functions [44]. For example, functionalization with upper rim sulfonamide substituents leads to a three-dimensional cavity that binds the tetrahedral
. Anion recognition can be performed by calixarenes substituted with coordinated metal ions and amidic or urea functions [44]. For example, functionalization with upper rim sulfonamide substituents leads to a three-dimensional cavity that binds the tetrahedral ![]() ion stronger than the spherical Cl− or the planar
ion stronger than the spherical Cl− or the planar ![]() ions.
ions.
10.9.6 Macrocyclic Cation Receptors
Macrocyclic cation receptors are synthetic analogs of natural cyclic ionophores. However, while natural ionophores display selectivity to only a couple of ions, synthetic macrocyclic receptors have been developed for a very broad range of both cations and anions.
Macrocyclic ethers (also termed crown ethers, Figure 10.21) were the first compounds of this type to be synthesized and tested. The name of a crown compound includes the term “crown” and two numbers; the first one indicates the total number of atoms in the cycle and the second one, the number of heteroatoms. Besides oxygen in oxa groups (ߝOߝ), aza groups (ߝNߝ) or thia groups (ߝSߝ) can be included in the macrocycle. The selectivity of such receptors is determined by size fitting and the nature and multiplicity of the analyte–binding-site interactions.
Figure 10.21 Macrocyclic ethers as alkali ion receptors.
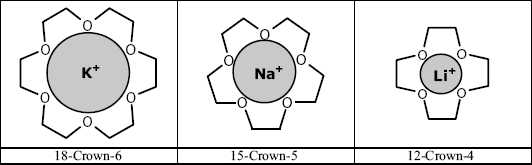
The importance of the steric factor is illustrates in Figure 10.21 that shows that a good match of the ion and receptor size allows the maximum possible ion–dipole interactions to occur, thus imparting maximum stability to the complex.
Beside the size matching, the nature of the heteroatoms in the crown receptor contributes substantially to the selectivity in accordance with the HSAB principle. Thus, the macrocyclic ether in Figure 10.22A includes strong Lewis bases and displays selectivity to the K+ strong Lewis acid. Introduction of tertiary nitrogen groups that are much softer bases (Figure 10.22B) imparts selectivity for the Hg2+ ion. The Lewis base hardness of the nitrogen site depends on the electronic effect applied by the substituent R that allows variation in the polarity of the CߝN group.
Figure 10.22 Effect of the Lewis base hardness of the binding sites in macrocyclic ion receptors.
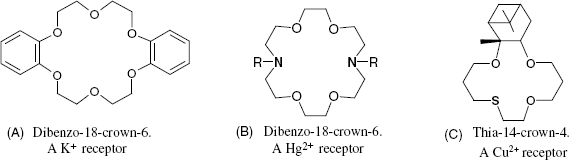
The soft base thia group in Figure 10.22C imparts very good selectivity for the Ag+ soft acid, although its ionic diameter (2.00 Å) is somewhat larger than the cavity diameter (1.1–1.4 Å). Binding of more voluminous soft acid ions (e.g., Hg2+) is prevented by the bulky substituent in the upper part of the molecule. The combined effect of size fitting and steric hindrance leads to a selectivity coefficient ![]() of about 0.03 for an Ag+ sensor based on this receptor [45].
of about 0.03 for an Ag+ sensor based on this receptor [45].
A higher degree of organization of the receptor molecule is expected to impart more selectivity. This is demonstrated by examining the binding energy of alkali ions to the receptors in Figure 10.23. The compound in A is a spherand, that is, a macrocyclic molecule in which binding groups are not constituents of the macrocycle itself but are appended to the cycle. The binding energy sequence demonstrates a clear preference for small ions, particularly for Li+. Compounds in Figures 10.23B–D are cryptands (macrobicycles). In such compounds, one or more additional chains link distant atoms in the macrocycle. As a result, the conformation of the molecule is very rigid, which prevents the molecule from adapting the cavity size to that of the ion upon binding. Consequently, the compound in Figure 10.23B displays exceptional selectivity for the Li+ ion due to the size-exclusion effect. The larger molecule in Figure 10.23C can bind all alkali ions (except Cs+) but with a clear preference for Na+. An even larger bicyclic molecule like that in Figure 10.23D prefers more voluminous ions but does not bind to the small Li+ ion at all
Figure 10.23 Highly organized alkali ion receptors. (A) A spherand; (B–D) cryptands. The series of binding energy for alkali ions is indicated for each compound. Adapted with permission from [46]. Copyright 1986 Wiley-VCH Verlag GmBH & Co. KGaA.
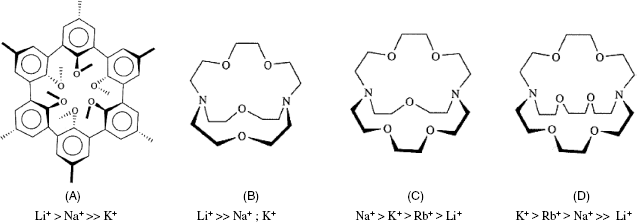
An enhancement of the binding strength is obtained by adding to the macrocycle a side arm that contains an ion binding site to form a lariat crown ether. The side arm could position itself over the cavity of the crown compound and contribute to ion encapsulation (Figure 10.24A). Alternatively, the macrocycle can function only as a scaffold for assembling binding sites in a suitable spatial arrangement (Figure 10.24B). Proper selection of the side arm binding group leads to enhanced selectivity in accordance with the HSAB principle.
Figure 10.24 Metal ion binding to lariat crown compounds. (A) Binding to both the macrocycle and the side arm; (B) binding to two or more side arms; (C) binding to the side arm and a site at the macrocycle. Thin arrows indicate binding to the macrocycle heteroatoms; thick arrows indicate the displacement of the binding group (L) at the side arm in order to bind to the ion.

10.9.7 Macrocyclic Anion Receptors
Macrocyclic receptors for anions are of great interest because, with the notable exception of the solid membrane fluoride sensor, other anion sensors based on solid-state membranes are generally plagued by poor selectivity. Anion receptors and potentiometric anion sensors are comprehensively reviewed in refs. [34, 40, 47]. Several macrocyclic anion receptors are shown in Figure 10.25. Thus, the aza crown compounds in Figure 10.25A can form hydrogen bonds with an oxygenated anion such as ![]() . Hydrogen bonding with anionic receptors can also take place with imidazolium moieties [48]. For example, Figure 10.25B shows a calixpyrrole compound that can adopt either a cone or 1,3-alternate conformation like calyx[4]arenes. Upon F− binding by hydrogen bonding, it is fixed in a cone conformation and preferentially discriminates well against the Cl− ion. The compound in Figure 10.25C includes an organometallic aluminum substituent. As aluminum is a hard Lewis acid, it interacts strongly with the hard Lewis base F−. Metal centers can also be included by coordination as shown in Figure 10.25C for a cyanide ion receptor. Each Cu2+ ion, which is a soft Lewis acid, can form coordinate bonds with a cyanide ion (soft Lewis base) which imparts selectivity for this ion.
. Hydrogen bonding with anionic receptors can also take place with imidazolium moieties [48]. For example, Figure 10.25B shows a calixpyrrole compound that can adopt either a cone or 1,3-alternate conformation like calyx[4]arenes. Upon F− binding by hydrogen bonding, it is fixed in a cone conformation and preferentially discriminates well against the Cl− ion. The compound in Figure 10.25C includes an organometallic aluminum substituent. As aluminum is a hard Lewis acid, it interacts strongly with the hard Lewis base F−. Metal centers can also be included by coordination as shown in Figure 10.25C for a cyanide ion receptor. Each Cu2+ ion, which is a soft Lewis acid, can form coordinate bonds with a cyanide ion (soft Lewis base) which imparts selectivity for this ion.
Figure 10.25 Macrocyclic anion receptors. Me in (C) stands for methyl.
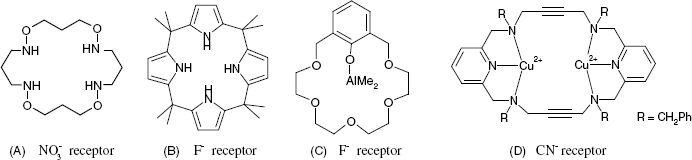
10.9.8 Neutral Receptors for Organic Ions
The interest in determining organic ions arises from the great importance of such ions in physiological processes as well as from the great number of pharmaceutical products of this type. Cationic receptors have been extensively employed for such applications, but, in a search for better selectivity, the attention has shifted to cyclic or noncyclic neutral receptors [34].
Of great interest is the determination of organic ammonium ions. It was found that aliphatic quaternary ammonium ions interact by ion dipole interactions with strongly polarized oxa groups such as that in the crown ether in Figure 10.22A but much better discrimination of alkali-metal ions has been found with the podand-type receptor shown in Figure 10.26A. Primary ammonium ions can form hydrogen bonds with oxa sites in macrocycles such as that shown in Figure 10.26B. This compound, which is rich in phenyl residues displays particular selectivity for aromatic derivatives such as amphetamine due to the ![]() stacking interactions.
stacking interactions.
Figure 10.26 Receptor for organic ions (up) and corresponding analytes (down). (A) A podand-type receptor for cationic surfactants; (B) a macrocyclic receptor for a primary ammonium aromatic compound; (C) a phthalocyanine-type receptor for salicylic acid and its derivatives.
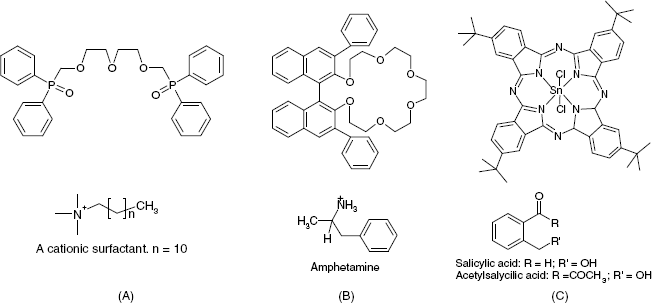
Determination of carboxylates and their esters is of particular interest due to the broad applications of salicylic acid and its derivatives in therapy. Figure 10.26C shows a receptor for compounds of this type. This receptor is a tin phthalocyanine and the analyte interacts with the metal center according to the HSAB principle.
Calixarenes present particular advantages because such receptors can form inclusion compounds with organic ions, thus providing an additional selectivity element in addition to the ion–dipole interactions.
Of particular interest is the chiral recognition of ions [49]. Chiral recognition requires at least three simultaneous interactions between the receptor and one of the enantiomers, with at least one of these interactions being stereochemically dependent.
10.9.9 Porphyrins and Phthalocyanines as Anion Receptors
Both porphyrins (Figure 10.27A) and phthalocyanines (Figure 10.27B) feature four pyrrole-like subunits linked to form a 16-membered ring which can bind a transition-metal ion at the center. Closely related to porphyrins are vitamin B12 derivatives. The metal ion in such compounds can be coordinated axially with either one or two ligands P and X−. In the first case (Figure 10.27C) the receptor is positively charged and the interaction with the analyte anion A− involves an ion-exchange process. This process is formally like that in scheme (10.64) except for the fact that the analyte in the membrane phase is not included in an ion pair but is bound to the metal center in the receptor. In the second case (Figure 10.27D), the receptor is doubly coordinated, hence it is a neutral species. Therefore, the exchange of the neutral ligand P with the ionic analyte results in a charge modification. Therefore, the membrane should contain an ionic additive to preserve the charge balance.
Figure 10.27 (A) A porphyrin (hemoglobin); (B) a phthalocyanine; (C) anion recognition by the ion-exchange mechanism; (D) anion recognition by ion-neutral ligand exchange.
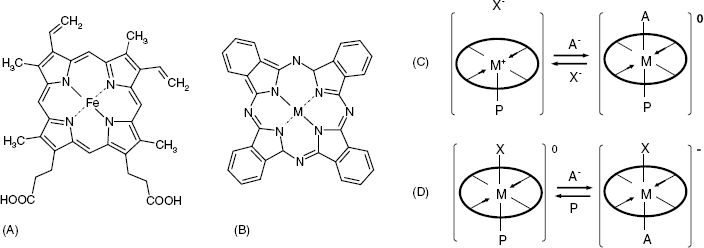
The strength of the analyte–receptor interaction depends on the nature of the metal center, its oxidation state, and the number and the nature of axial ligands. There are, therefore, various possibilities for tuning the receptor selectivity for various anion sensing applications [47].
10.9.10 Outlook
Neutral ion receptors offer endless possibilities for developing ion sensors with suitable selectivity for both inorganic and organic ions by rational design of the molecular structure and conformation. Selectivity arises from the multiple bonding character of the analyte–receptor interaction and also from the careful selection of the most suitable type of chemical bonding.
Currently, neutral receptors are dominating the field of ion sensor science and practical applications are restricted only by their availability. The single drawback of these devices is the relatively short lifetime of the membrane that is often limited to several months due to the leaking of the membrane components. The major advantage of neutral receptor-based ion sensors is their exceptional selectivity, which, in general, is not matched by any other type of ion-selective membrane.
10.10 Molecularly Imprinted Polymers as Ion-Sensing Materials
Molecularly imprinted polymers (introduced in Section 6.7.3) provide excellent opportunities for ion sensing and are of particular interest when no suitable molecular receptor is available [50–52]. In order to obtain an ion-selective polymer, specific ion ligands, appended with monomer tails are subjected to copolymerization with another monomer that produces the polymer matrix (Figure 10.28). The polymerization is conducted in the presence of the ion of interest and this acts as a template and obliges the ligand to adopt the spatial orientation specific to the metal-ion complex (Figure 10.28). After leaching out the ion from the resulting polymer, the matrix includes small pores that provide specific binding sites for the ion. In the case shown in Figure 10.8, two binding sites covalently bonded to the polymer backbone are present. However, binding sites can also be included by trapping receptor molecules in the matrix along with covalently bound ligands.
Figure 10.28 Molecular imprinting of a Pd2+ ion receptor. Left: template ion complex including monomers; right: imprinted receptor site. Adapted with permission from [50]. Copyright 2006 Elsevier.

Once formed, the polymer is ground down to some 60 mesh and cast in PVC membranes. Alternatively, the polymerization can be conducted at a solid support surface. Electrochemical polymerization at a conducting material surface represents yet a further method of preparing molecularly imprinted polymer membranes. Ion binding sites can be obtained using well-established metal ion reagents developed in classical analytical chemistry. Molecular receptors (ionophores) can also be appended to the polymer backbone; this results in enhanced selectivity compared with that of the free receptor.
Molecularly imprinted polymers have been used to produce sensors for more exotic ions such as uranyl, or dysprosium as well as for common ions such as Ca2+. A nitrate ion sensor with good selectivity has been obtained by electrochemical polymerization of pyrrole onto carbon electrodes in a NaNO3 solution [53]. This appears as a remarkable achievement if we remember that molecular receptor-based membranes for the nitrate ion are not particularly selective.
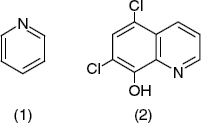
10.11 Conducting Polymers as Ion-Sensing Materials
The general characteristics of conducting polymers have been emphasized already in Section 5.9. In summary, a conducting polymer molecule includes an extended ![]() -orbital system. By oxidation, positive charges (in the form of bound radical ions) can be imparted to the polymer backbone. Charge balance is secured by mobile counterions (dopants) associated with the polymer in a gel-like structure. Various substituents can be appended to the polymer backbone in order to enhance the selectivity.
-orbital system. By oxidation, positive charges (in the form of bound radical ions) can be imparted to the polymer backbone. Charge balance is secured by mobile counterions (dopants) associated with the polymer in a gel-like structure. Various substituents can be appended to the polymer backbone in order to enhance the selectivity.
In potentiometric ion sensors, conducting polymers can be used as (a) ion-exchanger recognition materials, and (b) immobilization matrices for molecular ion receptors [13, 54, 55].
As a conducting polymer molecule contains fixed positive charges, it can act as an anion exchanger, as shown in Figure 10.29A. In order to perform cation exchange, the dopant should be a lipophilic anion (e.g., tetraphenylborate or dodecylsulfate) that is entrapped within the polymer matrix and acts as a fixed anionic site (Figure 10.29B). Sensors for various cations (including the hydrogen ion) have been developed in this way. Moreover, as conducting polymers can perform electron exchange with redox species in solution (see Figure 10.29C), this allows ionic and neutral redox species to be determined. However, redox reactions involving a conducting polymer can be a source of interference in ion determinations based on ion-exchange processes.
Figure 10.29 Charge-exchange processes involving polypyrrole. (A) Anion exchange involving the polymer counterion; (B) cation exchange involving a bulky anion embedded in the polymer. (C) electron exchange with a redox species.
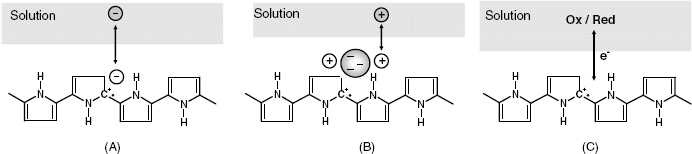
Improved selectivity over the ion-exchange recognition method is obtained by including a molecular ion receptor within the polymer matrix by entrapment or covalent binding. This alternative is characterized by enhanced stability of the sensing material.
10.12 Solid Contact Potentiometric Ion Sensors
Direct electrical contact of the sensing membrane with the external circuit can be achieved using a solid contact, thus avoiding the use of an internal reference electrode and internal solution. This simplifies the fabrication process, allows for miniaturization and imparts enhanced robustness. The solid contact should be designed such as to allow for facile transition from ionic conduction in the membrane to electronic conduction in the connecting wire. As a potential difference develops at the contact, it is included as an additional term in the cell voltage and, therefore, it should be very stable.
In the case of solid membranes, solid contact can be achieved by means of conductive epoxy resins. Better contact is achieved by depositing a thin metal layer over the top side of the membrane.
Direct contact to polymeric sensing membranes can be achieved simply by casting the membrane over the surface of a metal wire in order to obtain a so-called coated wire potentiometric sensor. However, the contact potential difference is not sufficiently stable. Nevertheless, such devices have proved useful as indicator electrodes in potentiometric titrations [56, 57]. It has been proved recently that a very stable response can be obtained by including highly porous materials (such as nanostructured macroporous carbon, porous silicon or carbon nanotubes) between the membrane and the metal wire [58].
Another approach is based on the inclusion of the receptor into a carbon paste mixture that functions as the sensing membrane [59–61]. Thus, perchlorate and tetrafluoroborate ion-selective carbon paste sensors demonstrated similar characteristics to those obtained with commercial polymer membranes [62]. However, the carbon pastes provide a faster response and have very low ohmic resistance.
Screen-printing technology is very convenient for developing direct contact potentiometric sensors [63]. In this design, the sensing membrane is formed as a thin layer over the surface of the working electrode. A molecular ion receptor can be incorporated into the conventional insulating paste. Alternatively, a carbon paste containing metal sulfide can act as a metal ion-sensing element. Ag/AgCl reference electrodes can also be fabricated by screen printing. Arrays of millimeter-sized sensors assembled on the same strip have been developed.
A good membrane-metal contact is obtained by means of materials that elicit both ionic and electronic conductivity, such as conducting polymers [13, 55]. In such materials, ionic conductance is provided by counterion displacement, whereas electronic conductance arises from electrons jumping from filled to empty orbitals in the polymer (hole conduction mechanism). The transition from ionic to electronic conductivity is achieved according to reaction (10.80) in which P+ is a positive site in the polymer backbone and A− is the counteranion:
It is easy to realize that this process is analogous to that occurring in a standard reference electrode (reaction (10.22)).
Direct contact with a metal conductor can be achieved if the sensing membrane is made of a conducting polymer that is deposited by electrochemical polymerization onto a metal or carbon electrode surface. When using a sensing polymer membrane, a layer of conducting polymer between the top side of the membrane and a metal contact piece secures the contact. Direct contact between the membrane and a metal conductor can also be obtained by including a small amount of conducting polymer in the membrane material. In order to secure a stable potential, it is important to avoid the formation of a solution layer between the sensing membrane and the conducting polymer plug.
10.13 Miniaturization of Potentiometric Ion Sensors
Potentiometric microsensors are used in the biosciences for the measurement of ion activity in individual cells or in intracellular liquid and also in environmental science for ion determination in pore water samples or for profiling the ion concentration in sediments. Such sensors have also proved useful as detectors in liquid chromatography and capillary zone electrophoresis and are promising for applications in micro-total analysis systems. Principles and applications of potentiometric microsensors are amply reviewed in refs. [64, 65] and the applications in plant-cell biology are surveyed in ref. [66].
Microsensors for common ions (such as H+, K+, Ca2+, Cl−) have been developed using suitable recognition materials or reagents. They are manufactured from glass micropipettes with a tip diameter down in the micrometer range. pH microsensors of this type rely on glass membranes, whereas metal cation sensors are based on molecular receptors dissolved in a nonaqueous solvent to form a liquid membrane, as shown in Figure 10.30. In order to prevent water penetration through the micropipette tip, the inner surface should be made hydrophobic by silanization. The internal filling solution can be replaced by a hydrogel layer.
Figure 10.30 Cross section of micropipette potentiometric ion sensors. (A) Single-barreled sensor; (B) double-barreled sensor. A septum separates the reference (right) and the measuring (left) compartments.
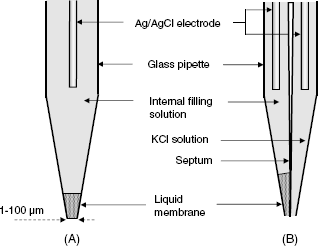
When a single-barreled sensor (Figure 10.30A) is used in a single cell, the reference electrode is placed outside the cell. This procedure needs additional corrections to the voltage read-out and is not suitable for electrically excitable cells. Such problems can be circumvented by using double-barreled microsensors (Figure 10.30B). Although micropipette-type sensors are physically large devices, they can be used with sample volumes down to the picoliter range. However, such microelectrodes are fragile, difficult to prepare, and the lifetime is only in the order of days.
Recent research work has been focused on improving the ruggedness and stability of microsensors by using new materials and design principles [13, 58]. A more robust design relies on direct internal contact achieved by means of conducting polymers, which exclude the need for reference electrodes. The fragile glass micropipette can be replaced by monolithic capillaries as holders for the ion-sensing membrane [67].
An alternative to the micropipette design is based on sensing membranes attached over a recessed gold microelectrode of some 25 μm diameter [68]. Advanced miniaturization has been achieved by using conducting polymers as sensing materials. Thus, a sub-μm-sized pH sensor with excellent selectivity has been developed by coating polyaniline over the surface of a carbon microdisk [69].
Microfabrication technology has been successfully employed to fabricate sensor microarrays for application in physiological research (Figure 10.31) [70]. Also, silicon nitride micropipette arrays have been obtained by microfabrication.
Figure 10.31 Microfabricated nine-sensor array (12 × 4 mm) for ionic distribution measurements in porcine beating hearts. Adapted with permission from [15]. Copyright 2001 American Chemical Society.

Miniaturized reference electrodes that are needed in such applications can be obtained from an ion microsensor that responds to an ion with constant concentration (e.g., Na+ in blood serum analysis). Ion-independent miniaturized reference electrodes have also been obtained on the basis of hydrophobic membrane materials [71].
As in vivo determination of pH is a matter of great concern, much attention has been paid to the development of miniature pH sensors. A comprehensive overview of this topic is give in ref. [72].
10.14 Analysis with Potentiometric Ion Sensors
Direct determinations by means of potentiometric ion sensors can be performed by means of a calibration graph like that shown in Figure 10.8. Care should be taken to secure identical pH and ionic strength in all the standard and sample solutions. IUPAC recommended procedures for calibration of potentiometric sensors are presented in ref. [73]
Ion determination can also be performed by resorting to controlled modification of the analyte concentration in what is known as the Gran method [74, 75]. In this method, the analyte concentration is varied by adding a known volume Va of an analyte solution with known concentration ca (standard solution). If c0 and V0 are the sample concentration and volume, respectively, then, the total concentration after performing the standard addition is:
(10.81) ![]()
where the total volume is ![]() . On substituting ct for the analyte concentration in Equation (10.34) and rearranging one obtains:
. On substituting ct for the analyte concentration in Equation (10.34) and rearranging one obtains:
where s is the empirical sensitivity of the sensor and ![]() . By plotting y vs. Va a straight line is obtained whose intersection with the horizontal axis (y = 0) allows the calculation of the unknown concentration c0. This concentration can also be obtained numerically from the quotient of the slope and the intercept of the regression equation.
. By plotting y vs. Va a straight line is obtained whose intersection with the horizontal axis (y = 0) allows the calculation of the unknown concentration c0. This concentration can also be obtained numerically from the quotient of the slope and the intercept of the regression equation.
A simple alternative to the above method is the single standard addition method. After several algebraic manipulations of Equation (10.82) one obtains the following equation that allows the unknown concentration to be calculated:
(10.83) ![]()
where ![]() is the difference between the cell voltage before and after performing the standard addition.
is the difference between the cell voltage before and after performing the standard addition.
The above methods are advantageous when the sample matrix cannot be reproduced in the standard solutions. If Va ![]() V0, only negligible modification in the matrix component concentration occurs. Thus, the effects of variations in the activity coefficient ant the junction potential are minimized. Moreover, the analyte concentration can be changed by adding a ligand that forms a very stable complex with the analyte. In this case, the
V0, only negligible modification in the matrix component concentration occurs. Thus, the effects of variations in the activity coefficient ant the junction potential are minimized. Moreover, the analyte concentration can be changed by adding a ligand that forms a very stable complex with the analyte. In this case, the ![]() term in the Equation (10.82) is negative.
term in the Equation (10.82) is negative.
Potentiometric ion sensors are also commonly used for detecting endpoints in titrimetric analysis. More details on analytical methods based on potentiometric sensors can be found in refs. [5, 7, 76].
10.15 Recent Advances in Potentiometric Ion Sensors
For a long time, it has been assumed that the lower limit of detection of potentiometric sensors is around 10−6 M, which makes such sensors unsuitable for use in most environmental monitoring applications. However, recent theoretical and experimental research efforts led to the conclusion that the lower detection limit can be lessened to about 10−10 M by means of straightforward modifications in the sensor design and the operational procedure [77–81]. Such performances emerged from a careful analysis of the factors that determine the limit of detection.
The standard definition of the lower limit of detection assumes that this limit is determined by analyte competition with interfering ions for the recognition sites. However, for highly selective membranes, the calculated limit of detection may be in the femtomolar region. Such a low limit has been approached in the presence of a strong complexing agent that produces only a very low free ion activity in the solution even if the total ion concentration is much larger.
What actually restricts the limit of detection is the contamination of the sample by analyte ions originating from the internal solution of the sensor. At first glance, this is not possible because the sensor is operated at zero current and the ion flux through the membrane should also be zero. However, even under zero-current conditions, ion diffusion across the membrane is possible to some extent, either by codiffusion with a counterion or by letting an ion with the same charge sign diffuse in the opposite direction from the test-solution.
Therefore, the limit of detection can be improved by minimizing the analyte ion flux across the membrane. The ion flux can be minimized by adjusting the analyte activity in the internal solution to a value close to that in the sample. By this means, a low concentration gradient across the membrane is achieved. A very low ion activity in the internal solution is obtained by adding a strong complexing agent (such as EDTA) along with the ion in the normal concentration range. As a result, most of the ions in the internal solution are bound in a hydrophilic complexe and only the extremely low free ion activity contributes to the activity gradient across the membrane. The same effect can be obtained by including a solid ion-exchanger in the internal system of the sensor.
Alternatively, the analyte flux can be reduced by decreasing the diffusion coefficient in the membrane. This can be achieved by decreasing the plasticizer content, by using a polymer other than PVC as the membrane matrix, or by using covalently bound receptors that are not free to diffuse and carry the ion across the membrane.
Another method for minimizing the detection limit is based on operating the sensor under strong convection conditions. In this way, accumulation of analyte arising from the internal solution at the sample/membrane interface is prevented. Strong convection can be secured with a rotating sensor setup in which the membrane is placed off-center from the rotation axis. Similarly, good results have been obtained in wall-jet systems, in which a sample stream impinges with a high velocity on the sensor surface. This approach has proved to be effective not only with polymeric membrane sensors, but also with solid salt membranes. Thus, impressive detection limits for Cu2+ have been obtained with a jalpaite (Ag3CuS2) polycrystalline membrane operated under rotating sensor conditions.
Diffusion of the analyte from the internal solution is completely eliminated when an internal solid contact is set up. However, possible instability in the contact potential will result in random potential fluctuations that represent a noise signal and can drastically degrade the limit of detection. As already emphasized, conducting polymers represent very convenient materials for contacting the sensing membrane with a metallic conductor. However, if a solution layer forms between the metal and the polymer, this layer leads to a cell voltage instability. Such a problem has been mitigated by using solvent casting to form the conducting polymer layer. For a Pb2+ sensor this led to a detection limit in the nanomolar range.
As far as glass membranes are concerned, exceptionally low detection limits for Hg2+ in sea water have been obtained with a chalcogenide glass sensor [82].
Potentiometric sensor arrays combined with multivariate data analysis appears to be a promising approach to trace analysis by potentiometric ion sensors. Thus, an array of chalcogenide glass and polymeric membrane sensors enabled the determination of Cu2+, Zn2+, Pb2+, and Cd2+ activity at the nanomolar level in artificial seawater [83].
Clearly, recent progress in potentiometric ion-sensor technology has led to devices and methods that show extremely low limits of detection that compare favorably with those characteristic of advanced spectrometric methods. This opens up new opportunities for ion sensor applications to environment monitoring, particularly in speciation analysis.
New applications of potentiometric ion sensors have arisen from operations under nonequilibrium conditions [84]. This operation method provides a means of solving analytical problems that cannot be approached by equilibrium potentiometry. Such problems are the determination of the total ion concentration in a sample or determination of polyanions. In the case of polyanion determination, the high charge of the analyte renders the Nernstian response sensitivity extremely low. Operation under nonequilibrium conditions alters substantially the response function and makes possible the potentiometric determination of such ions.
10.16 Potentiometric Gas Sensors
Potentiometric ion sensors can function as transducers in neutral-molecule sensors provided that the recognition of the neutral species results in ions being produced or consumed. The best-known application of this kind is the potentiometric carbon dioxide sensor (the Severinghaus sensor) which was initially developed in 1958 for the determination of the partial pressure of carbon dioxide in blood [85, 86]. Determination of carbon dioxide is also of interest to food industry [87]. This sensor is based on the property of carbon dioxide to dissolve in water forming carbonic acid. By dissociation, carbonic acid gives rise to hydrogen cations and hydrogen carbonate anions. The activity of the hydrogen ion can be assessed by a suitable pH sensor (e.g., a pH glass electrode) and correlated with the carbon dioxide concentration in the sample. The pH sensor is immersed in the internal solution (int) separated from the external, test solution (ext), by a CO2-permeable membrane (Figure 10.32). In order to form a galvanic cell, a reference electrode is immersed in the test-solution.
For a liquid sample, the overall process is described by the following sequence:
The membrane can be made of a microporous hydrophobic polymer (e.g., polypropylene) that allows the gas molecule to cross it by effusion through the pores. Alternatively, the membrane can be formed of a compact polymer (such as silicone rubber) that dissolves the gas and lets it diffuse across from the sample to the internal solution. Carbon dioxide dissolution and carbonic acid dissociation occurs to a large extent within a thin solution layer located between the membrane and the pH sensor surface.
Figure 10.32 The lower section of a potentiometric CO2 sensor. Adapted with permission from [88]. Copyright 2002 A. B![]() nic
nic![]() and F.G. B
and F.G. B![]() nic
nic![]() .
.
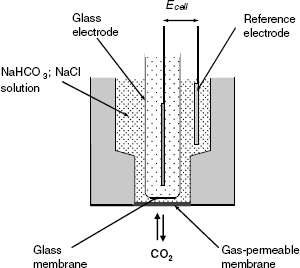
The response signal of this sensor is the potential difference between the internal reference electrode of the pH sensor glass electrode and the external reference electrode (Ecell). The response time depends on the time needed to attain the equilibrium and is hence determined by the rate of gas transfer through the membrane. In order to obtain a convenient response time, the membrane should be very thin. It should be about 0.1 mm thick if the transfer proceeds by effusion, but it can be thicker (0.1 to 0.3 mm) when the transfer is achieved by diffusion.
As is typical for the pH glass electrode, the cell voltage depends on the hydrogen ion activity in the internal solution as follows:
where b is the response slope. In turn, the pH depends on the carbon dioxide activity that can be expressed by taking into account the following equilibrium constant of the overall process in (10.84):
(10.86) ![]()
Here, ![]() and
and ![]() are ion activities in the internal solution and
are ion activities in the internal solution and ![]() is the gas activity in the sample. On substituting into Equation (10.85) the
is the gas activity in the sample. On substituting into Equation (10.85) the ![]() expression derived from (10.3), one obtains the response function as:
expression derived from (10.3), one obtains the response function as:
(10.87) ![]()
In order to obtain a nonequivocal response, the internal solution contains NaHCO3 at a sufficiently high concentration so that the concentration of hydrogen carbonate ion remains practically constant. Under these conditions the response is a linear function of ![]() with a slope similar to the glass electrode sensitivity. This gas sensor works equally well with both liquid and gas samples.
with a slope similar to the glass electrode sensitivity. This gas sensor works equally well with both liquid and gas samples.
Carbon dioxide determination is of great interest to food industry and attempts at improving the design and developing new sensing procedures is a subject of current research interest [89]. In order to perform miniaturization and impart enhanced robustness, the pH glass electrode can be replaced by a more convenient sensor. By using an iridium/iridium dioxide pH sensor and a solid, gas-permeable film of nonaqueous polymer-gel electrolyte, a planar CO2 sensor has been fabricated by the screen-print method [90].
A CO2 sensor has been obtained by using a polypyrrole polymer matrix impregnated with hydrogen carbonate as the counterion [91]. Miniaturized design has allowed integration of multiple sensors (for oxygen, pH and carbon dioxide) within a catheter-type sensor [92].
Similar sensors for other reactive gases have been developed using suitable potentiometric ion sensors as transducers [3]. Thus, pH monitoring can be used in sulfur dioxide or ammonia sensors. A more selective ammonia sensor can be obtained by using an ammonium sensor as transducer. Hydrofluoric acid, hydrogen cyanide and hydrogen sulfur sensor have been obtained using fluoride, cyanide or sulfide ion sensors, respectively, as transducers. Nitrogen dioxide, which yields nitrate and nitrite on dissolution, can be detected by potentiometric monitoring of nitrate ions.
Due to the slow diffusion of the gas analyte through the membrane, potentiometric gas sensors can display a slow response and long recovery times at low gas concentrations. When using a pH sensor as the transduction element, the selectivity over other acidic or basic gases is poor.
10.17 Solid Electrolyte Potentiometric Gas Sensors
10.17.1 General Principles
Gas determination at elevated temperatures is a critical problem in combustion control and various industrial processes. Potentiometric gas sensors for such applications have been developed using solid electrolytes instead of aqueous electrolyte solutions [93].
Solid electrolytes are ionic crystalline compounds containing mobile ions that impart electrical conductivity. In a gas sensor, the solid electrolyte surface is coated with a thin, porous layer of a metal with catalytic properties, which also forms the electrode. In this way, triple phase boundaries are formed, as shown in Figure 10.33. At elevated temperatures, neutral gas molecules X, can undergo an electrochemical reaction leading to an ionic product Yz+/− that appears in the solid phase. This process is accompanied by electron exchange with the metal phase. At equilibrium, the potential of the metal electrode indicates the extent of the gas reaction and, hence, the analyte concentration in the gas phase.
Figure 10.33 Electrochemical reaction of a gas X at the triple phase boundary gas-metal-solid electrolyte. Yz+/− is an ion produced by the electrochemical reaction of the gas molecule X.
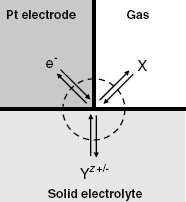
A simple reaction of this type is the reversible electrochemical reaction of hydrogen gas that results in hydrogen ions in a proton-conducting solid:
This reaction occurs at the contact point of three phases: the gas, the platinum electrode and the solid electrolyte (Figure 10.33). At equilibrium, the electrical potential of the Pt electrode is determined by the ratio of H2 activity in the gas phase and H+ activity in the solid electrolyte.
Determination of oxygen in a gas mixture can be performed by means of a solid electrolyte containing mobile O2− ions, such as zirconia (ZrO2). In the pure state, the crystal lattice of this compound is formed of Zr4+ and O2− ions located at fixed positions so that no ionic conductivity is present. However, if ZrO2 is doped with a divalent (e.g., Ca2+) or a trivalent (e.g., Y3+) ion, O2− vacancies appear in the lattice in order to preserve the charge balance. Consequently, O2− ions can jump from their position in the lattice to an adjacent vacancy, thus providing electrical conductivity. If yttrium is used as dopant, the material thus obtained is called yttrium-stabilized zirconia (in short, YSZ). The term “stabilized” indicates the fact that yttrium stabilizes the tetragonal crystal structure of ZrO2.
Oxygen determination by means of a YSZ-based sensor is made possible by the following electrochemical reaction:
A high reaction rate in each direction is important in order to provide fast equilibration of the chemical system, which secures a short response time. This condition is fulfilled at temperatures over 800 °C. This is not a drawback because the sensor is designed for applications involving high-temperature samples, such as exhaust gases or molten metals and alloys.
In order to obtain a gas sensor, an indicator half-cell, such as that in Figure 10.33 should be combined with a reference half-cell to form a galvanic cell. The reference half-cell can be similar to the indicator one, except for the fact that the content of the analyte gas is held constant in the adjacent gas phase. The electromotive force of this cell depends on the gas concentration according to the Nernst equation.
10.17.2 Solid Electrolyte Potentiometric Oxygen Sensors
An oxygen sensor can be produced by combining two half-cells in order to obtain the galvanic cell below, which consists of a reference and an indicator half-cell:
(10.90) ![]()
![]() indicates the partial pressure of oxygen in the gas phase. In electrochemistry, such a cell is called a concentration cell because the sole difference between the half-cells resides in the concentration of the electrochemically active species. The electromotive force of this cell can be derived from thermodynamic considerations and is given by the following Nernst-type equation [94, 95]:
indicates the partial pressure of oxygen in the gas phase. In electrochemistry, such a cell is called a concentration cell because the sole difference between the half-cells resides in the concentration of the electrochemically active species. The electromotive force of this cell can be derived from thermodynamic considerations and is given by the following Nernst-type equation [94, 95]:
(10.91) ![]()
As the partial pressure of oxygen in the reference half-cell is constant, the actual response function is:
(10.92) ![]()
where the constant term is ![]() . However, the actual cell voltage differs from the EMF due to additional terms arising from the secondary effects. Therefore, the sensor response function in terms of measured cell voltage (V) is:
. However, the actual cell voltage differs from the EMF due to additional terms arising from the secondary effects. Therefore, the sensor response function in terms of measured cell voltage (V) is:
where the V0 term includes the E0 constant as well as contributions from secondary effects.
Commonly used reference gases are pure oxygen and air. A rigorous control of the oxygen partial pressure in the reference chamber can be achieved by means of a sealed-in mixture of metal and metal oxide that forms a reference electrode [96].
Equation (10.93) indicates that a decade change in the partial pressure of oxygen results in a change in EMF of approximately 50 mV at 1000 K. This kind of response occurs over a very broad range of oxygen partial pressure. As the response depends on temperature, this parameter should be rigorously controlled; to this end, a temperature probe is included in the sensor design. Temperature can also affect other parameters included in the V0 term in Equation (10.93) [96].
The limits of the response range are determined by possible electronic conduction at extreme oxygen contents in the sample. At very low oxygen content, electron conductivity becomes significant, while at high oxygen content, hole conduction plays an important role. In both cases, the response deviates from Equation (10.93)[96]. In addition, interferences can be produced by other gases that can react with either the oxygen gas (thus depleting it at the sensor surface) or with O2− ions in the solid electrolyte. Thus, oxygen depletion can be caused by catalytic oxidation of NO, CO, or hydrocarbon. The same compounds can undergo electrochemical reactions that modify the O2− concentration in the solid electrolyte, such as:
Under suitable conditions, the error in oxygen partial pressure is not greater than 2%.
A possible configuration of potentiometric oxygen sensors is shown in Figure 10.34A. It includes a YSZ membrane coated on each side with a porous platinum layer or a platinum net. The operating temperature is controlled by means of an electrical heater winding and measured by a temperature probe (not shown).
Figure 10.34 (A) Basic structure of a solid electrolyte oxygen sensor. A temperature probe (not shown) is also included in the sensor design. (B) Cross section of a potentiometric oxygen sensor in the planar configuration
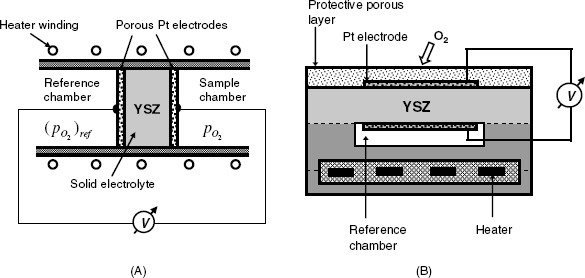
In addition to the sensor design in Figure 10.34, other configurations have been implemented. A planar (Figure 10.34B) is advantageous due to its compact form, small size, and compatibility with thick-film microfabrication technology [97].
10.17.3 Applications of Potentiometric Oxygen Sensors
The most common application of the potentiometric oxygen sensor is combustion control in motor vehicles in order to optimize fuel consumption and avoid air pollution by carbon monoxide [98–100]. The oxygen content in the exhaust gas indicates the degree of completion of the combustion reaction. From the chemical standpoint, the combustion process can be characterized by the nondimensional combustion parameter λ defined in Equation (10.96), which indicates to what extent the combustion of an air–fuel mixture approaches stoichiometric conditions. It is defined as:
A stoichiometric mixture is characterized by ![]() ; for a fuel-lean mixture,
; for a fuel-lean mixture, ![]() , whereas for a fuel-rich mixture,
, whereas for a fuel-rich mixture, ![]() . Figure 10.35 demonstrates the typical response of the potentiometric oxygen sensor in terms of the combustion parameter. Near the stoichiometric condition there is an abrupt drop in the sensor response due to a several orders of magnitude change in the oxygen concentration. Therefore, in practice, this kind of sensor works with a binary response, assuming only two possible states that correspond to either air excess or fuel excess. In order to achieve complete combustion, a slight excess of air should be maintained. In the case of complete combustion, no carbon monoxide is present in the exhaust gas.
. Figure 10.35 demonstrates the typical response of the potentiometric oxygen sensor in terms of the combustion parameter. Near the stoichiometric condition there is an abrupt drop in the sensor response due to a several orders of magnitude change in the oxygen concentration. Therefore, in practice, this kind of sensor works with a binary response, assuming only two possible states that correspond to either air excess or fuel excess. In order to achieve complete combustion, a slight excess of air should be maintained. In the case of complete combustion, no carbon monoxide is present in the exhaust gas.
Figure 10.35 Typical response of a lambda sensor.
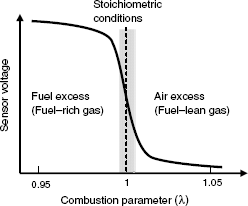
Also known as the lambda sensor, this device was first introduced in 1961 by Peters and Möbius [101] and Weissbart and Ruka [102] and became commercially available by 1965. It is currently produced on an industrial scale and is widely used due to the strict regulations concerning traffic pollution.
10.17.4 Types of Solid Electrolyte Potentiometric Gas Sensors
Solid electrolyte potentiometric gas sensors can be classified according to the relation between the gas analyte and the ions present in the structure of the solid electrolyte. Three types of sensors can be distinguished, as summarized in Figure 10.36.
Figure 10.36 Various types of potentiometric gas sensor based on solid electrolytes. XY = solid electrolyte; Xz+/− = mobile ion (charge carrier). Adapted with permission from [106]. Copyright 1992 Kluwer Academic Publishers.
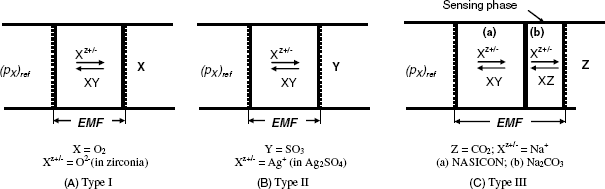
In this arrangement, Na+ is the common, mobile ion. The reaction of carbon dioxide (10.100) alters the concentration of Na+ in phase (b) and hence, this ion concentration in phase (a). As a result, the cell voltage changes owing to the implication of the Na+ ion in reaction (10.101) that occurs at the reference half-cell. Consequently, the EMF is dependent on the carbon dioxide concentration according to the Nernst equation:
(10.102) ![]()
where EC is a constant term and ![]() is the partial pressure of CO2 in the sample gas.
is the partial pressure of CO2 in the sample gas.
Similarly, a solid metal nitrate or nitrite salt can be used as the sensing phase for NO2 in combination with NASICON as reference phase. However, much better performances have been obtained with a sensor composed of nickel oxide as sensing phase, in conjunction with YSZ as reference phase. Both electrolytes share the same mobile ion, namely O2−, and nitrogen dioxide reacts as follows:
(10.103) ![]()
Sulfur oxides can be determined by means of sodium sulfate as sensing phase in conjunction with a proton-conducting reference phase [103].
It is clear that type III potentiometric gas sensors offer a large variety of applications using a given solid electrolyte as the phase (a) in combination with various metal salts as sensing phases.
10.17.5 Mixed Potential Potentiometric Gas Sensors
It has already been mentioned that certain gases that can undergo electrochemical reactions at the triple phase boundary affect the response of the potentiometric oxygen sensor and cause the response to deviate from that predicted by the Nernst equation. This arises because, in addition to the oxygen electrochemical reaction, additional reactions such as (10.94) or (10.95) contribute to the establishment of a certain electron density at the metal electrode and thus affect its electrical potential. Thus, oxygen reduction depletes the electrode of electrons, while carbon monoxide oxidation supplies electrons to the metal. At equilibrium, the electrode potential assumes a value determined by the extent of each of the above reactions and the electrode potential thus developed is a mixed potential [94, 108].
The effect of each reaction on the mixed potential is determined by its reaction rate. In turn, the reaction rate depends on three factors: gas concentration, temperature, and the nature of the metal catalyst. Optimal selection of these factors can render a given solid electrolyte suitable for detection of a particular gas with minor interferences from other gases present in the sample. For example, if gold is used as electrode catalyst, carbon monoxide oxidation (reaction (10.94)) is much faster than oxygen reduction (reaction (10.89)) and the sensor responds reliably to carbon monoxide at constant oxygen content in the gas mixture. The mixed potential is in this case a logarithmic function of the partial pressure of carbon monoxide. Such a response has been noted when the difference between equilibrium potentials of the pertinent reactions is small. Conversely, a linear dependence on the carbon monoxide content has been found when the electrode was made of platinum, which allows oxygen reduction to occur faster.
The effect of the operating temperature can be illustrated by the case of the mixed potential sensor for nitrogen oxide based on the Pt/YSZ system. At temperatures above 800 °C, oxygen reaction is very fast and the mixed potential is determined mainly by this gas. Conversely, at temperatures below 600 °C, the oxygen reaction is much slower and the mixed potential depends on the nitrogen oxide concentration.
10.17.6 Outlook
Solid-state potentiometric gas sensors have been developed as a consequence of the demand for monitoring the content of hazardous compounds in hot gases. These sensors make use of a solid electrolytes containing ions that are in equilibrium with the gas phase through an electrochemical reaction that occurs at the triple contact phase gas–solid electrolyte–metal electrode. In this reaction, the metal electrode functions as an electron sink. In order to obtain a sensor, two systems of this kind can be assembled to form a galvanic cell. The above-mentioned reactions give rise to a nonuniform distribution of mobile ions across the solid electrolyte, which determine the electromotive force of the cell. The electromotive force is related to the gas content by a Nernst-type equation.
The best-known sensor of this type is the oxygen sensor used to monitor the combustion process in car engines. It is the prototype of the type I sensor. Other detection schemes have been designed in order to develop type II and III solid electrolyte potentiometric gas sensors.
If a reducing gas is present along with oxygen, this gas undergoes oxidation with electrons being released to the electrode. In this case, the electrode reaction depends on two processes: electron donation by the reducing gas and electron withdrawing by oxygen. Proper selection of the catalyst and the operating conditions allows the reducing gas to be determined. Owing to the involvement of two gases in the potential-determining processes, the name mixed potential sensor is used to denote this kind of sensor. Very often, the response of mixed potential sensors deviates from the Nernst equation.
Application of solid electrolytes allowed the development of sensors of particular relevance to the monitoring of air quality as well as for industrial applications. Because these sensors operate at high temperatures, they can be designed so as to allow dissolved gases to be determined in molten metals or alloys.
The principles and applications of solid electrolyte gas sensors are amply reviewed in the literature (see, e.g., see above references and also [109–111]).
10.18 Potentiometric Biocatalytic Sensors
Many enzyme-catalyzed reactions involve an ionic species either as reactant or product. Therefore, a potentiometric enzyme sensor can be obtained by integrating a biocatalytic layer with a potentiometric ion sensor. In such a device, the ion sensor functions as a transducer that monitors the concentration of the relevant ion. The enzyme can be immobilized over the ion sensor surface by crosslinking within nylon netting followed by entrapment under a dialysis membrane. Alternatively, the enzyme can be attached by covalent immobilization.
Further, certain enzymatic reaction yield gaseous products, such as ![]() or
or ![]() that makes possible the application of potentiometric gas probes for performing the transduction.
that makes possible the application of potentiometric gas probes for performing the transduction.
Due to the simple configuration and wide availability of potentiometric sensors, a great number of potentiometric transduction schemes for enzymatic sensors have been developed [112–114]. A good example is the application of urease in potentiometric urea sensors. The enzymatic hydrolysis of urea leads to carbon dioxide and ammonia (Figure 3.11). At pH values around 7, both these products undergo protonation to ![]() and
and ![]() , respectively. Therefore, the transduction can be performed either by ion sensors (H+ or
, respectively. Therefore, the transduction can be performed either by ion sensors (H+ or ![]() ) or by gas sensors (
) or by gas sensors (![]() or
or ![]() ).
).
The configuration of a gas probe-based enzyme sensor is shown in Figure 10.37. The potentiometric gas probe consists of a potentiometric ion sensor 5 (a pH glass electrode in this case) in contact with a thin layer of internal solution 4 confined by means of a gas-permeable membrane 3. A biocatalytic layer is immobilized below and is separated from the solution by a semipermeable membrane 1. The substrate crosses the external membrane 1 and undergoes enzymatic conversion within the biocatalytic layer 2. The gas product thus formed diffuses across membrane 3 and, by reaction within the internal solution, modifies the pH in the thin layer adjacent to the pH sensor surface and, therefore, the response of the pH sensor. Consequently, the voltage developed between the two reference electrodes is dependent on the substrate concentration in the sample.
Figure 10.37 A biocatalytic potentiometric sensor based on a gas sensor as transducer element. (1) External membrane; (2) biocatalytic layer; (3) gas-permeable membrane; (4) gas sensor internal solution; (5) combination pH glass electrode; (6) reference electrodes; (7) liquid junction. Adapted with permission from [88]. Copyright 2002 A. B![]() nic
nic![]() and F.G. B
and F.G. B![]() nic
nic![]() .
.
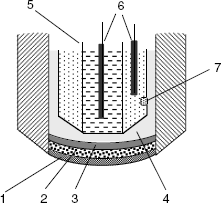
As many enzyme reactions involve hydrogen ions, the use of a pH glass electrode as transducer is probably the most straightforward alternative. However, in order to detect the pH change produced by enzymatic reaction, a buffer system with a low buffer capacity should be used (e.g., 1 mM phosphate buffer). This may bring about some difficulties in sensor operation as possible pH variation may affect enzyme activity. However, this method has proved useful, particularly with both oxidase- and hydrolase-type enzymes. An example of this kind is the potentiometric penicillin sensor based on penicillase (beta-lactamase) that catalyzes the hydrolysis of the substrate to penicilloic acid. Hence, the pH increases in proportion to the substrate concentration. Analogously, acetylcholine sensors have been developed using acetylcholine esterase that gives rise to acetic acid by substrate hydrolysis.
Oxidase-type enzymes that give rise to hydrogen peroxide as a side product can be used in combination with an iodide ion sensor provided that the iodide ion is present in the solution. In the presence of horseradish peroxidase, hydrogen peroxide oxidizes iodide ion to iodine and the transducer monitors the decrease of the iodide concentration at the surface. Sensors for glucose and amino acids based on this principle have been developed. An alternative approach relies on the reaction of hydrogen peroxide with 4–fluorophenol that releases fluoride ions. A potentiometric fluoride sensor can therefore be used as the transducer in this case.
The response function of a potentiometric enzyme sensor can be derived using the results of the approach in Chapter 4. The response function will be derived next for the case of external diffusion control and first-order kinetics. In this case, the rate-determining step is the substrate diffusion across the external membrane and the product is uniformly distributed within the biocatalytic layer. If the product does not undergo further conversion or is totally converted to another detectable species, the response of the potentiometric transducer is determined by the product concentration pe which is given by Equation (4.17). Assuming that the potentiometric transducer provides a Nernstian response to the product concentration with the sensitivity b:
(10.104) ![]()
the response to the substrate concentration is:
Here, ![]() and
and ![]() are the mass-transfer coefficients of the substrate and product, respectively, in the external membrane, s is the substrate concentration in the test solution and
are the mass-transfer coefficients of the substrate and product, respectively, in the external membrane, s is the substrate concentration in the test solution and ![]() is the substrate modulus for external diffusion. The
is the substrate modulus for external diffusion. The ![]() parameter is directly proportional to the enzyme loading (that is, the product of the enzyme concentration and the thickness of the biocatalytic film). At the same time,
parameter is directly proportional to the enzyme loading (that is, the product of the enzyme concentration and the thickness of the biocatalytic film). At the same time, ![]() is inversely proportional to the mass transfer coefficient of the substrate in the external membrane. Therefore, the
is inversely proportional to the mass transfer coefficient of the substrate in the external membrane. Therefore, the ![]() value can be adjusted by the selection of the enzyme loading and the characteristics of the external membrane (thickness and permeability to the substrate).
value can be adjusted by the selection of the enzyme loading and the characteristics of the external membrane (thickness and permeability to the substrate).
According to Equation (10.105), the sensor sensitivity relative to substrate concentration is determined by the sensitivity of the ion sensor used as transducer. However, the second term in this equation is a function of the ![]() parameter and, therefore, is not sufficiently reliable. The response function assumes a more convenient form if
parameter and, therefore, is not sufficiently reliable. The response function assumes a more convenient form if ![]() ; in this case, the term in
; in this case, the term in ![]() vanishes and the response becomes independent of the parameters of the enzyme layer.
vanishes and the response becomes independent of the parameters of the enzyme layer.
If the enzyme sensor is devised for applications in inhibitor determination, it is convenient to design it such as to function under the zero-order kinetic regime in order to secure maximum sensitivity.
Potentiometric enzyme sensors are simple and inexpensive devices that can be fabricated readily even in the disposable form [115]. The range of applications encompasses the determination of various substrate compounds for diagnostic purposes as well as monitoring of toxic species in the environment by enzyme inhibition.
10.19 Potentiometric Affinity Sensors
Potentiometric transduction in affinity sensors can be achieved either by means of redox electrodes or by means of potentiometric ion or gas sensors [116–118].
A very simple approach to potentiometric transduction in immunosensors rests on redox electrodes combined with a redox enzyme as label. A platinum electrode in contact with a solution containing a redox couple develops an electrode potential according to the Nernst equation (10.21). Substrate oxidation leads to a change in the molar ratio of the oxidized/reduced forms of the redox couple that induces a change in the electrode potential. The sensor can be assembled by immobilizing the receptor at the surface of a platinum electrode. In a competition assay, the enzyme label is brought to the electrode surface by the reaction of the receptor with the enzyme-labeled analyte analog, while in the sandwich assay the enzyme is held by the formation of a ternary complex involving a labeled entity.
Ion-selective membranes can be used as transducer in conjunction with enzyme labels that catalyze ion-releasing reactions. Gaseous products, such as ammonia or carbon dioxide can also function in the transduction process when the sensing layer is build up at the surface of a potentiometric gas sensor.
Immunorecognition, as well as other affinity interactions, involves charged particles and the recognition event results in a partial neutralization of the charges on the reactants. The change in the electric charge density can be detected by means of a PVC-coated electrode to which the receptor is appended [119]. This is an example of a potentiometric label-free immunoassay based on the polyelectrolyte character of the involved reactants.
An interesting approach to potentiometric immunosensors is based on ion channels formed by the interaction of the immunocomplex with a complementary species supplied by a blood serum sample [120]. In this approach, a marker-ion solution is entrapped in liposomes that act as a label integrated with the receptor antigen. The analyte–receptor complex forms at the liposome surface and interacts further with the complementary species. The ternary complex thus formed gives rise to ion channels that allow the marker ion to leak out and makes it available for detection by an ion sensor. This method has been designed for assays with a very small sample volume (down to 1 μL) in the form of a thin solution layer. A protocol for manufacturing and assay applications of such a sensor is available in ref. [121].
10.20 Summary
Potentiometric ion sensors are based on specific interactions between a selective membrane and the analyte ion that leads to the alteration of the charge density at the membrane surface and, consequently, a change in the membrane potential. The membrane potential is monitored by means of two reference electrodes placed in the solution on each side of the membrane. By keeping the ion concentration constant in one of the two solutions, the measured potential difference depends only on the ion concentration in the second solution. The potential difference is a logarithmic function of the analyte concentration, this being called a Nernstian response.
Ion-responsive membranes can be prepared in the solid-state form (e.g., sparingly soluble salts or glass materials) or can consist of a polymeric matrix embedding ionic or neutral ion receptors. Solid membranes are very rugged, whereas polymeric membranes are less resilient but allow achieving excellent selectivity and limit of detection.
Potentiometric ion sensors are simple, robust, and inexpensive analytical devices, well suited for either laboratory of field applications as well as for continuous concentration monitoring and in vivo chemical analysis. The accuracy of potentiometric ion sensors, which is commonly of ![]() , compares favorably with analytical techniques that require more complex and expensive instrumentation.
, compares favorably with analytical techniques that require more complex and expensive instrumentation.
It should be made clear that, as far as the field of applications is concerned, potentiometric ion sensors have competition from other widely used analytical methods such as atomic spectrometry, mass spectrometry and ion chromatography. Such methods are superior to potentiometry since they allow for simultaneous determination of more than one analyte. In addition, the limits of detection of the above methods are generally better than that of standard potentiometry. That is why potentiometric ion sensors appeared early in their development not to be suitable for monitoring toxic ions in biological and environment samples. However, recent progress in this field led to developments that allowed potentiometric sensor to achieve detection limits that compare favorably with those characteristic of mass spectrometry.
Further, the problem of multianalyte potentiometric determination can be addressed by means of potentiometric sensor arrays. Although the degree of generality achieved by contending methods cannot be achieved in this way, potentiometric sensor arrays represent the method of choice in certain specialized applications.
Potentiometric sensor arrays can provide a relief of the problems of interferences arising from crossreactivity. Indeed, data from an array of multiresponsive sensors can be processed so as to extract the unbiased concentration of each ion of interest.
As far as ruggedness is concerned, best performances are achieved by solid-state membranes. However, the selectivity of such sensors is not of the best. Polymer membranes that can be made very selective by using suitable ion receptors are less resilient. Nevertheless, this apparent drawback can be offset by including replaceable membranes in the sensor design.
As certain biochemical and nonbiochemical reactions produce ions, potentiometric ion sensors can function as transduction elements in various kinds of sensor based on ion-generating reactions. This principle is used in the development of gas sensors, biocatalytic sensors, and immunosensors.
As ions are found in both environmental and biological samples, as well as in certain industrial products, the potential field of application of ion sensors is very broad. Comprehensive surveys of potentiometric ion sensor application in various areas can be found in the literature as follows: monitoring of environment pollution [83, 122]; ion determination in marine chemistry [123]; biomedical sciences [11, 124, 125]; pharmaceutical analysis [126]; petroleum industry [127]. A spectacular example of field analysis application is the in situ determination of ions leached from Mars planet soil by an automatic wet laboratory fitted with an array of potentiometric ion sensors [128]
Last but not least, the large volume of expertise developed in the framework of potentiometric ion sensor research has proved invaluable in the development of other types of ion sensors such as field effect transistor ion sensors (Chapter 11) and optical ion sensors (Chapter 19).
In addition to the applications outlined above, potentiometry principles have allowed development of high-temperature gas sensors based on solid electrolytes as gas-sensitive materials. Sensors of this kind are widely used in monitoring burning processes as well as for the determination of various hazardous gases in the atmosphere.
1. Morf, W.E. (1981) The Principles of Ion-Selective Electrodes and of Membrane Transport, Elsevier, Amsterdam.
2. Koryta, J. and Štulík, K. (1983) Ion-Selective Electrodes, Cambridge University Press, Cambridge.
3. Lakshminarayanaiah, N. (1976) Membrane Electrodes, Academic Press, New York.
4. Fawcett, W.R. (2004) Liquids, Solutions, and Interfaces: From Classical Macroscopic Descriptions to Modern Microscopic Details, Oxford University Press, New York, pp. 383–505.
5. Cammann, K. (1979) Working with Ion-Selective Electrodes: Chemical Laboratory Practice, Springer, Berlin.
6. Covington, A.K. (1979) Ion-Selective Electrode Methodology, CRC Press, Boca Raton, Fla.
7. Cammann, K. and Galster, H. (1996) Working with Ion-Selective Electrodes (in German), Springer, Berlin.
8. Ma, T.S. and Hassan, S.S.M. (1982) Organic Analysis using Ion-Selective Electrodes, Academic Press, London.
9. Cosofret, V.V. (1977) Applications of Ion-Selective Membrane Electrodes in Organic Analysis, Ellis Horwood, Chichester.
10. Havas, J. (1985) Ion- and Molecule-Selective Electrodes in Biological Systems, Springer, Berlin.
11. Spichiger-Keller, U.E. (1998) Chemical Sensors and Biosensors for Medical and Biological Applications, Wiley-VCH, Weinheim.
12. Buck, R.P. and Lindner, E. (1994) Recommendations for nomenclature of ion-selective electrodes – (IUPAC Recommendations 1994). Pure Appl. Chem., 66, 2527–2536.
13. Bobacka, J., Ivaska, A., and Lewenstam, A. (2008) Potentiometric ion sensors. Chem. Rev., 108, 329–351.
14. Bakker, E. and Pretsch, E. (2007) Modern potentiometry. Angew. Chem. Int. Edit., 46, 5660–5668.
15. Buck, R.P. and Lindner, E. (2001) Tracing the history of selective ion sensors. Anal. Chem., 73, 88A–97A.
16. Shinwari, M.W., Zhitomirsky, D., Deen, I.A. et al. (2010) Microfabricated reference electrodes and their biosensing applications. Sensors, 10, 1679–1715.
17. Kahlert, H. (2010) Reference electrodes, in Electroanalytical Methods: Guide to Experiments and Applications (ed. F. Scholz), Springer-Verlag, Berlin, pp. 291–308.
18. Bakker, E., Pretsch, E., and Buhlmann, P. (2000) Selectivity of potentiometric ion sensors. Anal. Chem., 72, 1127–1133.
19. Bakker, E., Buhlmann, P., and Pretsch, E. (1997) Carrier-based ion-selective electrodes and bulk optodes. 1. General characteristics. Chem. Rev., 97, 3083–3132.
20. Umezawa, Y., Umezawa, K., and Sato, H. (1995) Selectivity coefficients for ion-selective electrodes – recommended methods for reporting KA,BPot values. Pure Appl. Chem., 67, 507–518.
21. Umezawa, Y., Buhlmann, P., Umezawa, K. et al. (2000) Potentiometric selectivity coefficients of ion-selective electrodes Part I. Inorganic cations. Pure Appl. Chem., 72, 1851–2082.
22. Gadzekpo, V.P.Y. and Christian, G.D. (1984) Determination of selectivity coefficients of ion-selective electrodes by a matched-potential method. Anal. Chim. Acta, 164, 279–282.
23. Umezawa, Y., Umezawa, K., Buhlmann, P. et al. (2002) Potentiometric selectivity coefficients of ion-selective electrodes. Part II. Inorganic anions (IUPAC technical report). Pure Appl. Chem., 74, 923–994.
24. Umezawa, Y., Buhlmann, P., Umezawa, K. et al. (2002) Potentiometric selectivity coefficients of ion-selective electrodes Part III. Organic ions – (IUPAC technical report). Pure Appl. Chem., 74, 995–1099.
25. Lindner, E. and Umezawa, Y. (2008) Performance evaluation criteria for preparation and measurement of macro- and microfabricated ion-selective electrodes (IUPAC technical report). Pure Appl. Chem., 80, 85–104.
26. Bates, R.G. (1973) Determination of pH: Theory and Practice, Wiley, New York.
27. Galster, H. (1991) pH Measurement: Fundamentals, Methods, Applications, Instrumentation, VCH, Weinheim.
28. Tanaka, K. (2001) Chalcogenide glasses, in The Encyclopedia of Materials: Science and Technology (eds K.H.J. Buschow, R.W. Cahn, M.C. Flemings, B. Ilschner, E.J. Kramer and S. Mahajan), Elsevier, Amsterdam.
29. Vassilev, V.S. and Boycheva, S.V. (2005) Chemical sensors with chalcogenide glassy membranes. Talanta, 67, 20–27.
30. Schoning, M.J. and Kloock, J.P. (2007) About 20 years of silicon-based thin-film sensors with chalcogenide glass materials for heavy metal analysis: Technological aspects of fabrication and miniaturization. Electroanalysis, 19, 2029–2038.
31. Vlasov, Y.G., Bychkov, E.A., and Legin, A.V. (1994) Chalcogenide glass chemical sensors – research and analytical applications. Talanta, 41, 1059–1063.
32. Steed, J.W. and Atwood, J.L. (2009) Supramolecular Chemistry, Wiley, Chichester.
33. Johnson, R.D. and Bachas, L.G. (2003) Ionophore-based ion-selective potentiometric and optical sensors. Anal. Bioanal. Chem., 376, 328–341.
34. Buhlmann, P., Bakker, E., and Pretsch, E. (1998) Carrier-based ion-selective electrodes and bulk optodes. 2. Ionophores for potentiometric and optical sensors. Chem. Rev., 98, 1593–1687.
35. Sanchez, J. and del Valle, M. (2005) Determination of anionic surfactants employing potentiometric sensors – A review. Crit. Rev. Anal. Chem., 35, 15–29.
36. Craggs, A., Moody, G.J., and Thomas, J.D.R. (1974) PVC matrix membrane ion-selective electrodes – construction and laboratory experiments. J. Chem. Educ., 51, 541–544.
37. Eugster, R., Rosatzin, T., Rusterholz, B. et al. (1994) Plasticizers for liquid polymeric membranes of ion-selective chemical sensors. Anal. Chim. Acta, 289, 1–13.
38. Pearson, R.G. (1967) Hard and soft acids and bases. Chem. Br., 3, 103–107.
39. Pearson, R.G. (1997) Chemical Hardness, Wiley-VCH, New York.
40. Antonisse, M.M.G. and Reinhoudt, D.N. (1998) Neutral anion receptors: Design and application. Chem. Commun., 443–448.
41. Snowden, T.S. and Anslyn, E.V. (1999) Anion recognition: synthetic receptors for anions and their application in sensors. Curr. Opin. Chem. Biol., 3, 740–746.
42. Bianchi, A., Bowman-James, K., and García-España, E. (1997) Supramolecular Chemistry of Anions, Wiley-VCH, New York.
43. Upreti, P., Metzger, L.E., and Bühlmann, P. (2004) Glass and polymeric membrane electrodes for the measurement of pH in milk and cheese. Talanta, 63, 139–148.
44. Lhotak, P. (2005) Anion receptors based on calixarenes, in Anion Sensing, Springer-Verlag Berlin, Berlin, pp. 65–95.
45. Siswanta, D., Nagatsuka, K., Yamada, N. et al. (1996) Structural ion selectivity of thia crown ether compounds with a bulky block subunit and their application as an ion-sensing component for an ion-selective electrode. Anal. Chem., 68, 4166–4172.
46. Cram, D.J. (1986) Preorganization – from solvents to spherands. Angew. Chem. Int. Edit. Engl., 25, 1039–1057.
47. Antonisse, M.M.G. and Reinhoudt, D.N. (1999) Potentiometric anion selective sensors. Electroanalysis, 11, 1035–1048.
48. Xu, Z., Kim, S.K., and Yoon, J. (2010) Revisit to imidazolium receptors for the recognition of anions: highlighted research during 2006–2009. Chem. Soc. Rev., 39, 1457–1466.
49. Stibor, I. and Zlatuskova, P. (2005) Chiral recognition of anions, in Anion Sensing (ed. P. Stibor), Springer-Verlag, Berlin, pp. 31–63.
50. Rao, T.P., Kala, R., and Daniel, S. (2006) Metal ion-imprinted polymers – novel materials for selective recognition of inorganics. Anal. Chim. Acta, 578, 105–116.
51. Rao, T.P. and Kala, R. (2008) Potentiometric transducer based biomimetic sensors for priority envirotoxic markers – an overview. Talanta, 76, 485–496.
52. Suryanarayanan, V., Wu, C.T., and Ho, K.C. (2010) Molecularly imprinted electrochemical sensors. Electroanalysis, 22, 1795–1811.
53. Hutchins, R.S. and Bachas, L.G. (1995) Nitrate-selective electrode developed by electrochemically mediated imprinting/doping of polypyrrole. Anal. Chem., 67, 1654–1660.
54. Bobacka, J., Ivaska, A., and Lewenstam, A. (2003) Potentiometric ion sensors based on conducting polymers. Electroanalysis, 15, 366–374.
55. Bobacka, J. (2006) Conducting polymer-based solid-state ion-selective electrodes. Electroanalysis, 18, 7–18.
56. Vytras, K. (1991) Coated wire electrodes in the analysis of surfactants of various types – an overview. Electroanalysis, 3, 343–347.
57. Vytras, K. (1984) Determination of some pharmaceuticals using simple potentiometric sensors of coated-wire type. Mikrochim. Acta, 3, 139–148.
58. Bakker, E. and Pretsch, E. (2008) Nanoscale potentiometry. TrAc-Trends Anal. Chem., 27, 612–618.
59. Kalcher, K., Švancara, I., Metelka, R. et al. (2006) Heterogeneous carbon electrochemical sensors, in Encyclopedia of Sensors (eds C.A. Grimes, E.C. Dickey, and M.V. Pishko,), American Scientific Publishers, Stevenson Ranch, CA, pp. 283–429.
60. Švancara, I., Kalcher, K., Walcarius, A. et al. (2012) Electroanalysis with Carbon Paste Electrodes, CRC Press, New York.
61. Kalcher, K., Švancara, I., Buzuk, M. et al. (2009) Electrochemical sensors and biosensors based on heterogeneous carbon materials. Monatshefte für Chemie, 140, 861–889.
62. Jezkova, J., Musilova, J., and Vytras, K. (1997) Potentiometry with perchlorate and fluoroborate ion-selective carbon paste electrodes. Electroanalysis, 9, 1433–1436.
63. Tymecki, L., Glab, S., and Koncki, R. (2006) Miniaturized, planar ion-selective electrodes fabricated by means of thick-film technology. Sensors, 6, 390–396.
64. Ammann, D. (1986) Ion-Selective Microelectrodes: Principles, Design and Application, Springer, Berlin.
65. Dierkes, P.W., Neumann, S., Müller, A. et al. (2002) Multi-barrelled ion-selective microelectrodes. Measurements of cell volume, membrane potential, and intracellular ion concentrations in invertebrate nerve cells, in Electrochemical Microsystem Technologies (eds J.W. Schultze, T. Osaka, and M. Datta), CRC Press, Boca Raton.
66. Felle, H.H. (1993) Ion-selective microelectrodes – their use and importance in modern plant-cell biology. Botanica Acta, 106, 5–12.
67. Vigassy, T., Huber, C.G., Wintringer, R. et al. (2005) Monolithic capillary-based ion-selective electrodes. Anal. Chem., 77, 3966–3970.
68. Sundfors, F., Bereczki, R., Bobacka, J. et al. (2006) Microcavity based solid-contact ion-selective microelectrodes. Electroanalysis, 18, 1372–1378.
69. Zhang, X.J., Ogorevc, B., and Wang, J. (2002) Solid-state pH nanoelectrode based on polyaniline thin film electrodeposited onto ion-beam etched carbon fiber. Anal. Chim. Acta, 452, 1–10.
70. Cosofret, V.V., Erdosy, M., Johnson, T.A. et al. (1995) Microfabricated sensor arrays sensitive to pH and K+ for ionic distribution measurements in the beating heart. Anal. Chem., 67, 1647–1653.
71. Bakker, E. (1999) Hydrophobic membranes as liquid junction-free reference electrodes. Electroanalysis, 11, 788–792.
72. Zhang, X., Ju, H., and Wang, J. (2008) Microelectrodes for in-vivo determination of pH, in Electrochemical Sensors, Biosensors, and their Biomedical Applications (eds X. Zhang, H. Ju, and J. Wang), Academic Press, Amsterdam, pp. 261–305.
73. Buck, R.P. and Cosofret, V.V. (1993) Recommended procedures for calibration of ion-selective electrodes. Pure Appl. Chem., 65, 1849–1858.
74. Gran, G. (1988) Equivalence volumes in potentiometric titrations. Anal. Chim. Acta, 206, 111–123.
75. Michalowski, T., Toporek, M., and Rymanowski, M. (2005) Overview on the Gran and other linearisation methods applied in titrimetric analyses. Talanta, 65, 1241–1253.
76. Bailey, P.L. (1980) Analysis with Ion-Selective Electrodes, Heyden, London.
77. Bakker, E. and Pretsch, E. (2002) The new wave of ion-selective electrodes. Anal. Chem., 74, 420A–426A.
78. Pretsch, E. (2007) The new wave of ion-selective electrodes. TrAc-Trends Anal. Chem., 26, 46–51.
79. Bakker, E. and Pretsch, E. (2005) Potentiometric sensors for trace-level analysis. TrAc-Trends Anal. Chem., 24, 199–207.
80. Bakker, E., Bühlmann, P., and Pretsch, E. (2004) The phase-boundary potential model. Talanta, 63, 3–20.
81. Bakker, E., Buhlmann, P., and Pretsch, E. (1999) Polymer membrane ion-selective electrodes – What are the limits? Electroanalysis, 11, 1088–1088.
82. De Marco, R. and Shackleton, J. (1999) Calibration of the Hg chalcogenide glass membrane ion-selective electrode in seawater media. Talanta, 49, 385–391.
83. Rudnitskaya, A., Legin, A., Seleznev, B. et al. (2008) Detection of ultra-low activities of heavy metal ions by an array of potentiometric chemical sensors. Microchim. Acta, 163, 71–80.
84. Bakker, E. and Meyerhoff, M.E. (2000) Ionophore-based membrane electrodes: new analytical concepts and non-classical response mechanisms. Anal. Chim. Acta, 416, 121–137.
85. Severinghaus, J.W. and Bradley, A.F. (1958) Electrodes for blood pO2 and pCO2 determination. J. Appl. Physiol., 13, 515–520.
86. Severinghaus, J.W., Astrup, P., and Murray, J.F. (1998) Blood gas analysis and critical care medicine. Am. J. Respir. Crit. Care Med., 157, S114–S122.
87. Neethirajan, S., Jayas, D.S., and Sadistap, S. (2009) Carbon dioxide (CO2) sensors for the agri-food industry-a review. Food Bioprocess Technol., 2, 115–121.
88. Ion, A. and B![]() nic
nic![]() , F.G. (2002) Electrochemical Methods in Analytical Chemistry, Ars Docendi, Bucharest.
, F.G. (2002) Electrochemical Methods in Analytical Chemistry, Ars Docendi, Bucharest.
89. Senorans, F.J., Ibanez, E. and Cifuentes, A. (2003) New trends in food processing. Crit. Rev. Food Sci. Nutr. 43, 507–526.
90. McMurray, H.N., Lewis, M.J., and Brinz, T. (2003) Planar solid-state potentiometric carbon dioxide sensors incorporating polymer-gel electrolyte. Electrochem. Solid State Lett., 6, H5–H7.
91. Tongol, B.J.V., Binag, C.A., and Sevilla, F.B. (2003) Surface and electrochemical studies of a carbon dioxide probe based on conducting polypyrrole. Sens. Actuators B-Chem., 93, 187–196.
92. Meruva, R.K. and Meyerhoff, M.E. (1998) Catheter-type sensor for potentiometric monitoring of oxygen, pH and carbon dioxide. Biosens. Bioelectron., 13, 201–212.
93. Zhuiykov, S. (2008) Electrochemistry of Zirconia Gas Sensors, CRC Press, Boca Raton, FL.
94. Park, C.O., Akbar, S.A., and Weppner, W. (2003) Ceramic electrolytes and electrochemical sensors. J. Mater. Sci., 38, 4639–4660.
95. Weppner, W. (1987) Solid-state electrochemical gas sensors. Sens. Actuators, 12, 107–119.
96. Maskell, W.C. and Steele, B.C.H. (1986) Solid-state potentiometric oxygen gas sensors. J. Appl. Electrochem., 16, 475–489.
97. Riegel, J., Neumann, H., and Wiedenmann, H.M. (2002) Exhaust gas sensors for automotive emission control. Solid State Ion., 152, 783–800.
98. Docquier, N. and Candel, S. (2002) Combustion control and sensors: a review. Prog. Energy Combus. Sci., 28, 107–150.
99. Lopez-Gandara, C., Ramos, F.M., and Cirera, A. (2009) YSZ-based oxygen sensors and the use of nanomaterials: A review from classical models to current trends. J. Sensors, Article ID 258489.
100. Lee, J.H. (2003) Review on zirconia air-fuel ratio sensors for automotive applications. J. Mater. Sci., 38, 4247–4257.
101. Peters, H. and Möbius, H.H. (1961) Procedure for the gas analysis at elevated temperatures using galvanic solid electrolyte elements. DDR-Patent 21673. 20.5.1958.
102. Weissbart, J. and Ruka, R. (1961) Oxygen gauge. Rev. Sci. Instrum., 32, 593–&.
103. Jacob, K.T. and Mathews, T. (1990) Solid-state electrochemical sensors in process control. Indian J. Technol., 28, 413–427.
104. Traversa, E. (1995) Ceramic sensors for humidity detection – the state-of-the-art and future-developments. Sens. Actuators B-Chem., 23, 135–156.
105. Hotzel, G. and Weppner, W. (1986) Application of fast ionic conductors in solid-state galvanic cells for gas sensors. Solid State Ion., 18–9, 1223–1227.
106. Yamazoe, N. and Miura, N. (1992) New approaches in the design of gas sensors, in Gas Sensors: Principles, Operation and Developments (ed. G. Sberveglieri), Kluwer, Dordrecht, pp. 1–42.
107. Maruyama, T., Sasaki, S., and Saito, Y. (1987) Potentiometric gas sensor for carbon-dioxide using solid electrolytes. Solid State Ion., 23, 107–112.
108. Park, C.O., Fergus, J.W., Miura, N. et al. (2009) Solid-state electrochemical gas sensors. Ionics, 15, 261–284.
109. Miura, N., Elumalai, P., Plashnitsa, V.V. et al. (2009) Solid-state electrochemical gas sensing, in Solid State Gas Sensing (eds E. Comini, G. Faglia, and G. Sberveglieri), Springer Science+Business Media, LLC, Boston, MA, pp. 181–207.
110. Zhuiykov, S. and Miura, N. (2007) Development of zirconia-based potentiometric NOx sensors for automotive and energy industries in the early 21st century: What are the prospects for sensors? Sens. Actuators B: Chem., 121, 639–651.
111. Garzon, F.H., Mukundan, R., Lujan, R. et al. (2004) Solid state ionic devices for combustion gas sensing. Solid State Ion., 175, 487–490.
112. Koncki, R. and Glab, S. (1991) Potentiometric enzymatic sensors. Chem. Anal., 36, 423–446.
113. Kauffmann, J.M. and Guilbault, G.G. (1992) Enzyme electrode biosensors – theory and applications. Method Biochem. Anal., 36, 63–113.
114. Guilbault, G.G. (1988) Enzyme electrode probes. Method Enzymol., 137, 14–29.
115. Gaberlein, S., Knoll, M., Spener, F. et al. (2000) Disposable potentiometric enzyme sensor for direct determination of organophosphorus insecticides. Analyst, 125, 2274–2279.
116. Janata, J. and Blackburn, G.F. (1984) Immunochemical potentiometric sensors. Ann. NY Acad. Sci., 428, 286–292.
117. Yakovleva, J. and Emneus, J. (2008) Electrochemical immunoassay, in Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications (ed. P.N. Bartlett) Wiley, Chichester, pp. 377–410.
118. Stefan, R.I., van Staden, J.F., and Aboul-Enein, H.Y. (2000) Immunosensors in clinical analysis. Fresenius J. Anal. Chem., 366, 659–668.
119. Janata, J. (1975) Immunoelectrode. J. Am. Chem. Soc., 97, 2914–2916.
120. Umezawa, Y., Sofue, S., and Takamoto, Y. (1984) Thin-layer ion-selective electrode detection of anticardiolipin antibodies in syphilis serology. Talanta, 31, 375–378.
121. Radecka, H. and Umezawa, Y. (1998) Immunosensors based on ion-selective electrodes, in Affinity Biosensors: Techniques and Protocols (eds K.R. Rogers and A. Mulchandani), Humana Press, Totowa, pp. 149–160.
122. De Marco, R., Clarke, G., and Pejcic, B. (2007) Ion-selective electrode potentiometry in environmental analysis. Electroanalysis, 19, 1987–2001.
123. Denuault, G. (2009) Electrochemical techniques and sensors for ocean research. Ocean Sci., 5, 697–710.
124. Solsky, R.L. (1982) Ion-selective electrodes in biomedical analysis. Crit. Rev. Anal. Chem., 14, 1–52.
125. Lunte, C.E. and Heineman, W.R. (1988) Electrochemical techniques in bioanalysis. Top. Curr. Chem., 143, 1–48.
126. Campanella, L. and Tomassetti, M. (1989) Sensors in pharmaceutical analysis. Sel. Electrode Rev., 11, 69–110.
127. Badoni, R.P. and Jayaraman, A. (1988) Use of ion-selective membrane electrodes in the petroleum-industry. Erdol & Kohle Erdgas Petrochemie, 41, 23–30.
128. Kounaves, S.P., Lukow, S.R., Comeau, B.P. et al. (2003) Mars surveyor program '01 mars environmental compatibility assessment wet chemistry lab: A sensor array for chemical analysis of the martian soil. J. Geophys. Res. -Planets, 108, Article Number: 5077.