![Chapter 5: Ionic [Cu(NN)(PP)]+ TAD9727 F Complexes with Pyridine‐based Diimine Chelating Ligands and Their Use in OLEDs TAD9727 F Complexes with Pyridine‐based Diimine Chelating Ligands and Their Use in OLEDs](https://imgdetail.ebookreading.net/cover/cover/20200215/EB9783527339006.jpg)
5
Ionic [Cu(NN)(PP)]+ TAD9727 F Complexes with Pyridine‐based Diimine Chelating Ligands and Their Use in OLEDs
Rongmin Yu and Can‐Zhong Lu
Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, Fujian, PR China
5.1 Introduction
In 1998, Forrest and coworkers discovered that all the excitons (both singlet and triplet) can be harvested and transferred into light in an electroluminescent device with a Pt phosphorescent complex as the emitting dopant [1]. Since then, phosphorescent materials, especially those of the third‐row transition metals with strong spin–orbit interaction, have been studied extensively, driven majorly by their wide range of applications in electroluminescent devices, such as organic light‐emitting diodes (OLEDs) and light‐emitting electrochemical cells (LEECs) [2–11]. The phosphorescent materials of Ir(III) and Pt(II) complexes are the most attractive because their high emission quantum yields of up to almost 100%, and short emission decay times of 2 µs can be achieved. For these materials, the central metals can induce strong spin–orbit coupling (SOC), resulting in the fast intersystem crossing (ISC) and the effective radiative transition from the first triplet excited state to the ground state. The emission mechanism of these phosphors allows the use of all excitons for the generation of light in OLEDs. However, there still exist materials limitations, for example, low efficient blue‐light emission for d6 and d8 transition metal complexes and material costs of the precious metal sources. Thus, it is desirable to look for alternative materials to solve these problems. Cuprous emissive complexes have been paid great attentions for their cost‐effectiveness, attractive emissions, and the d10 configuration of Cu(I) [12–14].
Cuprous emissive complexes have been investigated intensively since 1970 and become an important class of the luminescent materials [15–18]. Except a few examples of three‐coordinate emissive Cu(I) complexes [19–25], the majority of the Cu(I) luminescent complexes have a distorted tetrahedral geometry around the Cu(I) atoms defined by two diimine ligands ([Cu(NN)2]+) or a diimine ligand and a diphosphine ligand ([Cu(NN)(PP)]+) [25–36]. As compared with the homoleptic [Cu(NN)2]+ complexes, the heteroleptic [Cu(NN)(PP)]+ complexes with phosphine ligands exhibit much higher photoluminescence quantum yields (PLQYs).
In contrast to the heavy‐metal elements, the SOC of the copper element is much less efficient. Consequently, the cuprous phosphorescent complexes usually possess long emission lifetimes of up to hundred microseconds. The long decay times of triplet states facilitate triplet–triplet annihilation (TTA) in electroluminescence devices. However, this detrimental effect can be avoided by the fact that many emissive cuprous complexes can emit efficient thermally activated delayed fluorescence (TADF) at room temperature because of the existence of a small energy gap between the S1 and T1 states [7, 13, 18]. Displaying TADF at ambient temperature, the cuprous complexes have an observed lifetime as short as comparable with those of the heavy‐metal phosphorescent materials. Capable of harvesting both singlet and triplet excitons as well as having compatible lifetimes, the TADF cuprous complexes are believed to have realistic potential application in OLEDs that are currently dominant by the heavy‐metal phosphorescent materials [12, 37].
5.2 The Influence of Molecular and Electronic Structure on Emissive Properties of Cu(I) Complexes
Copper has two common oxidation states: Cu(I) and Cu(II). The Cu(II) ion has a d9 electronic configuration. Its compounds have relatively intense colors due to metal‐centered (MC) absorption bands. The deactivation of the MC excited states is usually through nonradiative pathways, so the luminescence of Cu(II) compounds is very rare and unattractive. In contrast to the Cu(II) complexes, the Cu(I) complexes with a d10 electronic configuration have much more interesting photophysical properties that have caused considerable attention and have been investigated intensively for decades. Many emissive Cu(I) complexes with different types of ligands have been reported, among which, four‐coordinate complexes with homoleptic [Cu(NN)2]+ or heteroleptic [Cu(NN)(PP)]+ basic configuration, are the most extensively investigated. Much effort has been made to elucidate fundamentally the relationship between the emissive performance of the Cu(I) complexes and their structures. It has been found that the emission performance of the Cu(I) complexes is strongly affected by the steric and electronic structures of the ligands, as well as the overall structural rigidity and steric protection of the Cu(I) complex structures.
For the cationic [Cu(NN)2]+ and [Cu(NN)(PP)]+ complexes, their highest occupied molecular orbital (HOMO) contains usually substantial contribution from d orbitals of Cu atom due to the low oxidation potential of Cu(I) ion. As a result, the lowest excited states have the character of MLCT, leading to the broad and unstructured emission spectra that are very common for the emissive Cu(I) complexes. Upon excitation, the lowest MLCT excited state is populated; thus the metal center changes its formal oxidation state from Cu(I) to Cu(II). Because the structural preference for Cu(I) and Cu(II) is different, structural rearrangement undergoes after excitation occurs. The emissive copper complex performs a flattening distortion from the Cu(I)‐preferred tetrahedral structure toward the Cu(II)‐favorite square‐planar structure. The structural distortion leads to a better overlap of the ground state and the excited‐state vibrational wave functions, which causes an increase of the radiationless deactivation and a smaller energy gap between the ground state and excited state. Meanwhile, the resulting smaller energy gap induces a further increase of the radiationless decay rate according to energy gap law. Furthermore, in the excited‐state flattened tetrahedral structure, a fifth coordination site becomes available for the Cu(II) ion. The interaction of the available site with solvent molecules or counterions, forming exciplex, often causes severely nonradiative quenching [38]. Such structural rearrangement is especially severe in a nonrigid environment. Many Cu(I) complexes exhibit poor emissive properties because of the structural rearrangement. Thus, in the design of highly luminescent Cu(I) complexes, it is important to choose appropriate ligands that can construct a rigid and sterically protected environment around the Cu(I) ion that can keep the complex from structural distortion and exciplex quenching upon excitation.
The homoleptic [Cu(NN)2]+ complexes have been investigated continually for decades. The influence of the steric factors on the emissive properties of the Cu(I) complexes has been demonstrated in many studies. The homoleptic [Cu(phen)2]+ complex with pristine phenanthroline ligand does not emit luminescence in CH2Cl2. The substitutions on the 2 and 9 positions with a simple methyl group in [Cu(dmp)2]+ have been shown to improve the emissive properties of the [Cu(NN)2]+ complexes [39]. In the solution of [Cu(dmp)2]+ in degassed CH2Cl2, a luminescence with Φ ≈ 0.1% is observed. The introduction of the methyl group into the pristine phenanthroline ligands gives a more rigid [Cu(NN)2]+ structure that suppresses the degree of the structural distortion upon excitation, so the emissive performance is improved. A better result (Φ = 1%) has been found in the heteroleptic complex, [Cu(dmp)(dtbp)]+ (dtbp = 2,9‐di‐tert‐butyl‐1,10‐phenanthroline), in which the excited‐state distortion is further limited by the bulky tert‐butyl substituent [40]. Even though the PLQYs in these examples are very small, these examples show the importance of the steric factors in affecting the emissive properties of the Cu(I) complexes. Many phenanthroline‐based ligands (as shown in Figure 5.1) with various substituents have been designed and used in the synthesis of the homoleptic [Cu(NN)2]+ complexes [15]. Some of them, such as the sterically encumbered phenanthroline ligands with bulky aryl substituents (3,5‐di‐tert‐butyl‐4‐methoxyphenyl, 2,4,6‐trimethylphenyl) in the 2,9‐position, have been shown to be very effective in protecting the Cu(I) excited states from the exciplex quenching, and the homoleptic [Cu(NN)2]+ complexes with these robust ligands have emissive lifetimes that are almost unaffected by the presence of molecular oxygen and only slightly shortened in the nucleophilic solvents [41]. However, the emission performance of the [Cu(NN)2]+ complexes is generally poor. In CH2Cl2 at ambient temperature, the emission quantum yields of the complexes are typically below 0.001, which limited strongly their application in OLEDs.
![Chemical structures of many phenanthroline-based ligands with various substituents that have been designed and used in the synthesis of the homoleptic [Cu(NN)2]+ complexes.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f001.jpg)
Figure 5.1 Phenanthroline‐based ligands.
In recent years increasing attention has been paid to the heteroleptic [Cu(NN)(PP)]+ complexes because they exhibit greatly increased PLQYs as compared with the homoleptic [Cu(NN)2]+ complexes due to the electronic and steric effect of the P ligands [42–44]. The history of the heteroleptic [Cu(NN)(PP)]+ complexes can be dated back to late 1970s when McMillin and coworkers reported the spectral studies of a series of Cu(I) complexes with mixed ligands of diimine ligands and triphenylphosphine [42, 45]. However, these studies have not caused much attention until 2002 when McMillin and coworkers obtained the first example of the highly emissive heteroleptic [Cu(NN)(PP)]+ complexes, [Cu(dmp)(POP)]+ (dmp = 2,9‐dimethyl‐1,10‐phenanthroline; POP = bis[2‐(diphenylphosphino)phenyl]ether) [46, 47]. The emission quantum yield and lifetime of the complex in deaerated CH2Cl2 are reported to be 0.15 and 14.3 µs, respectively. In this compound, the combination of the diimine ligand with alkyl substituents at 2,9 positions and the chelating diphosphine POP ligand imparts the Cu(I) complexes with an exceptionally sterically protected and rigid structure, which is necessary for a highly emissive Cu(I) complexes. In addition, the electronic effect of the POP ligand stabilizes the HOMO and thus leads to the higher‐energy MLCT excited states, compared with [Cu(NN)2]+ complexes, which disfavors nonradiative deactivations according to the energy law. The structural rigidity and effective steric protection of the complex obstruct efficiently the structural flattening distortion and the exciplex quenching at the excited states, along with the electronic effect of the POP ligand, which leads to the improvement of the emissive efficiencies for the heteroleptic Cu(I) complex. The uses of this complex and its analogs in electroluminescent devices display very promising results [48–51]. Ever since, the research on the emissive Cu(I) complexes has reclaimed tremendous interest, especially in the area of the electroluminescence devices.
A variety of phosphine ligands has been used to prepare Cu(I) complexes together with phenanthroline‐type units: bis[2‐(diphenylphosphino)phenyl]ether (POP), triphenylphosphine (PPh3), bis(diphenylphosphino)ethane (dppe), and bis(diphenylphosphino)methane (dppm) (Figure 5.2) [26, 46, 47, 50, 52–55]. Meanwhile, various diimine ligands other than phenanthroline have been designed and utilized [26, 28–30, 35, 56–59]. Currently, it is not uncommon that cuprous complexes exhibit high PLQYs over 80% in the solid state [30, 57, 60–62]. The application of these materials as the emitting dopants in the fabrication of OLEDs and LEECs has also been studied [35, 47, 51, 63–67]. The importance of the ligand choice for the emissive Cu(I) complexes has been shown in many cases. Properly tuning the steric and electronic properties of the ligands has been shown effective to improve emission efficiency somehow. However, it is not easy to draw a general rule to predict the photophysical behavior of the emissive cuprous complexes. Hard work on trial and errors in the optimization of the emissive properties of the Cu(I) complexes is still unavoidable.
![Chemical structures of phosphine ligands used in the synthesis of heteroleptic [Cu(NN)(PP)]+ complexes: bis[2-(diphenylphosphino)phenyl] ether (POP), triphenylphosphine (PPh3), bis(diphenylphosphino)ethane (dppe), and bis(diphenylphosphino)methane (dppm).](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f002.jpg)
Figure 5.2 Examples of phosphine ligands used in the synthesis of heteroleptic [Cu(NN)(PP)]+ complexes.
Next we will introduce the recent development of the cationic emissive complexes Cu(NN)(PP)+ with pyridyl derivative diimine ligands and their uses in the fabrication of OLEDs.
5.3 Heteroleptic Diimine/Diphosphine [Cu(NN)(PP)]+ Complexes with Pyridine‐Based Ligand
5.3.1 [Cu(NN)(PP)]+ Complexes with 2,2′‐bipyridyl‐based Ligands
Bipyridyl derivative ligands have been used extensively in the preparation of emissive complexes of Ru(II) and Ir(III) [68, 69]. In contrast, the emissive Cu(I) complexes with bipyridyl derivative ligands have been seldom studied, possibly due to the typically lower quantum yields observed for Cu(I) complexes with these ligands in comparison with analogous phenanthroline Cu(I) complexes [42, 45, 70–72]. Yersin and coworkers reported a strongly emissive Cu(I) complex with 4,4′,6,6′‐tetramethyl‐2,2′‐bipyridyl ligand, complex 1, as shown in Figure 5.3 [33]. This complex displays a bright green emission (λmax = 555 nm, τ = 13 µs) at room temperature as powder with high quantum yield (Φ = 74%). In comparison, complex 2 emits much weakly (λmax = 575 nm, Φ = 9%) under the same conditions. The improvement of the emission for complex 1 is ascribed to the steric effect of 6,6′‐methyl groups on the bipyridyl ligand. The 4,4′,6,6′‐tetramethyl‐2,2′‐bipyridyl ligand joining with POP ligand imparts complex 1 a more rigid and sterically protected structure than the 4,4′‐dimethyl‐2,2′‐bipyridyl ligand does in complex 2. A detailed analysis of the temperature‐dependent decay behavior of 1 reveals that this complex emits efficient TADF at room temperature. At T = 77 K, the emission decay time is 87 µs, and the decay time decreases drastically to 11 µs at ambient temperature. The energy separation between the first excited singlet and the first triplet state (ΔEST) estimated from the shift of the emission spectra from 77 to 300 K is 630 cm−1 that is consistent well to the fit value of 770 cm−1 from Eq. 5.1 [33]:
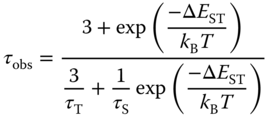
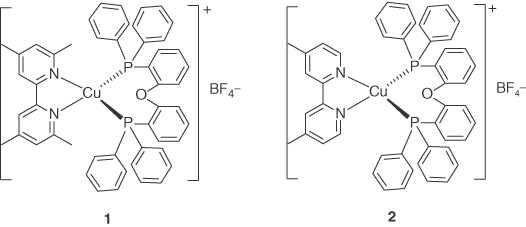
Figure 5.3 Chemical structures of heteroleptic Cu(I) complexes 1 and 2 with 2,2′‐bipyridyl diimine ligands.
5.3.1.1 [Cu(NN)(PP)]+ Complexes with 2‐(2′‐pyridyl)benzimidazole and 2‐(2′‐pyridyl)imidazole‐based Ligands
In 2005, Wang and coworkers reported a series of binuclear or trinuclear Cu(I) emissive complexes with 2‐(2′‐pyridyl)imidazolyl (pbb) derivatives as the bridging ligand and triphenylphosphine as the terminal ligands, as shown in Figure 5.4 [56]. The emission of these complexes in THF or CH2Cl2 solution is very weak. The films of these complexes in PVK or PMMA display yellow‐orange luminescence (λmax = 535–550 nm) when irradiated by UV light at ambient temperature. The absolute emission quantum yield of complexes 3–6 (doped in PMMA, 20 : 80 wt%) was measured to be 11%, 17%, 12%, and 15%, respectively, which are comparable with that of [Cu(Phen)(PPh3)2][BF4] (14%). Complex 6 was used as emitting dopant in the fabrication of OLED:ITO/PEDOT/PVK:20 wt% Cu(I) (60 nm)/F‐TBB (15 nm)/Alq3 (15 nm)/LiF (1 nm)/Al. The turn‐on voltage of the OLED is 9 V. The OLED emits weak yellow‐orange light with the highest current efficiency of 0.2 cd A−1 and a maximum brightness less than 60 cd m−2. The poor performance of the OLED is attributed to the poor quantum yield of complex 6 and its long decay time.
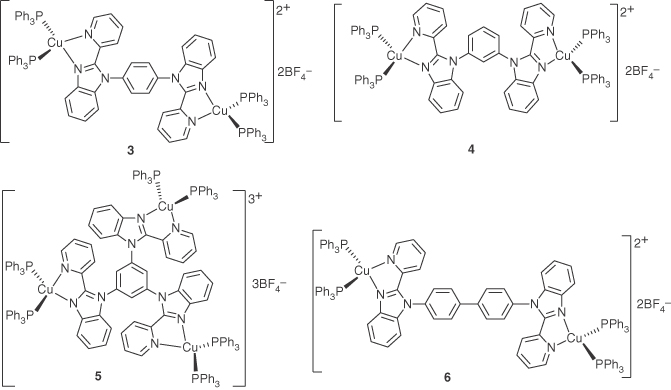
Figure 5.4 Chemical structures of heteroleptic Cu(I) complexes 3–6 with 2‐(2′‐pyridyl)benzimidazolyl diimine ligands.
The impact of the phosphine ligands, PPh3, dppe, POP, and DPPMB, on the electronic properties of the Cu(I) complexes (7–10) containing the 2‐(2′‐pyridyl)benzimidazolylbenzene ligand, has been studied (Figure 5.5) [56]. The optical bandgap between HOMO and lowest unoccupied molecular orbital (LUMO) using the edge of the lowest‐energy absorption band was determined to be 2.80, 2.50, 2.60, and 2.10 eV for complexes 7–10, respectively. Density function calculation for these complexes shows that the HOMO is dominated by the d orbital of the copper(I) ion, together with the significant contributions from the phosphine ligands and a small contribution from the pbb ligand. The LUMO is almost totally composed of the atomic orbitals of the pbb ligand. The molecular orbital (MO) calculation results are consistent that the lowest electronic transition in complexes 7–10 is MLCT/ILCT in nature. The complexes 7–10 are not emissive neither in solution nor as pure solids at ambient temperature, but emit when doped into PMMA polymer with the maximum values at 536∼593 nm and very long decay time (>200 µs). It is surprising that the complex with PPh3 (7) has the best emissive performance among these complexes. In contrast to the phenanthroline‐based Cu(I) complexes, where the ligands increase substantially the emission quantum efficiency as compared with the PPh3 ligand, the POP ligand decreases evidently the emission quantum efficiency in this series. The PLQY for 7 and 9 is 13% and 1.04%, respectively. This study shows again that the phosphine ligands have great influence on the emissive properties of Cu(I) complexes, but it is difficult to draw a general rule to predict the effect of a phosphine ligand beforehand not considering the diimine ligand. Systematic check and optimization of the PP ligand is necessary to find a suitable combination of the ligands for a specified NN ligand and vice versa.
![The general structure of [Cu2(pbb)(P)2]+/0 and the chemical structures of heteroleptic Cu(I) complexes 7-10 with 2-(2′-pyridyl)benzimidazolyl benzene. (PPh3)2 (7); dppe (8); POP (9); DPPMB (10).](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f005.jpg)
Figure 5.5 Chemical structures of heteroleptic Cu(I) complexes 7–10 with 2‐(2′‐pyridyl)benzimidazolylbenzene.
Si et al. modified the 2‐(2′‐pyridyl)benzimidazolyl ligand with hole‐transporting or electron‐transporting group and obtained Cu(I) complexes (11–13) as shown in Figure 5.6 [73]. The complexes are emissive in a CH2Cl2 solution with the maximum value at 532 nm for 11, 552 nm for 12, and 521 nm for 13. OLEDs with the structure of ITO/m‐ MTDATA(30 nm)/NPB(20 nm)/dopant:CBP(30 nm)/Bphen(20 nm)/Alq3(20 nm)/LiF(0.8 nm)/Al(200 nm) were fabricated. The device efficiency turns out to be poor, but the brightness of the devices is impressive. The maximum brightness achieved from the devices based on 1 wt% of complex 12 and 7 wt% of complex 13 reaches 5543 cd m−2 at 16 V and 8669 cd m−2 at 14 V, respectively.

Figure 5.6 Chemical structures of heteroleptic Cu(I) complexes 11–13 with 2‐(2′‐pyridyl)benzimidazolyl diimine ligands.
Adachi and coworkers reported a series of emissive Cu(I) complexes with pyridyl‐ or quinolyl‐imidazole ligands as shown in Figure 5.7 [74]. DFT calculations for these complexes indicate that the electron density in the HOMOs is mainly localized at the Cu(I) ion and Cu–P σ‐bonding orbital, while that in the LUMOs is localized over the diimine ligands. The calculations confirm that the lowest excited states of these complexes contain the contributions of MLCT. Films containing 10 wt% of these complexes in 2,6‐dicarbazolo–1,5‐pyridine (PYD2) emit green (λmax = 530 nm for 14) or yellow (λmax = 549 nm for 15 and λmax = 544 nm for 16) luminescence when irradiated with light of 365 nm, and the PLQYs are 25% for 14, 27% for 15, and 36% for 16, respectively. The decay lifetimes for 15 and 16 are very long, 174.7 µs for 15 and 243.0 µs for 16, respectively. The Stokes‐like shifts for 14, 15, and 16 are 178, 156, and 157 nm, respectively, which indicate that the excited‐state distortion in 15 and 16 is suppressed more effectively. The electroluminescent performance of these complexes was tested in OLEDs with structure ITO/PEDOT:PSS (40 nm)/10 wt% of Cu(I): PYD2 (30 nm)/DPEPO (50 nm)/LiF (0.7 nm)/Al (100 nm). The maximum EQE is 2.0% for 14, 6.1% for 15, and 7.4% for 16, respectively. The enhancement of the PLQY and the electroluminescence efficiency for complex 16 is contributed to the suppression of not only the excited‐state distortion but also C–H vibrational quenching.
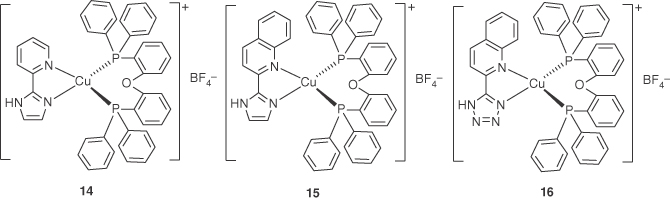
Figure 5.7 Chemical structures of heteroleptic Cu(I) complexes 14–16.
5.3.2 [Cu(NN)(PP)]+ Complexes with 5‐(2‐pyridyl)tetrazole‐based Ligands
The mononuclear neutral and ionic Cu(I) complexes with 5‐(2‐pyridyl)tetrazolate and various phosphine ligands were reported by Bräse and coworkers [60] (Figure 5.8). Their photophysical properties were studied and compared in the solid state (powder). The neutral complexes show strong green luminescence in the region from 502 to 545 nm with very high PLQYs (76–89%) and lifetimes of 17.8–26.6 µs. The cationic complexes exhibit redshifted emission bands from 518 to 569 nm, significantly smaller PLQYs of 4–46%, and shortened lifetimes (5.2–15.3 µs). The less efficient luminescence of the ionic complexes is ascribed to the vibrational quenching of the N–H bond on the tetrazole ring, the negative influence of counterions, and the relatively weaker binding of the neutral diimine ligand. The influence of the PP ligands on the PLQYs is not substantial in the neutral species, but is evident in the ionic species. The PPh3 ligand has the best emission performance when combined with neutral 5‐(2‐pyridyl)tetrazolate rather than the chelating diphosphine ligands. DFT calculations reveal that the localization of the frontier orbitals of the ionic complexes is very different from that of the neutral complexes. The HOMO of ionic complex 17b is mainly located on the Cu(I) atoms and the phosphorous atoms, while the LUMO is located on the neutral diimine ligand. In contrast, in the neutral complex 17a, the HOMO is mainly confined to the Cu(I) atom and the tetrazole moiety together with small contributions from the pyridine N atom, and the LUMO is located primarily on the pyridine ring and to a smaller extent on the tetrazole ring. The phosphines do not contribute to the frontier orbitals, i.e. they should not influence the emission wavelength directly. The calculation results are consistent to the experimental observation that the phosphine ligands have different effect on the photophysical properties of the ionic complexes and the neutral complexes.
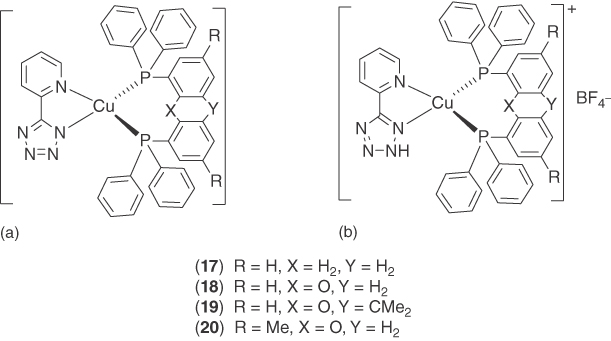
Figure 5.8 Chemical structures of heteroleptic Cu(I) complexes 17–20 with 5‐(2‐pyridyl)tetrazolate ligand.
Three mononuclear or binuclear Cu(I) heteroleptic complexes with 2‐(2‐tert‐butyl‐2H‐tetrazol‐5‐yl)pyridine and various P^P ligands as shown in Figure 5.9 were reported [75]. These complexes are weakly luminescent in CH2Cl2 solution but emit bright green light in a rigid matrix. The emission maxima and PLQYs of the PMMA films containing 25% wt of the complexes are 495 nm and 35% for 20, 515 nm and 7% for 21, and 515 nm and 17% for 22, respectively.
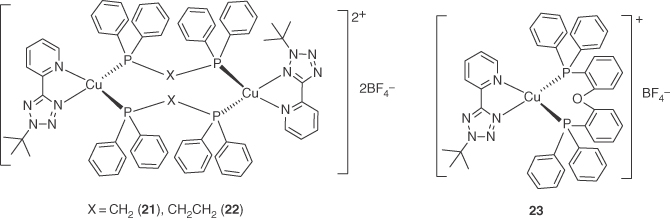
Figure 5.9 Chemical structures of heteroleptic Cu(I) complexes 21–23 with 5‐(2‐pyridyl)tetrazolate derivative ligand.
5.3.3 [Cu(NN)(PP)]+ Complexes with 3‐(2′‐pyridyl)‐1,2,4‐triazole‐based Ligands
A series of heteroleptic Cu(I) complexes based on 3‐(2′‐pyridyl)‐1,2,4‐triazole diimine ligands are shown in Figure 5.10 [76]. Complexes 24–26 are ionic and contain 5‐tert‐butyl‐3‐(2‐pyridyl)‐1H‐1,2,4‐triazole as diimine ligand together with various phosphine ligands. In the solid state at room temperature, complex 25 emits a green luminescence (λmax = 525 nm, τ = 15.9 µs, Φ = 49.1%), and complexes 24 and 26 emit strongly a blue luminescence (λmax = 456 nm, τ = 107 µs, Φ = 41.8% for 24 and λmax = 468 nm, τ = 26.9 µs, Φ = 40.4% for 26, respectively). In degassed CH2Cl2 solution at ambient temperature, complexes 24–26 show a broad single emission band centered at 504, 581, and 525 nm with quantum yields of 9.7%, 1.6,% and 14%, respectively. The quantum yield of 24 is comparable with that of the [Cu(dmp)(POP)]+ [47]. Neutral complex 27 and ionic complex 28 are a pair of Cu(I) complexes based on 5‐trifluoromethyl‐3‐(2‐pyridyl)‐1,2,4‐triazole ligand that can readily be interconverted through protonation or deprotonation. It is interesting that the coordinating pattern of the ligand changes upon protonation or deprotonation, as shown in Figure 5.9. Complex 27 exhibits a slightly higher emission quantum yields than complex 28 both in the solid state and in degassed CH2Cl2 solution. The redshifts of the emission, caused by a change of the environment from powder (crystalline environment) to solution, are 65 and 33 nm for 27 and 28, respectively. This result shows that the coordinating pattern in 28 imparts the Cu(I) complex a more rigid molecular framework than that in 27. Similar to many TADF complexes, a blueshift of the emission occurs as temperature increases for 27 and 28. Surprisingly, a remarkable blueshift of 64 nm (from 536 to 472 nm) as temperature increases from 77 to 298 K was observed for 27 in the solid state, whereas a blueshift of only 4 nm (from 500 to 496 nm) was found for 28 under the same conditions, indicating that the ring inversion and N–H deprotonation of the 1,2,4‐triazolyl have a significant impact on the emissive properties. DFT calculations reveal that the frontier orbitals of the neutral 27 and ionic 28 have very similar orbital composition. In 27 and 28, the HOMO is primarily composed of the contribution from Cu and two PPh3, and the LUMO is essentially localized on the diimine ligand. Unlike in similar neutral Cu(I) complex 17a, the phosphines in 27 contribute largely to the frontier orbitals.
![Chemical structures of neutral and ionic heteroleptic [CuNN)(PP)]+ complexes 24-28 with 3-(2′-pyridyl)-1,2,4-triazole diimine ligands.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f010.jpg)
Figure 5.10 Chemical formula of heteroleptic Cu(I) complexes 24–28 with 3‐(2′‐pyridyl)‐1,2,4‐triazole diimine ligands.
5.3.4 [Cu(NN)(PP)] Complexes with 2‐(2‐pyridyl)‐pyrrolide‐based Ligands
Two neutral [Cu(NN)(PP)] complexes based on 2‐pyridyl pyrrolide‐diimine ligands are shown in Figure 5.11 [76]. In the degassed CH2Cl2 solutions at RT, complexes 29 and 30 show a single emission band maximized at 559 and 551 nm with quantum yields of 34% and 28%, respectively. In solid state at room temperature, 78 and 60 nm blueshifts of the emission as compared with that in the CH2Cl2 solutions are observed for 29 and 30, respectively. For most of the [Cu(NN)(PP)]+ complexes, the PLQY is improved as the rigidity of the matrix increases; however, the PLQY of 30 in the solid state (5%) is much smaller than that in CH2Cl2 solution. More interestingly, the emission spectra of these complexes in the 77 K rigid medium have well‐resolved vibronic progressions. This is very uncommon for the [Cu(NN)(PP)]+ complexes in which the S1 and T1 states have MLCT character. Theoretical calculations reveal that the S1 state for these complexes has a substantial MLCT(>20%) contribution, while the T1 state has ππ* configuration in nature. The calculated ππ* character of the T1 state is consistent to the well‐resolved vibronic progressions of the emission spectra of these complexes in the 77 K rigid medium. Complex 29 was utilized in the fabrication of OLEDs with structure as ITO/TAPC(40 nm)/mCP doped with 8 wt% of 29 (30 nm)/3TPyMB (3 nm)/TmPyPB(37 nm)/LiF (0.8 nm)/Al (150 nm), attaining peak EL efficiencies of 6.6%, 20.0 cd A−1, and 14.9 lm W−1 for the forward directions.

Figure 5.11 Chemical structures of heteroleptic Cu(I) complexes 29 and 30 with 2‐pyridyl pyrrolide‐ diimine ligands.
5.3.5 [Cu(NN)(PP)]+ Complexes with 1‐(2‐pyridyl)‐pyrazole‐based Ligands
Over the past decade, the uses of the pyrazole derivatives as diimine ligands instead of traditional 2,2‐bipyridyl and 2,10‐phenanthroline derivatives in the syntheses of emissive complexes have caused attention [16, 18, 20, 68, 69, 77–84]. In 2008, Qiu and coworkers reported two blue‐emitting cationic iridium complexes with pypz ((pypz = 1‐(2‐pyridyl)pyrazole)) as ancillary diimine ligands and demonstrated that the pypz ligand destabilizes significantly the LUMO orbital of the complexes and, therefore, shows much blueshifted emission with respect to the complex with 2,2‐bipyridyl ligand [85]. With the thought that the pypz ligand is more electron rich than the ligands of phenanthrolines, the pypz ligand and its derivatives have been used to the synthesis of cationic Cu(NN)(POP)+ complexes 31–33, as shown in Figure 5.12 [30]. The energy gaps between the HOMO and LUMO of these complexes 31–33 estimated from the onset wavelengths of the absorption spectra measured in CH2Cl2 are 3.18, 3.23, and 3.04 eV that are indeed higher than the value found for [Cu(dmp)(POP)]+ (2.7 eV). These complexes have been found to be TADF materials and highly emissive with very high PLQYs both in the degassed CH2Cl2 solutions and in the solid states at ambient temperature. High performances of the complexes as emitting dopant have been realized in a series of cost‐effective solution‐processed OLEDs.
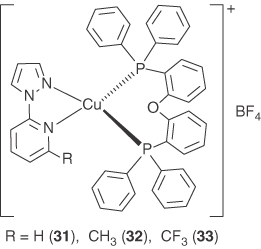
Figure 5.12 Chemical structures of heteroleptic Cu(I) complexes 31–33 with 1‐(2‐pyridyl)‐pyrazole‐diimine ligands.
Complexes of 31–33 in argon‐saturated CH2Cl2 display emissions with the maxima at 590, 536, and 540 nm with quantum yields of 2.1%, 45%, and 30% and decay times of 1.6, 11.9, and 13.3 µs, respectively. As compared with complex [Cu(phen)(POP)](BF4), complex 31 exhibits improved PLQY due to the electron‐donating effect of the pypz ligand [47]. For complex 32, an evident blueshift and a much higher PLQY are observed for its emission. This observation is due to the cooperative effect of the electron‐donating character and the steric demanding effect of the methyl group in the ligand. Complex 33 represents a typical example that shows the importance of steric effect in the Cu(I) emissive complexes. As compared with the pristine pypz ligand in 31, the electron‐withdrawing group of CF3 at the diimine ligand in 3 leads to a redshift effect and a lower efficiency of the emission, whereas the steric demand of the CF3 group leads to a smaller Stokes shift and a higher efficiency of the emission. Obviously, the steric effect of CF3 group dominates its electronic effect as indicated by the blueshift and higher PLQY of complex 33 as compared with complex 31.
In the solid state, complexes 31, 32, and 33 display the emission maxima of 490, 465, and 492 nm with quantum yields of 56%, 87%, and 75% and decay times of 20.4, 12.2, and 22.8 µs, respectively. Improved PLQYs are observed for these complexes in the solid states as compared with those in the CH2Cl2 solutions. With an increase of the matrix rigidity, the freedom for changes of the molecular geometries upon MLCT excitation is decreased, and the geometric distortion from the tetrahedral‐like ground state (d10) to the flattened excited state (d9) is constrained [22, 47, 86]. Consequently, shorter emission maxima, higher quantum yields, and longer emissive lifetime are observed. The largest redshift of the emission, caused by a change of the environment from solid state to the CH2Cl2 solution, is found for complex 31 that has the pristine pypz ligand, while for complexes 32 and 33 with relatively bulkier ligands, the shifts of the emission maxima are significantly smaller. This observation shows that the extent of the steric effect varies in different environments.
With temperature increasing from 77 to 298 K, a blueshift of the emission maximum by 18, 28, and 19 nm is observed for 31–33, respectively. Calculated from the onset energy of the emission spectra at 77 and 298 K, the energy gaps between S1 and T1 (ΔEST), 0.18, 0.17, and .018 eV, are obtained, for 31, 32, and 33, respectively. The observed emissive lifetimes of 33 reduce drastically by about 16 times from 358 µs at 77 K to 22.8 µs at 298 K. The emissive lifetimes at varied temperatures were measured and fit using Eq. 5.1. The fitted τ(T1) value (346 µs) approximates to the value (358 µs) observed at 77 K, and the ΔEST value (0.17 eV) agrees well with the energy differences (0.18 eV) between the onsets of emission spectra at 77 and 298 K. These observations are consistent with the properties of TADF complexes that are often observed in Cu(I) and Ag(I) complexes with small ΔEST [7, 19, 31, 32, 87].
Solution‐processed OLEDs containing these complexes were fabricated with the configuration ITO/PEDOT:PSS (40 nm)/20 wt% of Cu(I): 26mCPy (30 nm)/DPEPO (50 nm)/LiF (0.7 nm)/Al (100 nm). Using 33 as dopant, excellent efficiencies up to 23.68 cd A−1, 8.47%, and a peak brightness of 2033 cd cm−2 were realized. OLEDs based on 31 and 32 exhibited relatively inferior performances although the PL quantum yields of these complexes are comparable. The efficiencies of the OLEDs using 31 as dopant (CE = 8.82 cd A−1, EQE = 3.18%) and using 32 as dopant (CE = 4.07 cd A−1, EQE = 1.59%) are much lower than those of 33‐containing OLED. Better performances in the devices based on these complexes are expected to be obtained via further device optimization.
5.3.6 [Cu(NN)(PP)]+ Complexes with Carbazolyl‐modified 1‐(2‐pyridyl)‐pyrazole‐based Ligands
The carbazole group is a well‐known unit that has been used extensively as the host materials in the OLEDs, organometallic iridium(III) complexes, and pure organic TADF complexes for its high triplet level and good hole‐transporting property [88, 89]. However, highly emissive cuprous complexes with carbazole‐containing ligands have been far less studied. Liu and coworkers reported a codeposition approach in which CuI and a carbazole‐based compound were deposited simultaneously under vacuum to produce an emissive cuprous complex in situ in the resulting emitting layer [90, 91]. Highly efficient vacuum‐deposited OLEDs were fabricated, and the carbazole‐based compound served as both ligand and host matrix in the devices. Recently, the photophysical properties of two strongly luminescent Cu(I) complexes containing carbazole modifying 1‐(2‐pyridyl)‐pyrazole (czpypz)‐based diimine ligand as shown in Figure 5.13 have been synthesized and used in OLED fabrication [29].
![Chemical structures of heteroleptic [Cu(NN)(PP)]+ complexes 34 and 35 with carbazolyl-modified 1-(2-pyridyl)-pyrazole-based diimine ligands.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f013.jpg)
Figure 5.13 Chemical structures of heteroleptic Cu(I) complexes 34 and 35 with 1‐(2‐pyridyl)‐pyrazole‐based diimine ligands.
Complexes 34 and 35 are weakly luminescent in degassed CH2Cl2 at room temperature with λmax = 540 nm for 34 and λmax = 569 nm for 35, respectively. However, the complexes emit efficiently in the solid state at room temperature with quantum yields of 45% and 98%, emission maxima at 495 and 518 nm, and decay times of 134 and 23 µs, for 34 and 35, respectively. From the comparison between the emissive properties of complex 31 and 35, a conclusion that can be drawn is that the introduction of the carbazolyl substituent into pypz ligand causes a redshift of the emission maximum and an enhancement of the emission efficiency in the solid state. Complexes 34 and 35 display TADF. With temperature increasing from 77 to 298 K, a blueshift of the emission maximum and a decreased lifetime are observed. The energy gaps between the S1 and T1 (ΔEST) estimated from the onsets of emission spectra at 77 and 298 K are 0.15 eV for 34 and 0.12 eV for 35, respectively.
Interestingly, in addition to acting as chelating diimine ligand in the formation of the complexes 34 and 35, the czpypz can also be used as an excellent host material in the fabrication of OLEDs for the Cu(I) complexes. High efficient OLED was fabricated conveniently by preparing the emitting layer from directly spin‐coating a mixture of czpypz and the Cu(I) precursor, [Cu(CH3CN)2(POP)]BF4, in which the emissive dopant forms in situ and the excess czpypz acts as the host material. The device configuration is ITO/PEDOT:PSS (40 nm)/2 (20 wt%):czpzpy (30 nm)/DPEPO (10 nm)/TPBI (50 nm)/LiF (0.8 nm)/Al (100 nm). A comparison of the device performances with the emitting dopant prepared in advance and formed in situ shows essentially identical device properties. The maximum brightness, the peak efficiency, and the external quantum yield of the device with the czpypz ligand as the host and the Cu(I) complex as the dopant are 2939 cd m−2, 7.34 cd A−1 (CE), and 6.34% (EQE), respectively. The device with the formation of the Cu(I) complex in situ displays a maximum brightness of 3251 cd m−2, a peak efficiency of 17.53 cd A−1 (CE), and 6.36% (EQE).
5.3.7 [Cu(NN)(PP)]+ Complexes with 1‐phenyl‐3‐(2‐pyridyl)pyrazole‐based Ligands
Four cationic Cu(NN)(POP)]+ complexes 36–39 with various 1‐phenyl‐3‐(2‐pyridyl)pyrazole‐diimine ligands are shown in Figure 5.14 [26]. The energy gaps (ΔEg) between HOMO and LUMO for 36 (3.04 eV), 37 (3.08 eV), 38 (3.04 eV), and 39 (3.00 eV) estimated from the onset wavelengths of the absorption spectra in CH2Cl2 are very close, which indicates that the substituent groups on the phenyl ring have little influence on the electronic properties of these complexes. These complexes are highly emissive in the degassed CH2Cl2 solution, in the films of PMMA, and in the solid states. Blueshifted emission maximum, increased lifetime, and improved PLQYs are observed for these complexes as the matrix rigidity of the environment increases, which is very common for Cu(I) complexes because of the structural rearrangement upon excitation. In the CH2Cl2 solutions, these complexes emit bright yellow‐orange luminescence with maximum at 556–580 nm and lifetime in the range of 4.2–5.7 µs. In the PMMA films of 10% wt concentration, the complexes emit green light with maximum at 506–515 nm and PLQYs of 46–54%. In the solid states, the complexes are highly emissive with PLQYs up to 91% for complex 37. The redshift of the emission for these complexes is similar (62–86 nm), caused by a change of the environment from solid state to CH2Cl2 solution. From the onsets of the emission spectra of these complexes in PMMA at 77 and 298 K, small energy gaps ΔEST (0.16–0.24 eV) between S1 and T1 were obtained. The small ΔEST together with the dramatic decrease of the decay lifetime observed for complex 36 as the temperature increases indicates that the complexes are TADF materials. OLEDs with configuration as ITO/PEDOT:PSS(40 nm)/10 wt% of Cu(I): host(30 nm)/DPEPO(50 nm)/LiF(0.7 nm)/Al(100 nm) were made. The device with 36 as emitting dopant gives the best device performance with the peak current efficiency of 17.8 cd A−1 and EQE of 6.4%.
![Chemical structures of four cationic heteroleptic [Cu(NN)(POP)]+ complexes 36-39 with 1-phenyl-3-(2-pyridyl)pyrazole diimine ligands.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f014.jpg)
Figure 5.14 Chemical structures of heteroleptic Cu(I) complexes 36–39 with 1‐phenyl‐3‐(2‐pyridyl)pyrazole‐diimine ligands.
Figure 5.15 shows the chemical configuration of a series of ionic [Cu(NN)(POP)]+ complexes (40–45) with 1‐phenyl‐3‐(6‐methyl‐2‐pyridyl)pyrazole‐diimine ligands [92]. These complexes are highly emissive both in the CH2Cl2 solution and in the rigid matrices with the emission maximum in the blue‐ to bluish‐green region. It is remarkable that the PLQYs of these complexes are very high and up to 99% in the solid states. The PLQYs of the PMMA films of 10 wt% concentration of these complexes are also very impressive and up to 59% that is very high for the reported Cu(I) complexes. The blueshifted emission maximum and the improved PLQYs are also observed for these complexes as the matrix rigidity of the environment increases, but it is surprising that the lifetimes of these complexes in PMMA films are longer than those in the solid states. The redshifts of the emissions for these complexes caused by a change of the environment from solid powder to CH2Cl2 solutions are much smaller as compared with those for complexes 36–39. This result indicates that the diimine ligands in 40–44 combined with POP can suppress better the structural rearrangement and exciplex quenching of these Cu(I) complexes upon excitation than the ligands in 36–39. The smallest redshift is 16 nm observed for complex 42. For comparison, the redshift observed in 36 is 83 nm. The studies of varied temperature emission spectra and decay behaviors of these complexes indicate that these complexes are TADF complexes with small ΔEST (0.08–0.20 eV).
![Chemical structures of heteroleptic Cu(NN)(PP)]+ complexes 40-44 with 1-phenyl-3-(6-methyl-2-pyridyl)pyrazole diimine ligands.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f015.jpg)
Figure 5.15 Chemical structures of heteroleptic Cu(I) complexes 40–44 with 1‐phenyl‐3‐(6‐methyl‐2‐pyridyl)pyrazole‐diimine ligands.
5.3.8 [Cu(NN)(PP)]+ Complexes with 3‐phenyl‐5‐(2‐pyridyl)‐1H‐1,2,4‐triazole‐based Ligands
Four [Cu(NN)(POP)]+ Cu(I) complexes containing 3‐phenyl‐5‐(2‐pyridyl)‐1H‐1,2,4‐triazole derivatives as diimine ligands, 45–48, are shown in Figure 5.16 [27]. The energy gaps (ΔEg) between HOMO and LUMO estimated from the onset wavelengths of the absorption spectral of these complexes are 2.7 eV for 45 and 46 and 2.85 eV for 47 and 48. The redshifts of these caused by the change of matrix from PMMA films to degassed CH2Cl2 are 69, 64, 51, and 52 nm, for 45–48, respectively. These results demonstrate that the methyl group at the pyridine ring increases the energy gap of the frontier orbitals and the structural rigidity of the resulting Cu(I) complexes and thus improves the photoluminescence of the complexes. As compared, the carbazolyl group does not have obvious influence on the energy gap and the structural rigidity of the complexes and thus displays very limited influence on the photoluminescence of the complexes. The OLEDs with the configuration ITO/PEDOT:PSS (40 nm)/20 wt% of 47 or 48:PYD2 (2,6‐bis(N‐carbazolyl)pyridine, 30 nm)/DPEPO (bis[2‐(di‐(phenyl)phosphino)‐phenyl]ether oxide, 50 nm)/LiF (0.7 nm)/Al (100 nm) (where ITO denotes indium tin oxide, PEDOT:PSS is poly (3, 4‐ethylenedioxythiophene)–poly(styrenesulfonic acid) were fabricated. The device with 47 displays a maximum brightness of 2096 cd m−2, a peak efficiency of 14.2 cd A−1 (CE), and 5.9% (EQE). The device with 48 displays a maximum brightness of 2447 cd m−2, a peak efficiency of 16.7 cd A−1 (CE), and 6.7% (EQE). These results suggest that the dopant with carbazolyl group gives a better utilization of the excitons in the OLED.
![Chemical structures of heteroleptic [Cu(NN)(PP)]+ complexes 45-48 with 3-phenyl-5-(2-pyridyl)-1H-1,2,4-triazole-based diimine ligands.](http://images-20200215.ebookreading.net/4/1/1/9783527339006/9783527339006__highly-efficient-oleds__9783527339006__images__c05f016.jpg)
Figure 5.16 Chemical structures of heteroleptic Cu(I) complexes 45–48 with 3‐phenyl‐5‐(2‐pyridyl)‐1H‐1,2,4‐triazole‐based diimine ligands.
5.4 Conclusion and Perspective
In this chapter, the recent progress of the emissive [Cu(NN)(PP)]+ complexes with pyridine‐based diimine ligands has been briefly summarized. The rationalization of the relationship between the structure and the photophysical property of the [Cu(NN)(PP)]+ complex has made much progress over the last decade, and it has been shown that the electronic and steric effect of the ligands have substantial influence on the photophysical property of the [Cu(NN)(PP)]+ complexes. The key principle in designing the Cu(I) complexes is to construct an overall structure with rigid and steric conformation that can prevent the structure from distortion rearrangement and exciplex quenching upon excitation through judicious choice and adjustment of the ligands, including the ligand topology, bulk, and rigidity. Under the guide of designing principle, many [Cu(NN)(PP)]+ complexes based on pyridyl diimine ligands with interesting properties have been designed and optimized. Clearly, the present progress of the research on this field attests the ability of pyridyl diimine ligands to obtain highly emissive Cu(I) TADF materials, especially those highly blue‐emitting complexes that are problematic for Ir, Pt, and Os complexes because of the existence of dd* orbitals. Designing new diimine and diphosphine ligands, development of new TADF Cu(I) materials with high PLQY and short decay lifetime, and full assessment of their device stability and light output in OLEDs will be still the main tasks in this area in the near future [93].
References
- 1. Baldo, M.A., O'Brien, D.F., You, Y., Shoustikov, A., Sibley, S., Thompson, M.E., and Forrest, S.R. (1998). Nature 395: 151–154.
- 2. Song, G.F.W.M.X., Wang, J., Wang, Y.H., Bai, F.Q., and Qin, Z.K. (2015). Spectrochim. Acta. Part A 134: 406–412.
- 3. Moral, L.M.M., Son, W.J., Olivier, Y., and Sancho‐García, J.C. (2015). J. Chem. Theory Comput. 11: 168–177.
- 4. Kessler, F., Costa, R.D., Di Censo, D., Scopelliti, R., Orti, E., Bolink, H.J., Meier, S., Sarfert, W., Gratzel, M., Nazeeruddin, M.K., and Baranoff, E. (2012). Dalton Trans. 41: 180–191.
- 5. Du, B.‐S., Liao, J.‐L., Huang, M.‐H., Lin, C.‐H., Lin, H.‐W., Chi, Y., Pan, H.‐A., Fan, G.‐L., Wong, K.‐T., Lee, G.‐H., and Chou, P.‐T. (2012). Adv. Funct. Mater. 22: 3491–3499.
- 6. Lin, C.‐H., Hsu, C.‐W., Liao, J.‐L., Cheng, Y.‐M., Chi, Y., Lin, T.‐Y., Chung, M.‐W., Chou, P.‐T., Lee, G.‐H., Chang, C.‐H., Shih, C.‐Y., and Ho, C.‐L. (2012). J. Mater. Chem. 22: 10684.
- 7. Yersin, H., Rausch, A.F., Czerwieniec, R., Hofbeck, T., and Fischer, T. (2011). Coord. Chem. Rev. 255: 2622–2652.
- 8. Baranoff, E., Fantacci, S., De Angelis, F., Zhang, X., Scopelliti, R., Gratzel, M., and Nazeeruddin, M.K. (2011). Inorg. Chem. 50: 451–462.
- 9. Murawski, C., Liehm, P., Leo, K., and Gather, M.C. (2014). Adv. Funct. Mater. 24: 1117–1124.
- 10. Tang, D.P.T.M., Wong, Y., Chan, M., Wong, K., and Yam, V.W. (2014). J. Am. Chem. Soc. 136 (51): 17861–17868.
- 11. Yersin, H. and Finkenzeller, W.J. (2008). Highly Efficient OLEDs with Phosphorescent Materials, 1–97. Wiley‐VCH.
- 12. Volz, D., Wallesch, M., Flechon, C., Danz, M., Verma, A., Navarro, J.M., Zink, D.M., Brase, S., and Baumann, T. (2015). Green Chem. 17: 1988–2011.
- 13. Dumur, F. (2015). Org. Electron. 21: 27–39.
- 14. Czerwieniec, R., Leitl, M.J., Homeier, H.H.H., and Yersin, H. (2016). Coord. Chem. Rev. 325: 2–28.
- 15. Laviecambot, A., Cantuel, M., Leydet, Y., Jonusauskas, G., Bassani, D., and McClenaghan, N. (2008). Coord. Chem. Rev. 252: 2572–2584.
- 16. Xiang, H., Cheng, J., Ma, X., Zhou, X., and Chruma, J.J. (2013). Chem. Soc. Rev. 42: 6128–6185.
- 17. McMillin, D.R. and McNett, K.M. (1998). Chem. Rev. 98: 1201–1219.
- 18. Tao, Y., Yuan, K., Chen, T., Xu, P., Li, H., Chen, R., Zheng, C., Zhang, L., and Huang, W. (2014). Adv. Mater. 26: 7931–7958.
- 19. Osawa, M., Kawata, I., Ishii, R., Igawa, S., Hashimoto, M., and Hoshino, M. (2013). J. Mater. Chem. C 1: 4375.
- 20. Krylova, V.A., Djurovich, P.I., Whited, M.T., and Thompson, M.E. (2010). Chem. Commun. (Camb.) 46: 6696–6698.
- 21. Lotito, K.J. and Peters, J.C. (2010). Chem. Commun. (Camb.) 46: 3690–3692.
- 22. Hashimoto, M., Igawa, S., Yashima, M., Kawata, I., Hoshino, M., and Osawa, M. (2011). J. Am. Chem. Soc. 133: 10348–10351.
- 23. Krylova, V.A., Djurovich, P.I., Conley, B.L., Haiges, R., Whited, M.T., Williams, T.J., and Thompson, M.E. (2014). Chem. Commun. (Camb.) 50: 7176–7179.
- 24. Marion, R., Sguerra, F., Di Meo, F., Sauvageot, E., Lohier, J.F., Daniellou, R., Renaud, J.L., Linares, M., Hamel, M., and Gaillard, S. (2014). Inorg. Chem. 53: 9181–9191.
- 25. Leitl, M.J., Krylova, V.A., Djurovich, P.I., Thompson, M.E., and Yersin, H. (2014). J. Am. Chem. Soc. 136: 16032–16038.
- 26. Zhang, Q., Chen, X.‐L., Chen, J., Wu, X.‐Y., Yu, R., and Lu, C.‐Z. (2015). RSC Adv. 5: 34424–34431.
- 27. Zhang, Q., Chen, X.L., Chen, J., Wu, X.Y., Yu, R., and Lu, C.Z. (2015). Dalton Trans. 44: 10022–10029.
- 28. Qin, L., Zhang, Q., Sun, W., Wang, J., Lu, C., Cheng, Y., and Wang, L. (2009). Dalton Trans. 9388–9391.
- 29. Chen, X.‐L., Lin, C.‐S., Wu, X.‐Y., Yu, R., Teng, T., Zhang, Q.‐K., Zhang, Q., Yang, W.‐B., and Lu, C.‐Z. (2015). J. Mater. Chem. C 3: 1187–1195.
- 30. Chen, X.L., Yu, R.M., Zhang, Q.K., Zhou, L.J., Wu, C.Y., Zhang, Q., and Lu, C.Z. (2013). Chem. Mater. 25: 3910–3920.
- 31. Hofbeck, T., Monkowius, U., and Yersin, H. (2015). J. Am. Chem. Soc. 137: 399–404.
- 32. Czerwieniec, R. and Yersin, H. (2015). Inorg. Chem. 54: 4322–4327.
- 33. Linfoot, C.L., Leitl, M.J., Richardson, P., Rausch, A.F., Chepelin, O., White, F.J., Yersin, H., and Robertson, N. (2014). Inorg. Chem. 53: 10854–10861.
- 34. Yersin, H. Czerwieniec, R. and Hupfer, A. (2012). Proceedings of SPIE, Volume 8435, SPIE Photonics Europe, 16–19 April 2012, Brussels, Belgium.
- 35. Czerwieniec, R., Yu, J.B., and Yersin, H. (2011). Inorg. Chem. 50: 8293–8301.
- 36. Kang, L., Chen, J., Teng, T., Chen, X.L., Yu, R., and Lu, C.Z. (2015). Dalton Trans. 44: 11649–11659.
- 37. Volz, D., Chen, Y., Wallesch, M., Liu, R., Fléchon, C., Zink, D.M., Friedrichs, J., Flügge, H., Steininger, R., Göttlicher, J., Heske, C., Weinhardt, L., Bräse, S., So, F., and Baumann, T. (2015). Adv. Mater. 27: 2538–2543.
- 38. Chen, L.X., Jennings, G., Liu, T., Gosztola, D.J., Hessler, J.P., Scaltrito, D.V., and Meyer, G.J. (2002). J. Am. Chem. Soc. 124: 10861–10867.
- 39. Fasina, T.M., Collings, J.C., Burke, J.M., Batsanov, A.S., Ward, R.M., Albesa‐Jové, D., Porrès, L., Beeby, A., Howard, J.A.K., Scott, A.J., Clegg, W., Watt, S.W., Viney, C., and Marder, T.B. (2005). J. Mater. Chem. 15: 690–697.
- 40. Feng, D., Gu, Z.Y., Chen, Y.P., Park, J., Wei, Z., Sun, Y., Bosch, M., Yuan, S., and Zhou, H.C. (2014). J. Am. Chem. Soc. 136: 17714–17717.
- 41. Kalsani, V., Schmittel, M., Listorti, A., Accorsi, G., and Armaroli, N. (2006). Inorg. Chem. 45: 2061–2067.
- 42. Rader, R.A., McMillin, D.R., Buckner, M.T., Matthews, T.G., Casadonte, D.J., Lengel, R.K., Whittaker, S.B., Darmon, L.M., and Lytle, F.E. (1981). J. Am. Chem. Soc. 103: 5906–5912.
- 43. Breddels, P.A., Berdowski, P.A.M., Blasse, G., and McMillin, D.R. (1982). J. Chem. Soc., Faraday Trans. 2 78: 595–601.
- 44. Sheldrick, G.M. (2014). Institute for Inorganic Chemistry. Göttingen, Germany: University of Göttingen.
- 45. Dolbier, W.R., Xie, P., Zhang, L., Xu, W., Chang, Y., and Abboud, K.A. (2008). J. Org. Chem. 73: 2469–2472.
- 46. Kuang, S.M., Cuttell, D.G., McMillin, D.R., Fanwick, P.E., and Walton, R.A. (2002). Inorg. Chem. 41: 3313–3322.
- 47. Cuttell, D.G., Kuang, S.M., Fanwick, P.E., McMillin, D.R., and Walton, R.A. (2002). J. Am. Chem. Soc. 124: 6–7.
- 48. Zhang, Q., Zhou, Q., Cheng, Y., Wang, L., Ma, D., Jing, X., and Wang, F. (2006). Adv. Funct. Mater. 16: 1203–1208.
- 49. Zhang, Q., Li, J., Shizu, K., Huang, S., Hirata, S., Miyazaki, H., and Adachi, C. (2012). J. Am. Chem. Soc. 134: 14706–14709.
- 50. Armaroli, N., Accorsi, G., Holler, M., Moudam, O., Nierengarten, J.F., Zhou, Z., Wegh, R.T., and Welter, R. (2006). Adv. Mater. 18: 1313–1316.
- 51. Zhang, Q., Zhou, Q., Cheng, Y., Wang, L., Ma, D., Jing, X., and Wang, F. (2004). Adv. Mater. 16: 432–436.
- 52. Tsukuda, T., Nakamura, A., Arai, T., and Tsubomura, T. (2006). B. Chem. Soc. Jpn. 79: 288–290.
- 53. Saito, K., Arai, T., Takahashi, N., Tsukuda, T., and Tsubomura, T. (2006). Dalton Trans. 4444–4448.
- 54. Tsubomura, T., Takahashi, N., Saito, K., and Tsukuda, T. (2004). Chem. Lett. 33: 678–679.
- 55. Zhang, Q., Ding, J., Cheng, Y., Wang, L., Xie, Z., Jing, X., and Wang, F. (2007). Adv. Funct. Mater. 17: 2983–2990.
- 56. McCormick, T., Jia, W.L., and Wang, S.N. (2006). Inorg. Chem. 45: 147–155.
- 57. Chen, J.L., Cao, X.F., Wang, J.Y., He, L.H., Liu, Z.Y., Wen, H.R., and Chen, Z.N. (2013). Inorg. Chem. 52: 9727–9740.
- 58. Min, J., Zhang, Q., Sun, W., Cheng, Y., and Wang, L. (2011). Dalton Trans. 40: 686–693.
- 59. Sun, W., Zhang, Q., Qin, L., Cheng, Y., Xie, Z., Lu, C., and Wang, L. (2010). Eur. J. Inorg. Chem. 2010: 4009–4017.
- 60. Bergmann, L., Friedrichs, J., Mydlak, M., Baumann, T., Nieger, M., and Brase, S. (2013). Chem. Commun. (Camb.) 49: 6501–6503.
- 61. Volz, D., Zink, D.M., Bocksrocker, T., Friedrichs, J., Nieger, M., Baumann, T., Lemmer, U., and Bräse, S. (2013). Chem. Mater. 25: 3414–3426.
- 62. Czerwieniec, R., Kowalski, K., and Yersin, H. (2013). Dalton Trans. 42: 9826–9830.
- 63. Jia, W.L., McCormick, T., Tao, Y., Lu, J.‐P., and Wang, S. (2005). Inorg. Chem. 44: 5706–5712.
- 64. Tsuboyama, A., Kuge, K., Furugori, M., Okada, S., Hoshino, M., and Ueno, K. (2007). Inorg. Chem. 46: 1992–2001.
- 65. Zink, D.M., Bächle, M., Baumann, T., Nieger, M., Kühn, M., Wang, C., Klopper, W., Monkowius, U., Hofbeck, T., Yersin, H., and Bräse, S. (2012). Inorg. Chem. 52: 2292–2305.
- 66. Harkins, S.B. and Peters, J.C. (2005). J. Am. Chem. Soc. 127: 2030–2031.
- 67. Xin, X.L., Chen, M., Ai, Y.B., Yang, F.L., Li, X.L., and Li, F. (2014). Inorg. Chem. 53: 2922–2931.
- 68. Costa, R.D., Orti, E., Bolink, H.J., Monti, F., Accorsi, G., and Armaroli, N. (2012). Angew. Chem. Int. Ed. 51: 8178–8211.
- 69. Zhang, Z., Zhao, W., Ma, B., and Ding, Y. (2010). Catal. Commun. 12: 318–322.
- 70. Linfoot, C.L., Richardson, P., Hewat, T.E., Moudam, O., Forde, M.M., Collins, A., White, F., and Robertson, N. (2010). Dalton Trans. 39: 8945–8956.
- 71. Costa, R.D., Tordera, D., Ortí, E., Bolink, H.J., Schönle, J., Graber, S., Housecroft, C.E., Constable, E.C., and Zampese, J.A. (2011). J. Mater. Chem. 21: 16108.
- 72. Li, X.‐L., Ai, Y.‐B., Yang, B., Chen, J., Tan, M., Xin, X.‐L., and Shi, Y.‐H. (2012). Polyhedron 35: 47–54.
- 73. Si, Z., Li, J., Li, B., Liu, S., and Li, W. (2009). J. Lumin. 129: 181–186.
- 74. Wada, A., Zhang, Q., Yasuda, T., Takasu, I., Enomoto, S., and Adachi, C. (2012). Chem. Commun. (Camb.) 48: 5340–5342.
- 75. Femoni, C., Muzzioli, S., Palazzi, A., Stagni, S., Zacchini, S., Monti, F., Accorsi, G., Bolognesi, M., Armaroli, N., Massi, M., Valenti, G., and Marcaccio, M. (2013). Dalton Trans. 42: 997–1010.
- 76. Hsu, C.W., Lin, C.C., Chung, M.W., Chi, Y., Lee, G.H., Chou, P.T., Chang, C.H., and Chen, P.Y. (2011). J. Am. Chem. Soc. 133: 12085–12099.
- 77. Tung, Y.L., Chen, L.S., Chi, Y., Chou, P.T., Cheng, Y.M., Li, E.Y., Lee, G.H., Shu, C.F., Wu, F.I., and Carty, A.J. (2006). Adv. Funct. Mater. 16: 1615–1626.
- 78. You, Y. and Park, S.Y. (2009). Dalton Trans. 1267–1282.
- 79. Yang, C.H., Cheng, Y.M., Chi, Y., Hsu, C.J., Fang, F.C., Wong, K.T., Chou, P.T., Chang, C.H., Tsai, M.H., and Wu, C.C. (2007). Angew. Chemie Int. Ed. 46: 2418–2421.
- 80. Kalinowski, J., Fattori, V., Cocchi, M., and Williams, J.A.G. (2011). Coord. Chem. Rev. 255: 2401–2425.
- 81. Shavaleev, N.M., Monti, F., Scopelliti, R., Baschieri, A., Sambri, L., Armaroli, N., Grätzel, M., and Nazeeruddin, M.K. (2013). Organometallics 32: 460–467.
- 82. Chi, Y. and Chou, P.T. (2010). Chem. Soc. Rev. 39: 638–655.
- 83. Fan, C. and Yang, C. (2014). Chem. Soc. Rev. 43: 6439–6469.
- 84. Xu, H., Chen, R., Sun, Q., Lai, W., Su, Q., Huang, W., and Liu, X. (2014). Chem. Soc. Rev. 43: 3259–3302.
- 85. He, L., Duan, L., Qiao, J., Wang, R., Wei, P., Wang, L., and Qiu, Y. (2008). Adv. Funct. Mater. 18: 2123–2131.
- 86. Armaroli, N. (2001). Chem. Soc. Rev. 30: 113–124.
- 87. Gneuss, T., Leitl, M.J., Finger, L.H., Rau, N., Yersin, H., and Sundermeyer, J. (2015). Dalton Trans. 44: 8506–8520.
- 88. Wong, W.Y. and Ho, C.L. (2009). Coord. Chem. Rev. 253: 1709–1758.
- 89. Yook, K.S. and Lee, J.Y. (2012). Adv. Mater. 24: 3169–3190.
- 90. Liu, Z.W., Qayyum, M.F., Wu, C., Whited, M.T., Djurovich, P.I., Hodgson, K.O., Hedman, B., Solomon, E.I., and Thompson, M.E. (2011). J. Am. Chem. Soc. 133: 3700–3703.
- 91. Liu, Z., Qiu, J., Wei, F., Wang, J., Liu, X., Helander, M.G., Rodney, S., Wang, Z., Bian, Z., Lu, Z., Thompson, M.E., and Huang, C. (2014). Chem. Mater. 26: 2368–2373.
- 92. Zhang, Q., Chen, J., Wu, X.‐Y., Chen, X.‐L., Yu, R., and Lu, C.‐Z. (2015). Dalton Trans. 44: 6706–6710.
- 93. Leitl, M.J., Zink, D.M., Schinabeck, A., Baumann, T., Volz, D., and Yersin, H. (2016). Top. Curr. Chem. 374 (3): 1–34.