
CHAPTER 10
IN SILICO DRUG DESIGN USING A COMPUTATIONAL INTELLIGENCE TECHNIQUE
10.1 INTRODUCTION
Discovery of novel lead molecules has always been a long, time consuming, and expensive process in traditional drug discovery. It involves searching a chemical space of >1018 compounds [1] to find a suitable small molecule that can act as a drug and is safe to be administered. Finding a suitable molecule with the desired chemical property from such a large search space is a very complex problem. With the advent of new technologies and the abundance of three-dimensional (3D) structures of proteins, the scientific fraternity can exploit this structural information in order to design novel ligands possessing high-binding affinity to selective target proteins. This approach of finding novel ligand molecules using the structure information of the receptor is usually referred to as structure-based drug design.
Several Lead molecules have been discovered using the structure-based drug design approach. A few of them are approved drugs and many others are under clinical trials. Prostaglandin D synthase inhibitors [2] and X-linked inhibitor of apoptosis protein inhibitors [2] are most recent examples of drug design using fragment-based Lead design. The success of this approach inspired the scientists to apply computational technologies to aid the drug discovery process. The main objectives of the computational methods was to reduce the time and expense of the drug discovery process. One of the earliest such approaches for computer-aided drug design or rational drug design is DOCK [4]. This approach is a virtual screening method. It searches the database of 3D ligands to find the most suitable small molecule for a given target protein. To find the goodness of a ligand, it docks the ligand to the given receptor and gauges the stability of the complex. There is another approach to address the problem. Instead of searching among known 3D molecules to find a suitable molecule, algorithms can be developed to design molecules with desired chemical features.
The problem of Lead optimization, be it by searching known 3D molecules or by conceiving novel molecular scaffolds, is a search and optimization problem. In such a scenario, application of genetic algorithms (GAs) [5,6] seems natural and appropriate. These GAs are a family of search and optimization techniques inspired by the principles of evolution.
A few earlier applications of GAs for efficient ligand design are [7–9]. Budin {et al}., [10] developed a GA based approach for building peptides. However, a seed has to be provided to the program for building the final ligand. Globus {et al}., proposed another GA based approach to evolve small molecules represented as graphs, with atoms as nodes and bonds between the atoms as edges [11]. Goh and Foster [8] proposed a GA based ligand design framework that uses a tree structure encoding for representing the ligands. The trees representing the ligands contain a functional group, selected from a given library, at each leaf. But, it had an important limitation. This approach used a fixed tree length for encoding the ligands. Ligands can never be of the same size for every target, rather the ligand size would vary with the active site geometry of different targets. To overcome this, Bandyopadhyay {et al}., proposed a method based on variable length genetic algorithm (VGA) [7]. This approach was more realistic as it used a variable length tree for encoding ligand as chromosomes. Consideration of the variable tree length allowed ligand size to vary with different active site geometries [7]. But again this approach also builds two-dimensional (2D) ligands. Therefore, solutions provided by the approach may not be as good when conceived in 3Ds. This chapter endeavors to improve this work by mining the active site from a given protein structure, building 3D ligands, considering different energy components for optimization, a much larger suite of functional groups for constructing the ligand, both inter- and intramolecular interactions for optimization and domain specific crossover and mutation operators. Experiments have also been conducted to study the contribution of the intramolecular and the intermolecular energy components in virtual screening. The effectiveness of the algorithm is established through a comparative analysis of the results of proposed methods to VGA and two other existing approaches for ligand design, namely, NEWLEAD [12] and LigBuider [13].
10.2 PROPOSED METHODOLOGY
This chapter discusses a genetic algorithm-based approach to de novo ligand design and emphasizes the importance of the contribution of intramolecular and intermolecular interaction energy for optimizing the ligands. The program takes the protein data bank (PDB) structure files of the target proteins as input. From these input files, the active site of the target protein is mined. According to the geometry and chemistry of the target active site, the core ligand molecules are built while initialization. These molecules are evolved using specially tailored domain specific genetic operators according to the value of the optimization parameter. Intramolecular and intermolecular interaction energy are the optimization criteria computed by the fitness function. Intramolecular energy is a sum of bond stretching, angle bending, angle rotation, van der Waals, and electrostatic energy components. The intermolecular interaction energy is a sum of van der Waals and electrostatic energy components. We have obtained the fitness value as a weighted sum of the intramolecular and intermolecular energies to investigate the importance of their contribution for the Lead optimization problem. Further, to validate our results we have docked the ligands built using the proposed algorithm, VGA, NEWLEAD, and LigBuilder with their target proteins employing an already established software InsightII [ MSI/Accelrys, San Diego, CA] and have compared them to their similar molecules present in Cambridge structural database (CSD).
10.2.1 Active Site Processing
The steps involved in active site identification for ligand building are accounted for in Section 10.2.1.1–10.2.1.3.
10.2.1.1 Active Site Identification Preliminary information about the geometry and chemical composition of either the target active site or the natural ligand of the target is required to address the Lead molecule design problem. The program that we have developed requires the geometry and chemistry of the active site to build its corresponding ligands. The geometry of the target active site determines the size of the ligand and the chemical composition actuate the desired chemical property of the ligand. To mark the active site in the given protein, its coordinates obtained from the PDB are fit into a grid with a spacer of 0.5 Å. Grids containing protein coordinates are labeled as “full”. If the input file is a ligand–protein complex, then grids occupied by the ligand coordinates are labeled as “empty” and this group of grids is treated as the active site. If an isolated protein molecule is input, then the empty grids containing nothing are labeled “empty” and the largest assembly of such “empty” grids is considered as the active site.
10.2.1.2 Active Site Surface Marking After identifying the active site, the dimensions and chemical property of the active site is determined. Therefore, as the next step the surface of the active site is marked and the amino acids on its surface are also marked. To find the active site surface each “empty” grid comprising the active site is filled with a water molecule of radius 1.4 Å. After placing the water molecule in each grid, the distance between the water molecule and the nearest protein coordinate is calculated. If the distance is 0.5 Å or less, then that particular grid containing the coordinate of the protein molecule is labeled as “surface” and the atom on that coordinate is noted. The atoms on the surface of the active site are responsible for the biological functionality of the protein. So, these atoms are necessary to be considered while building a ligand that is supposed to bind to it. These atoms are responsible for the protein–ligand interaction. The center of the active site is detected by calculating the median of the set of grid points considered as the active site. The ligand building starts from this point.
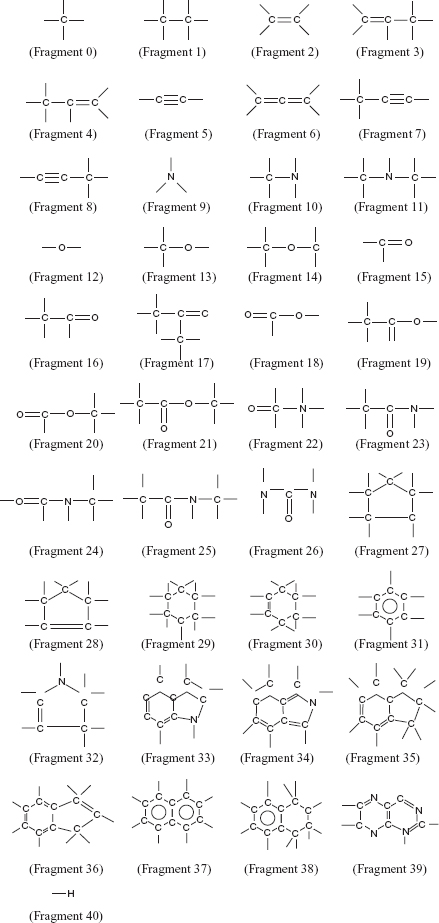
10.2.1.3 Building Preference Matrix The atoms on the surface of the active site are marked to design ligands that can bind efficiently to the target protein. A preference matrix is constructed to make a sequential list of every fragment being in the vicinity of a particular atom in the active site. The preference matrix contains atoms of the receptor protein comprising the active site in its rows while columns contain the estimation of the different fragments, as mentioned in Figure 10.1, of being in the vicinity of the given atom in the specific row.
FIGURE 10.1 Forty-one fragments (groups) were used to build the ligands using the proposed method.
To estimate the probability of a fragment being in the neighborhood of a particular active site atom, first the nonbonding interactions (NBI) between all the fragments and each of the atoms constituting the active site are calculated. These interactions are calculated as
Equation (10.1) considers van der Waals and electrostatic interactions. Here, Cn and Cm are constants, rij is the distance between the ith atom of the fragment f and jth interacting atom on the receptor active site, and qi, qj are charges on i and j, respectively.
Then the probability of each fragment f′, f′ = 0, 1, 2, …, 40, to be considered in the neighborhood of a given atom j on the receptor active site, is calculated as

Here, probj,f′ is the probability of the fragment f′ being in proximity of the atom j, in the active site of the receptor. The numerator is the NBI energy between atom j on the receptor active site and a fragment f′. The denominator is the sum of the NBI energies between each fragment and the atom j of the receptor active site.
10.2.2 Genetic Algorithm-Based Ligand Design
A GA based de novo ligand design algorithm is described here. This algorithm uses the fragment-based approach for designing ligands. The fragment library that it uses for ligand building is shown iny Figure 10.1. Chromosome representation, population initialization, fitness computation, and genetic operators of the proposed algorithm for ligand design are explained in Sections 10.2.2.1–10.2.2.4.
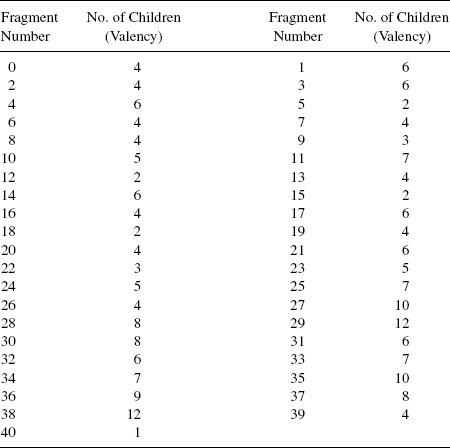
10.2.2.1 Chromosome Representation and Population Initialization The present work uses 41 fragments, as shown in Fig. 10.1, for constructing the ligands. Table 10.1 gives the valencies corresponding to each fragment. For example, fragment number 9 is a nitrogen atom and and it has a valency of 3 (i.e., it can form three more bonds to join itself to other fragments). The ligand size may vary according to the different target active sites. Therefore, to allow construction of ligands with varying length, a chromosome is encoded as a variable length tree like structure. Each gene of a chromosome or each node of the tree representing a ligand is a structure containing 3
integers and 12 pointers. The first integer is the gene number, the second contains the fragment number of the fragment present at that gene position, and the third denotes the valency of the fragment as given in Table 10.1. Twelve pointers are considered since the highest valency of a fragment in the fragment library considered for ligand building is 12. Each pointer can either be NULL or can point to any other node. The valency of a fragment decides the number of pointers pointing to any other node. For the convenience of implementation, a back pointer is kept from every node of the tree, except the root, which points back to its parent.
TABLE 10.1 Fragment Number and Their Corresponding Valencies
The scheme is defined as follows:

To initialize a chromosome i, the first gene position of the chromosome is filled by a fragment selected randomly from the fragment library, as mentioned in Figure 10.1. From the second gene position onward, each fragment is placed at every gene position and the fragment due to which the internal energy of the ligand is least is used for ligand extension. The ligand extension occurs as long as the ligand does not grow out of the active site. This incremental construction is done only for initializing the population with core ligands that are further evolved using the GA.
10.2.2.2 Fitness Evaluation The fitness value of a ligand gauges the goodness of the solution. The goodness of a probable ligand is calculated as a function of its chemical composition and proximity to the target active site. The fitness function computes the intramolecular ligand energy and the intermolecular nonbonding interaction energy of the ligand and protein. Low intramolecular energy ensures a stable ligand and low inter molecular interaction energy warrants a stable ligand–protein complex implying better binding affinity. In this work, we have taken a weighted sum of the intramolecular and intermolecular interaction energy to enunciate their contributions in selectivity of ligands.
The van der Waals (vdw) energy is calculated using the following Lennard-Jones 6–12 potential function [14]
Here, Evdw(x, y), Cn, Cm, and rxy are the van der Waals interaction energy, constants and the distance between the interacting atoms x and y, respectively.
The electrostatic interaction energy Eel(x,y) between two atoms x and y is calculated by [14]
Here, qx and qy are formal charges of the interacting atoms, ![]() 0 is a constant, and rxy is the distance between the interacting groups. Solvent water molecules are not considered for the calculations, so distance-dependent dielectric constant (
0 is a constant, and rxy is the distance between the interacting groups. Solvent water molecules are not considered for the calculations, so distance-dependent dielectric constant (![]() ) is used to mimic the solvent effect during calculation [15].
) is used to mimic the solvent effect during calculation [15].
Equation (10.5) is used for calculating the bond-stretching energy El(x,y) p14]
Here, lxy and lxy,0 are the calculated bond length and reference bond length between the two atoms x and y, respectively. The bond-stretching constant is kl.
The angle bending energy Eθ is calculated using the following equation [14]:
Here, θ and θ0 are the calculated angle and reference angle, respectively and kθ is the angle-bending constant.
The torsional energy EФ is calculated using the following expression [14]:
Here, Ф and Ф0 are the calculated and reference torsion angles, respectively, kФ is the torsion contribution constant, and n is the periodicity linked with the type of central bond of the torsion.
Therefore the intramolecular energy can be calculated by combining all the above mentioned energies as follows:
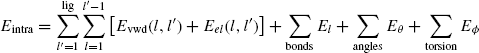
The intermolecular energy is
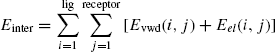
Here, lig and receptor are the number of groups in the ligand and the receptor, respectively. Sum of the intramolecular and intermolecular energy gives the total energy that determines the stability of the ligand, as well as the ligand–protein complex. But, as mentioned above, to enunciate the contributes of the Eintra and Einter in ligand selection pressure weighted sum of both the energy components are considered. Therefore the total energy is calculated as
Here, w, that varies between 0 and 1, controls the contributions of the two energy components toward the composite energy, Etotal. Different values of w are used in the experiments in order to evaluate the relative importance of the two factors and to design an effective fitness function.
As mentioned before, lower energy corresponds to more stable ligands. Therefore the fitness value of a chromosome is set to be inversely proportional to the total energy value. In other words, the fitness value, F, of a chromosome is defined as
such that individuals with higher fitness correspond to ligands having lower energy values, and hence greater stability.
For refining the results, a few domain specific constraints are applied. Nonbonding interaction energy are is calculated for the functional group of the ligand lying at a distance not >5 Å and not <0.65 Å from the protein receptor molecule to avoid steric hindrance. Three dimensional conformation and orientation of a ligand is very important to make bonds with its target. For example, a polar hydroxyl group should lie close to an amine group or acidic amino acids to make stable hydrogen bonds. Similarly, hydrophobic interactions will occur only when hydrophobic atoms of the ligand will face the hydrophobic amino acids on the active site of the target protein. Ligands violating these constraints are heavily penalized by adding a large positive integer to the total energy so that they will be automatically eliminated in the evolutionary process.
10.2.2.3 Genetic Operators The genetic operators used are selection, crossover, and mutation. Roulette wheel selection technique is employed. According to this selection strategy, more fit individuals are more probable to reproduce (i.e., a chromosome is more likely to be selected as a parent for reproduction if it has better fitness).
A crossover probability of cprob is adaptively set for application of the domain specific crossover operator, on a pair of parent chromosomes. For performing the crossover, the crossover points in both the parents are generated. A crossover point is a randomly generated gene number in a parent. The subtrees following these gene numbers on the crossover points are exchanged to obtain two new chromosomes. After the exchange, the pointers and the gene numbers are rearranged appropriately by following a breadth first traversal. According to the cprob, if the crossover is not to performed between a pair of parents, then they are simply copied to the next population. The adaptive crossover probability is calculated similarly to [1]. If fmax is the maximum fitness value of the current population,![]() is the average fitness value of the population and f′ is the larger of the fitness values of the solutions to be crossed. Then the probability of crossover, cprob, can be calculated as
is the average fitness value of the population and f′ is the larger of the fitness values of the solutions to be crossed. Then the probability of crossover, cprob, can be calculated as
Here, similar to [16], the values of k1 and k3 are both considered as 1.0. Note that, when ![]() and cprob attains the same value as k3. The impetus for this adaptation is to achieve a trade-off between exploration and exploitation in a different manner. When two chromosomes with poor fitness are to be crossed, the cprob increases, but it decreases if the two solutions under consideration for crossover are good solutions. This adaptation increases the likelihood of the bad solution to be evolved and decreases the likelihood of disrupting a good solution by crossover.
and cprob attains the same value as k3. The impetus for this adaptation is to achieve a trade-off between exploration and exploitation in a different manner. When two chromosomes with poor fitness are to be crossed, the cprob increases, but it decreases if the two solutions under consideration for crossover are good solutions. This adaptation increases the likelihood of the bad solution to be evolved and decreases the likelihood of disrupting a good solution by crossover.
Mutation is also performed with an adaptive mutation probability of mprob as described in [16]. A gene position is to be mutated or not is decided using the mutation probability mprob. If a gene position is found to be suitable for mutation then the preference matrix, described earlier, is consulted to replace the gene position with the most likely substitute. For finding the appropriate substitute, the nearest neighbor on the gene on the active site is located. Then, a roulette wheel selection scheme is employed to select an appropriate fragment from the preference matrix to replace the fragment in the selected gene position. The mprob is calculated using the following expression
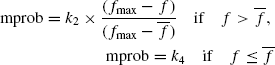
Here, values of k2 and k4 are set equal to 0.5. Adaptive mutation is an aid to GA for overcoming a local optimum.
Adaptive crossover and mutation probabilities are helpful in avoiding premature convergence of GA at local optima. When (![]() ) decreases indicating an optima, both cprob and mprob increases preventing the convergence of the GA. But due to this phenomenon, disruption of the near-optimal solutions can occur in order to prevent the convergence of the GA at even the global optimum. But as cprob and mprob attain lower values for more fit solutions and attain higher values for less fit solutions, individuals with high fitness values help in the convergence of the GA while the individuals with low fitness values prevent the GA from getting stuck at a local optimum. For solutions with the maximum fitness value, cprob and mprob are both zero.
) decreases indicating an optima, both cprob and mprob increases preventing the convergence of the GA. But due to this phenomenon, disruption of the near-optimal solutions can occur in order to prevent the convergence of the GA at even the global optimum. But as cprob and mprob attain lower values for more fit solutions and attain higher values for less fit solutions, individuals with high fitness values help in the convergence of the GA while the individuals with low fitness values prevent the GA from getting stuck at a local optimum. For solutions with the maximum fitness value, cprob and mprob are both zero.
10.2.2.4 Elitism The termination criterion for the algorithm is a specified number of generations. Elitism is incorporated in each generation. The best solution observed till a generation is stored in the next population, as well as in a location outside the population, is referred to as the elite population. Note that the size of the elite population is equal to the number of generations executed.
10.2.3 Postprocessing of the Ligand
The proposed algorithm is executed with the value of w as 0, 0.25, 0.50, 0.75, and 1. Elite and final populations are screened to identify the three top scoring individuals for each run of the proposed algorithm with w as 0, 0.25, 0.50, 0.75, and 1. A total of 15 high-scoring ligands were screened from the solution pool obtained using the proposed algorithm with various weights of inter- and intramolecular energy and are further analyzed for their goodness. For this analysis, the “Docking" module of Insight II [MSI/Accelrys, San Diego, CA] is used. Three ligands corresponding to the weight producing best results are chosen and are further compared with the output of VGA [7], NEWLEAD [12], and LigBuilder [13]. The optimized 3D structure of the 2D ligands designed by VGA are obtained by using a “Build” module of Insight II [MSI/Accelrys, San Diego, CA]. This module has a built in optimization routine to find the most stable conformation of the input ligand. Similar to comparing the activity of the ligands designed using the proposed algorithm, VGA, NEWLEAD, and LigBuilder, they are docked using the “Docking" module of Insight II [MSI/Accelrys, San Diego, CA]. To study if it was possible to synthesize the designed ligands, CSD, a library of >450,000 small molecules, is searched using ConQuest [17] to find molecules similar to the designed ligands. These are considered for further docking and comparative analyses. The idea behind the comparison of the real molecules, retrieved from CSD, to the designed molecules, is that if any derivative of the designed molecule has already been synthesized, then perhaps the designed molecules can also be synthesized. Therefore, the corresponding real molecule can also assist in finding out the activity of the designed molecules. Moreover, if the similar real molecule is found to posses high-binding affinity to the given target, then both the real molecule and the designed molecules could be investigation worthy.
10.3 EXPERIMENTAL RESULTS AND DISCUSSION
C programming language has been used to develop the proposed algorithm on the UNIX platform. The performance of GA depends on the choice of the control parameters. Hence, the mutation and crossover probability are adaptively set as suggested in [16]. The number of generations and the population size are set to 1000 and 100, respectively.
The drug targets considered for the study are HIV-1 Protease and Thrombin. The posttranscriptional processing of viral gag and gag-pol proteins for the production of functional viral proteins are assisted by HIV-l Protease. The structural proteins of virion core (i.e., p17, p24, p7, and p6) are transcribed and translated from the gag gene. Gag-pol is responsible for the release of viral replicative enzymes (protease, reverse transcriptase, and integrase), necessary for its retroviral life cycle. Thrombin is a serine protease prominently participating in blood coagulation. It hydrolyzes fibrinogen to fibrin for activating platelets to form the clot. Thrombin can be used as a tool to control coagulation cascade and ameliorate specific diseases [18]. A wide variety of binding modes and geometries of thrombin has been revealed by many scientists. According to [19] thrombin contains three principal interaction sites, namely, S1, D, and P. The S1 site contains an aspartic acid residue. Both D and P sites contain hydrophobic pockets. All three sites are responsible for the specificity of thrombin. The PDB entries 1AAQ and 1DWD [19, 20], the structure of HIV-1 Protease and Thrombin, respectively, are used as input files to the program. Both NAPAP and Ritonavir are the known synthetic inhibitors of Thrombin and HIV-1 Protease, respectively. Therefore, the ligands designed using the proposed algorithm, VGA, NEWLEAD, and LigBuilder are compared with these molecules to estimate their goodness.
10.3.1 Comparative Analyses of the Contribution of Intramolecular and Intermolecular Energy for Ligand Designing
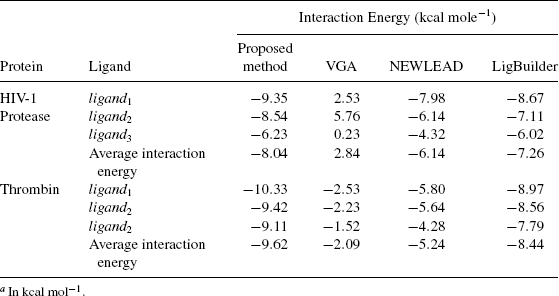
The three top scoring individuals obtained after each run of the proposed algorithm using the value of w as 0, 0.25, 0.50, 0.75, and 1 corresponding to each of the target proteins are docked to their respective receptors. Their corresponding energy values are given in Table 10.2. The results shows that the ligand target complexes possess less energy when the ligands are designed with w = 0.5. This observation indicates that the ligands designed using the equal contribution of intramolecular and intermolecular energy are better in comparison to the ligands designed with unequal contribution of the intermolecular and intramolecular energy. The reason behind such an observation could be that the ligands that are being designed need to have a stable conformation for itself and should be able to bond well to its target. It can also be observed from Table 10.2 that the ligands designed considering only intermolecular energy (w = 0) are better than the ligands designed using only intramolecular energy (w = 1). The reason for this is that while designing ligands for a given target, the interaction of the target to the protein must be considered so as to get an approximate estimate of the binding affinity of the designed molecule to its given target. In fact, the work in [7] used only this component of the energy. Though the intermolecular interaction energy is more important to be evaluated than de novo ligand design, the efficacy of the algorithm increases when intramolecular energy is incorporated. Therefore, we conclude that the fitness function for evolving small drug-like molecules should give equal importance to both intramolecular and intermolecular interaction. Further analysis of the results for the comparative study with other existing approaches are obtained using w = 0.5.
TABLE 10.2 Interaction Energies for the Ligands Designed Using the Proposed Method With the Weight as 0, 0.25, 0.50, 0.75, and 1 for HIV-1 Protease and Thrombina
10.3.2 Comparative Analyses of Interaction Energy and Hydrogen-Bond Interaction
The ligands designed by the proposed algorithm (VGA, NEWLEAD, and LigBuilder) for HIV-1 Protease and Thrombin are docked with their corresponding receptor proteins using the “Docking” module of Insight II [MSI/Accelrys, San Diego, CA]. The corresponding interaction energies are provided in Table 10.3. The significantly lower energy values of the molecules designed using the proposed algorithm indicate that these produce more stable receptor ligand complexes. The ligands designed for HIV-1 Protease by each of the de novo design scheme and their docked complexes with the protein are shown in Figure 10.2. The ligands designed by the proposed algorithm are generally smaller and comprise less aromatic groups (see Figure 10.2), causing less stearic hindrance and better interaction with the target protein, in comparison to the ligands designed by the other methods.
FIGURE 10.2 Structure of a ligand and the docked protein–ligand complex obtained by (a and b) the proposed method, (c and d) VGA, (e and f) NEWLEAD, and (g and h) LigBuilder for HIV-1 Protease (visualized using Insight II [ MSI/Accelrys, San Diego, CA], where the protein is represented in ribbons and the ligands are represented in CPK).
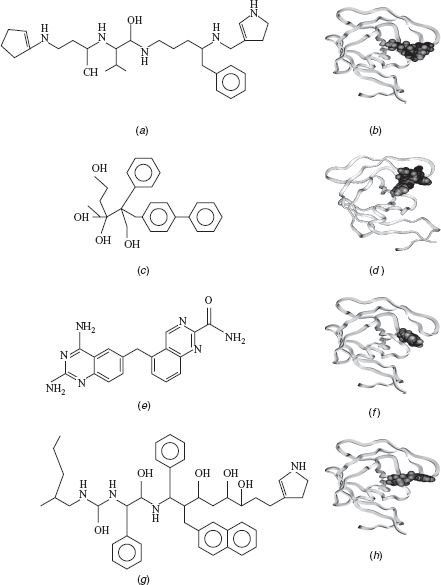
TABLE 10.3 Interaction Energies for the Ligands Designed Using the Proposed Method, VGA, NEWLEAD, and LigBuilder for HIV-1 Protease and Thrombina
Hydrogen bonds in a protein–ligand complex are very essential since the binding affinity between the protein and the ligand is dependent on them. Therefore, the interacting residues of the protein–ligand complex are observed to identify the hydrogen bonds.
Tables 10.4–10.6 give the hydrogen-bond details for the protein–ligand interaction. From these tables, it is evident that ligands designed by the proposed algorithm are in general forming more hydrogen bonds with their target proteins, implying a more stable docked complex.
TABLE 10.4 H-Bond Interactions of Ligand1 Designed by the Proposed Method, VGA, NEWLEAD, and LigBuilder With HIV-1 Protease and Thrombin
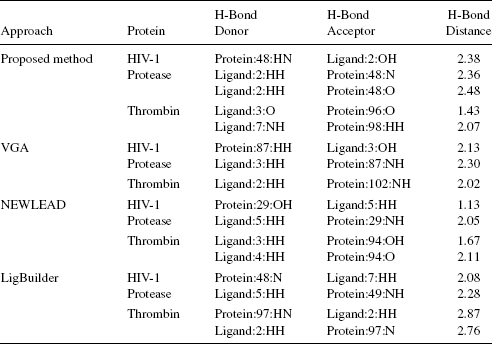
TABLE 10.5 H-Bond Interactions of Ligand2 Designed by the Proposed Method, VGA, NEWLEAD, and LigBuilder With HIV-1 Protease and Thrombin

TABLE 10.6 H-Bond Interactions of Ligand3 Designed by the Proposed Method, VGA, NEWLEAD, and LigBuilder With HIV-1 Protease and Thrombin
The rationale behind the formation of more and good hydrogen bonds between the target and the ligand designed by the proposed algorithm could be better genetic operators, bigger fragment libraries, and building 3D ligands. The proposed algorithm builds 3D ligands, therefore it can be expected that the ligands designed by it will always dominate results of VGA since 3D adaptation will allow better exploration of the search space. Consideration of preference matrix for mutation makes the process knowledge based and thus the algorithm is able to evolve a more fit individual in comparison to NEWLEAD and LigBuilder. The fragment library of the proposed algorithm is a balanced combination of the chemical fragments and atoms making the algorithm more flexible while designing small molecules. It is not compelled to grow ligands using only fragments, unlike LigBuilder, or only atoms, unlike [21, 22]. It can grow ligands using both fragments and atoms as per requirement. References [21, 22] have a comparatively large time complexity due to usage of atoms for building ligands. Therefore, the type of fragment library modeled for this algorithm increases the precision of the algorithm and reduces the time complexity.
To investigate the synthesizability of the ligands designed by the proposed method, VGA, NEWLEAD, and LigBuilder, analogous real molecules, are retrieved from CSD using the search module “ConQuest” [17]. When CSD is searched using the proposed ligands as query, several similar molecules are reported by ConQuest. Among these, the molecules that form a stable docked complex with their receptors are reported. The CSD Ref Codes, along with the interaction energy with their corresponding target proteins, HIV-1 Protease, and Thrombin are reported in Table 10.7. As seen earlier, the interaction energies are found to be smaller for the real CSD molecules that are similar to the ligands designed by the proposed algorithm in comparison to the molecules designed by VGA, NEWLEAD, and LigBuilder. The results point out that the molecules designed by the proposed algorithm form more stable docked complexes than the other three approaches.
TABLE 10.7 Comparison of the Interaction Energies for the Molecules Obtained from CSD for HIV-1 Protease and Thrombin
10.3.3 Comparative Study Using Root-Mean-Square Deviation Analysis
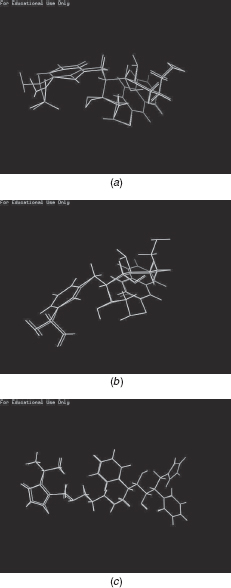
In order to compare the RMSD values between the known protein inhibitor and the ligands designed by the proposed method (VGA, NEWLEAD, and LigBuilder), the protein Thrombin is considered. Pymol [23] is used for aligning the two molecules appropriately and computing the RMSD values. The comparative results are reported in Table 10.8. As seen, the ligands designed using the proposed method and LigBuilder have small RMSD values, indicating their high similarity to the know inhibitor for Thrombin, NAPAP. The poor performance of VGA is expected since it builds the ligand in 2D space. Figures 10.3 (a) (b) show the ligands designed by LigBuilder and the proposed method, respectively, superimposed over NAPAP. For HIV-1 Protease, the known inhibitor is Ritonavir. A result for LigBuilder is unavailable for this protein. The ligand designed using the proposed method superimposed on Ritonavir is shown in Figure 10.3 (c). As seen, the proposed molecule is highly similar to the known inhibitor, with an RMSD value, computed using PyMol, of 1.97 Å.
TABLE 10.8 RMSD Values, (Å) between NAPAP and Ligands Designed Using the Computational Approaches for Thrombin

FIGURE 10.3 (a) Ligand built using grow module of LigBuilder (White) superimposed with NAPAP (Grey). (b) Ligand built using the proposed method (White) superimposed with NAPAP (Grey). (c) Ligand built using the proposed method (White) superimposed with Ritonavir (Grey).
10.4 CONCLUSION
Traditional drug discovery is a well-established process that involves large amounts of time and money. Though there is no substitute for the methodology, the advancement of science and technology have definitely found various ways to assist the procedure to reduce its time consumption and expense. Rational drug design tries to do the same thing (i.e., it helps reducing time and cost of the process by reducing the search space for wet experiments). This chapter describes a rational drug design technique for de novo ligand design. The program illustrated in this chapter takes the structure files of the target proteins in PDB file format and mines the active site from it. A preference matrix is built using the chemical property of the active site that is further used to evolve the ligands. It uses a library of 41 fragments for building ligands. The program uses genetic algorithm to search the chemical space and find an appropriate ligand for the given target. It uses a variable length tree-like structure for chromosome representation and domain-specific genetic operators. Though the chromosome representation involves memory overhead, it is manageable as the ligands designed are usually small. The fitness function of the algorithm computes bond stretching, dihedral angle, angle bending, electrostatic, and van der Waals energy components.
The results can be further improved if the algorithm amalgamates hydrophobic interaction and desolvation energies with the present fitness function for evaluating the goodness of the solutions. Usage of Lipinski rule of five, QSAR properties, and ADMET properties for ligand building can also improve results. Inclusion of these properties will assist in deciding the safety and efficacy of the ligands designed. The memory overhead also needs to be reduced using some other chromosome representation. The ligands designed using the proposed methodology are flexible, but while designing ligands, receptor proteins are considered rigid. To improve the algorithm, receptor flexibility needs to be incorporated.
REFERENCES
1. W. P. Walters, M. T. Stahl, and M. A. Murcko (1998), Virtual screening - an overview. Drug Disc. Today, 3:160–178.
2. M. Hohwy, L. Spadola, B. Lundquist, P. Hawtin, J. Dahmén, I. Groth-Clausen, E. Nilsson, S. Persdotter, K. v. Wachenfeldt, R. H. A. Folmer, and K. Edman (2008), Novel prostaglandin d synthase inhibitors generated by fragment-based drug design. J. Med. Chem., 51(7):2178–2186.
3. J-W. Huang, Z. Zhang, B. Wu, J. F. Cellitti, X. Zhang, R. Dahl, C-W. Shiau, K. Welsh, A. Emdadi, J. L. Stebbins, J. C. Reed, and M. Pellecchia (2008), Fragment-based design of small molecule x-linked inhibitor of apoptosis protein inhibitors. J. Med. Chem., 51(22):7111–7118.
4. I. D. Kuntz, E. C. Blaney, S. J. Oatley, R. Langridge, and T. E. Ferrin (1982), A Geometric Approach to Macromolecule-Ligand Interactions. J. Mol. Biol., 161:269–288.
5. D. E. Goldberg (1989), Genetic Algorithms in Search, Optimization and Machine Learning. Addison-Wesley, NY.
6. J. H. Holland (1975), Adaptation in Natural and Artificial Systems. The University of Michigan Press, AnnArbor, MI.
7. S. Bandyopadhyay, A. Bagchi, and U. Maulik (2005), Active Site Driven Ligand Design: An Evolutionary Approach. J. Bioinformatics Compu. Biol., 3:1053–1070.
8. G. Goh and J. A. Foster (2000), Evolving molecules for drug design using genetic algorithm via Molecular Tree. newblock Proceedings of the Genetic and Evolutionary Computation Conference (GECCO ‘00), Morgan Kaufmann, Las Vegas, Nevada, USA, pp. 27–33.
9. S. C. Pegg, J. J. Haresco, and I. D. Kuntz ( 2001), A Genetic Algorithm for Structure-based De Novo Design. J. Comput. Aided. Mol. Des., 15:911–933.
10. N. Budin, N. Majeux, C. Tenette Souaille, and A. Caflisch (2001), Structure-based Ligand Design by a Build-up Approach and Genetic Algorithm Search in Conformational Space. J. Comput. Chem., 22:1956–1970.
11. A. Globus, J. Lawton, and T. Wipke (1999), Automatic Molecular Design Using Evolutionary Techniques. Sixth Foresight Conference on Molecular Nanotechnology, 1998, Nanotechnology, 10:290–299.
12. V. Tschinke and N. C. Cohen (2006), The NEWLEAD program: a new method for the design of candidate structures from pharmacophoric hypothesis. J. Med. Chem., 45A:1834–1837.
13. R. Wang, T. Gao, and L. Lai (2000), Ligbuilder: A multi-purpose program for structure-based drug design. J. Mol. Model., 6:498–516.
14. A. R. Leach (2001), Molecular Modelling Principles and Applications. Prentice Hall, NY.
15. J. M. Yang and C. Y. Kao (2000), Flexible Ligand Docking Using a Robust Evolutionary J. Comput. Chem., 21:988–998.
16. M. Srinivas and L. M. Patnaik (1994), Adaptive Probabilities of Crossover and Mutation in Genetic IEEE Transactions on Syatem, Man And Cybernatics, 24:656–667.
17. I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson, and R. Taylor (2002), New software for searching the cambridge structural database and visualizing crystal structures. Acta Crystallogra. Sect. B, 58:389–397.
18. B. Blomback, M. Blomback, B. Hessel, and S. Iwanaga (1967), Structures of N-Terminal Fragments of Fibrinogen and Specificity of Thrombin. Nature (London), 215:1445–1448.
19. D. W. Banner and P. Hadvary (1991), Crystallographic analysis at 3.0-a resolution of the binding to human thrombin of four active site-directed inhibitors. J. Biol. Chem., 266:20085–20093.
20. G. B. Dreyer, D. M. Lambert, T. D. Meek, T. J. Carr, T. A. Tomaszek, Jr. A. V. Fernandez, H. Bartus, E. Cacciavillani, and A. M. Hassell (1992), Hydroxyethylene isostere inhibitors of human immunodeficiency virus-1 protease: structure-activity analysis using enzyme kinetics, x-ray crystallography, and infected T-cell assays. Biochemistry, 31:6646–6659.
21. R. S. Bohacek and C. McMartin (1994), Multiple highly diverse structures complementary to enzyme binding sites: Results of extensive application of a de novo design method incorporating combinatorial growth. J. Am. Chem. Soc., 116:5560–5571.
22. Y. Nishibata and A. Itai (1993), Confirmation of usefulness of a structure construction program based on three-dimensional receptor structure for rational lead generation. J. Med. Chem., 36:2921–2928.
23. W. L. DeLano (2002), The pymol molecular graphics system. Technical report, San Carlos, CA.