
CHAPTER 3
BIOLOGICAL INFORMATICS: DATA, TOOLS, AND APPLICATIONS
3.1 INTRODUCTION
Biological informatics (BI) has emerged as one of the most important interdisciplinary fields in the last decade. Major research efforts in BI include nucleotide sequence and structure alignment, gene finding and genome assembly, gene expression and regulation, protein and ribonucleic acid (RNA) structure prediction, biological networks and pathway analysis, genome-wide association studies, computational proteomics and biomarker detection, molecular evolution and population genetics, to name a few. These efforts are often concurrent with the development of software tools for data analysis, leading to new discoveries in diverse areas of the biological sciences
This chapter presents a case study in BI, focusing on locating noncoding RNAs (ncRNAs) in Drosophila genomes using software tools, in particular the Infernal package Noncoding RNAs are functional RNA transcripts that are not messenger RNAs (mRNAs) and therefore are not templates in protein biosynthesis. Recent experiments have shown that ncRNAs perform a wide range of cellular functions [8]. In particular, RNA on the X-1 (roX1) plays an essential role in the dosage compensation system, which increases the transcription level on the X chromosome in Drosophila males (XY) with respect to that of females (XX) [9]. Experiments have shown roX1 functionality in some species of Drosophila whose genomes have been annotated [10–12]. Our working hypothesis is that RNA transcripts of the roX1 genes across Drosophila species possess conserved secondary structures. Advances in genomic sequencing from 12 Drosophila species [13] will contribute to support this hypothesis.
The software package Infernal has implemented covariance models for the prediction of ncRNA functional conservation and it is considered to be one of the most accurate tools for this purpose [14]. A covariance model is a statistical representation or profile of a family of related RNAs that share a consensus RNA secondary structure [15]. Infernal [6,7] contains the utility cmbuild for the production of a covariance model from a multiple sequence alignment in Stockholm format. Another utility, cmsearch, is used to search for sequences that are similar to the model. The cmsearch process is computationally expensive when using a single-processor approach. However, by utilizing a parallel-processing approach, search results can be obtained expeditiously.
We have used a covariance model to demonstrate its capabilities in genome-scale searching. The roX1 sequences were experimentally obtained from eight Drosophila species, namely, D. ananassae, D. erecta, D. melanogaster, D. mojavensis, D. pseudoobscura, D. simulans, D. virilis, and D. yakuba. We focused on obtaining evidence of conserved RNA secondary structures within roX1 sequences on the entire genomes of these eight organisms To this effect, we used a covariance model derived from several roX1 sequences at hand. We found conserved RNA secondary structures within roX1 genes of six of the eight Drosophila species. We then used the same covariance model to search for evidence of conserved RNA secondary structures in the complete genomes of the four remaining sequenced Drosophila species for which we have no experimentally obtained roX1 genes. These four species are D. grimshawi, D. persimilis, D. sechellia, and D. willistoni. Our results show strong evidence for the presence of roX1 functional domains encoded in the genome of D. sechellia.
3.2 DATA
There is a wide variety of biological data stored in open access databases. Herbert et al. [16] surveyed the many bioinformatic databases accessible on the worldwide web. In this case study, we use two categories of data: roX1 genes and Drosophila genomes (cf. Tables 3.1 and 3.2). We used a “slide-and-fold” method to construct thermodynamically stable RNA secondary structures in the roX1 genes. Gene subsequences of 100 nucleotides (nt) long or less were folded according to thermodynamic properties using the Vienna RNA package [17,18]. Adjacent subsequences were overlapped by 50 nt. With this method, RNA secondary structures can be derived accurately and efficiently for two reasons: (1) predicting the formation of small secondary structures is more accurate and efficient than for large ones; and (2) secondary structures with size <50 nt are folded twice as subsequences of two different larger secondary structures, further increasing the chance of getting accurate RNA secondary structures. We also used the setting in the Vienna package that yielded multiple RNA secondary structures with the same minimum energy for a given sequence to further improve the folding accuracy. The number of predicted RNA secondary structures for each roX1 gene is shown in the last column of Table 3.1. A total of 773 RNA secondary structures was obtained for all eight species examined.
TABLE 3.1 Description of roX1 Genes Used in This Case Study
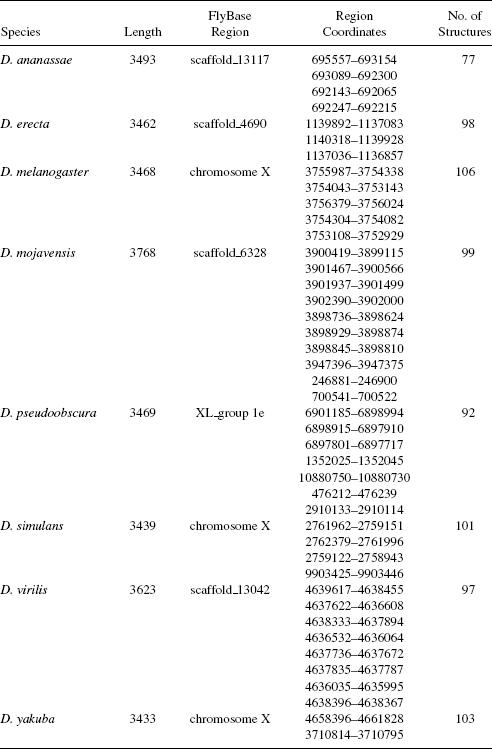
TABLE 3.2 Description of Drosophila Genomes Downloaded from FlyBase
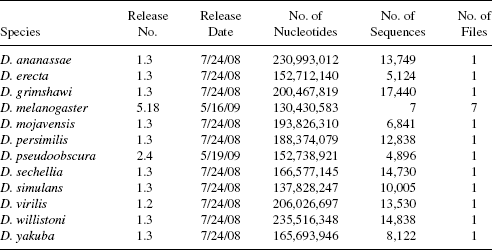
Table 3.2 presents details of the 12 Drosophila genomes used in our case study. Genome data were downloaded from FlyBase, accessible at http://flybase.org/. The number of files these sequences reside in is indicated in the last column. There is a total of 2,161,185,247 nucleotides in the 12 genomes, which collectively contain 122,120 sequences residing in 18 files. Most of the genomes have not been separated into clearly defined chromosomes. As sequencing efforts continue for species of Drosophila, we expect that in time all 12 specie genomes will be annotated regarding specific chromosome identification.
3.3 TOOLS
In the case study presented here, we used two software tools to analyze the genomic data at hand. The first tool, called RSmatch and developed in our lab [19–21], is capable of aligning structure-annotated RNA sequences so that both sequence and structure information are taken into consideration during the alignment process. The second tool, Infernal [6,7], was designed for genome-wide searching for conserved RNA secondary structures. Since the secondary structure of RNA determines its function, the Infernal tool is capable of predicting ncRNA functional conservation in genomes.
RSmatch [21] decomposes an RNA secondary structure into a set of components that are further organized by a tree model to capture the peculiarities of this RNAs framework. RSmatch can find the optimal global or local alignment between two RNA secondary structures using two scoring matrices, one for single-stranded regions and the other for double-stranded regions. The time complexity of RSmatch is O(mn), where m is the size of the query structure and n that of the subject structure.
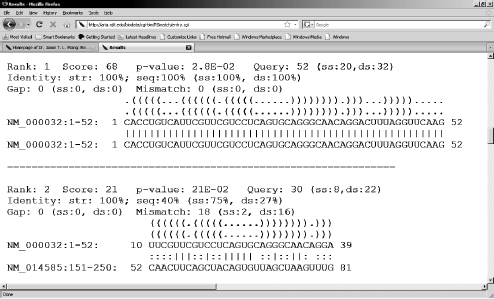
The RSmatch program is implemented into a web server called RADAR (acronym for RNA Data Analysis and Research) [19,22]. This web server can perform a multitude of functions related to RNA structure comparison, including pairwise structure alignment, constrained structure alignment, multiple structure alignment, database search, clustering, and consensus structure prediction. The goal behind establishing this web server is to develop a versatile tool that provides a computationally efficient platform for performing tasks related to RNA structure analysis. Figure 3.1 shows a partial output obtained from aligning a query RNA structure with a set of subject RNA structures using the local alignment function of RADAR. The figure lists the top two ranked subject structures that receive the largest alignment scores, where the structures are represented in the Vienna style dot bracket format [17, 18].
FIGURE 3.1 Screenshot showing a partial output from the local alignment function of RADAR.
A program component of Infernal, cmbuild, takes a structurally annotated multiple sequence alignment as input, and outputs a profile, whereas the program cmsearch uses the profile to search a nucleic acid sequence database for homologous RNAs. In addition, Infernal contains a program called cmalign, which uses the profile to align any number of unaligned RNA sequences to the profile, producing a structure-based multiple sequence alignment [6,7].
Infernal employs profile stochastic context-free grammars, which include both sequence and RNA secondary structure consensus information. This tool is used for constructing and maintaining the Rfam database of structurally annotated RNA multiple alignments [8]. The Rfam database contains hundreds of RNA sequence families, where each family has a hand-curated representative alignment, called a seed alignment. This seed alignment is used to make a profile, which can be aligned to new RNA sequences, obtained when nucleic sequence databases grow, to obtain a large, more complete alignment, called a full alignment. The Rfam sequence contains the seed alignments, full alignments, and consensus secondary structures of all the RNA families stored in its database.
3.4 APPLICATIONS
We applied RSmatch and Infernal to mining roX1 genes in Drosophila genomes as follows: We carried out species-against-species pairwise comparisons of all 773 RNA secondary structures obtained from the roX1 genes shown in Table 3.1 using the local alignment function of RSmatch. This required ∼520,000 pairwise comparisons of RNA secondary structures, each comparison yielding an alignment score. Then, we selected local matches across the species that received the largest alignment scores and that were the longest among all the local matches. We obtained one sequence from each of the following three species: D. melanogaster, D. simulans, and D. yakuba. Then, we used the MXSCARNA tool 23] to align the three sequences and obtain a multiple alignment in Stockholm format with predicted structure annotation. Figure 3.2 illustrates the multiple sequence alignment; the numeric range following the species code represents the portion of the roX1 gene from which the sequence was extracted. In Figure 3.3, the consensus secondary structure for the alignment in Figure 3.2 is portrayed using RNAz [24]. This structurally annotated alignment was input to the cmbuild program of Infernal 1.0 [7] to create a covariance model.
FIGURE 3.2 Illustration of the structurally annotated multiple sequence alignment used to build the covariance model in this case study.

FIGURE 3.3 Illustration of the consensus structure used to build the covariance model in this case study.

Then, we used the {cmsearch} program of Infernal 1.0 to locate homologs in the genomes of Drosophila species that have been sequenced to date (shown in Table 3.2) using the covariance model (CM) we constructed. Table 3.3 presents a summary of
TABLE 3.3 Summary of Homologs Found in the Drosophila Species Analyzed
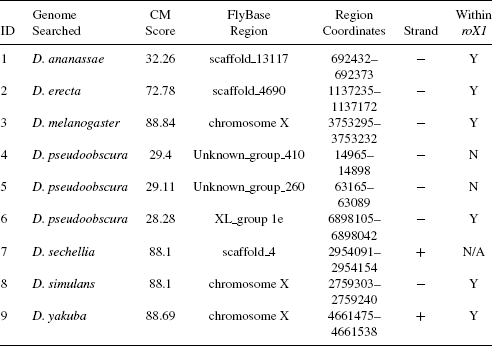
TABLE 3.4 Homologous RNA Sequences Found in the Drosophila Species Analyzed

the homologs with the largest scores found in each species, and Table ef{table4} lists the homologous RNA sequences.
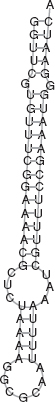
It was observed that for the three sequences used to build the covariance model, each of them received the largest score on its respective genome (D. melanogaster, D. simulans, and D. yakuba, respectively). In addition, we found high-score hits in three other species, namely, D. ananassae, D. erecta, and D. pseudoobscura. Note that in D. pseudoobscura, the homolog with the largest score is not within the roX1 gene in that species. Instead, we found a homolog with the third largest score that is within the roX1 gene in D. pseudoobscura. These homologs found in the above six species for which roX1 genes have been experimentally obtained fall within the known roX1 genes. The homologous RNAs are similar in their structure. This confirms our hypothesis that there are conserved RNA secondary structures within roX1 genes across these species. Finally, note that a homolog was found in the genome of D. sechellia for which we have not experimentally obtained a roX1 gene. Figure 3.4 illustrates the secondary structure of the homologous RNA discovered from D. sechellia; this secondary structure is similar in structure to the consensus structure in Figure 3.3. This uncovering may help compensation dosage researchers in locating roX1 functional domains in D. sechellia.
FIGURE 3.4 Secondary structure of the homologous RNA found in Drosophila sechellia
3.5 CONCLUSION
This chapter presents a case study of biological informatics, showing how software tools are applied to the analysis of genomic data arising in the biological sciences. In particular, this chapter showed how the widely used Infernal and RSmatch tools can be combined to mine roX1 genes in 12 species of Drosophila for which entire genomic sequencing data are available. As more genomes are sequenced, new techniques are needed to perform genome-wide discovery of functional elements that play essential roles in metabolic processes. This opens many new research directions in both the biological sciences and computing sciences, while bringing them together.
REFERENCES
1 1. S. Bandyopadhyay, U. Maulik, and J. T. L. Wang (Eds.) (2007), Analysis of Biological Data: A Soft Computing Approach, World Scientific, Singapore.
2 J. T. L. Wang, B. A. Shapiro, and D. Shasha (Eds.) (1999), Pattern Discovery in Biomolecular Data: Tools, Techniques and Applications, Oxford University Press, NY.
3. J. T. L. Wang, C. H. Wu, and P. P. Wang (Eds.) (2003), Computational Biology and Genome Informatics, World Scientific, Singapore.
4. J. T. L. Wang, M. J. Zaki, H. T. T. Toivonen, and D. Shasha (Eds.) (2005), Data Mining in Bioinformat., Springer, London.
5. S. R. Eddy (2002), A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure, BMC Bioinformat., 3:18.
6. S. Eddy and R. Durbin (1994), RNA sequence analysis using covariance models, Nucleic Acids Res., 22:2079–2088.
7. E. P. Nawrocki, D. L. Kolbe, and S. R. Eddy (2009), Infernal 1.0: inference of RNA alignments, Bioinformat., 25(10):1335–1337.
8. S. Griffiths-Jones, S. Moxon, M. Marshall, A. Khanna, S. R. Eddy, and A. Bateman (2005), Rfam: annotating non-coding RNAs in complete genomes, Nucleic Acids Res., 33:D121–D124.
9. Y. Park and M. I. Kuroda (2001), Epigenetic aspects of X-chromosome dosage compensation, Science, 293:1083–1085.
10. S. Park, Y. I. Kang, J. G. Sypula, J. Choi, H. Oh, and Y. Park (2007), An evolutionarily conserved domain of roX2 RNA is sufficient for induction of H4-Lys16 acetylation on the Drosophila X chromosome, Genetics, 177(3):1429–1437.
11. S. Park, M. I. Kuroda, and Y. Park (2008), Regulation of histone H4 Lys16 acetylation by predicted alternative secondary structures in roX noncoding RNAs, Mol. Cell. Biol., 28(16):4952–4962.
12. Y. Park, R. L. Kelley, H. Oh, M. I. Kuroda, and V. H. Meller (2002), Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins, Science, 298:1620–1623.
13. A. Stark, et al. (2007), Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures, Nature (London), 450:219–232.
14. A. Wang, W. Ruzzo, and M. Tompa (2007), How accurately is ncRNA aligned within whole-genome multiple alignments? BMC Bioinformat., 8:417.
15. R. Durbin, S. Eddy, A. Krogh, and G. Mitchison (1998), Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids, Cambridge University Press, Cambridge UK.
16. K. G. Herbert, J. Spirollari, J. T. L. Wang, W. H. Piel, J. Westbrook, W. C. Barker, Z. Z. Hu, and C. H. Wu (2008), Bioinformatic databases, Wiley Encyclopedia of Computer Science and Engineering, B. Wah (Ed.), John Wiley & Sons, Inc., NY, pp. 561: 1–561:10.
17. A. R. Gruber, R. Lorenz, S. H. Bernhart, R. Neubock, and I. L. Hofacker (2008), The Vienna RNA websuite, Nucleic Acids Res., 36:W70–W74.
18. I. L. Hofacker (2003), Vienna RNA secondary structure server, Nucleic Acids Res., 31:3429–3431.
19. M. Khaladkar, V. Bellofatto, J. T. L. Wang, B. Tian, and B. A. Shapiro (2007), RADAR: a web server for RNA data analysis and research, Nucleic Acids Res., 35:W300–W304.
20. M. Khaladkar, J. Liu, D. Wen, J. T. L. Wang, and B. Tian (2008), Mining small RNA structure elements in untranslated regions of human and mouse mRNAs using structure-based alignment, BMC Genomics, 9:189.
21. J. Liu, J. T. L. Wang, J. Hu, and B. Tian (2005), A method for aligning RNA secondary structures and its application to RNA motif detection, BMC Bioinformat., 6:89.
22. M. Khaladkar, V. Patel, V. Bellofatto, J. Wilusz, and J. T. L. Wang (2008), Detecting conserved secondary structures in RNA molecules using constrained structural alignment, Comput. Biol. Chem., 32:264–272.
23. Y. Tabei, H. Kiryu, T. Kin, and K. Asai (2008), A fast structural multiple alignment method for long RNA sequences, BMC Bioinformat., 9:33.
24. A. R. Gruber, R. Neubock, I. L. Hofacker, and S. Washietl (2007), The RNAz web server: prediction of thermodynamically stable and evolutionarily conserved RNA structures, Nucleic Acids Res., 35:W335–W338.