
CHAPTER 6
Influence of Functional Groups in Substituted Aromatic Molecules on the Selection of Reaction Channel in Semiconductor Surface Functionalization
6.1 INTRODUCTION
6.1.1 Scope of this Chapter
Aromaticity has long been a fascinating topic in the entire field of chemistry [1]. Since the discovery of benzene that in a few years will celebrate its bicentennial anniversary, scientists and engineers have put this concept to a good use. As the measurements and applications have evolved though, the definition of aromaticity has also undergone dramatic changes. The original formulations of Kekulé (1865) and Erlenmeyer (1866) have become much more general and are based mostly on experimental and computational observations of the exceptional stability of aromatic compounds. The unusually high degree of symmetry of their molecular and often crystal structures and, as a consequence, often a highly symmetric wave function describing their quantum mechanical behavior lead to unusual magnetic and spectroscopic characteristics of aromatic compounds. It should be pointed out that the initially accepted description of aromaticity as a characteristic of compounds whose molecules are built upon flat ring-like structures with 4n+2 π electrons has long been abandoned as the aromaticity in some species has been demonstrated to involve σ orbitals, or only 4n electrons instead of 4n+2, or even be observed in transition metal sandwich structures and in three-dimensional clusters.
Compared to such a long and storied history of aromaticity, chemistry of semiconductor surface modification is still in its infancy, despite a myriad of practical applications. This chapter will take a full advantage of the use of aromatic compounds for surface modification but we will mostly stick to the nearly two centuries old definitions of aromaticity to avoid the additional complexity. Most of what we will examine are the aromatic compounds that really are based on stable planar ring-like structures with reasonably well-characterized electronic properties. That said, we will also consider examples of heterocyclic compounds that are not aromatic within the classical definition but that may lead to the formation of aromatic structures in a reaction with the surface or that may help us understand the differences between reactivity of “aromatic” and “nonaromatic” compounds. One should hope that the amazing newly developed aromatic structures with extraordinary geometric, electronic, and magnetic properties will also at some point soon be used in realistic applications that are based on semiconductor substrates.
There are several main reasons why aromatic compounds are becoming such popular surface modifiers. With recent development of such fields as microelectronics, communication, and sensing, the interface between organic layers and semiconductor materials has truly become the focus of molecular-level research. The miniaturization of electronic components all the way down to subnanometer scale drives both fundamental research and engineering solutions to build such interfaces at a truly molecular level. Since silicon still remains a cornerstone of modern technology, it will be the main focus of this chapter. It is still one of the cheapest, most abundant, easily prepared semiconductor materials with electronic properties easily tuned by well-developed doping methods. The problem with this material is that it is very reactive, regardless of whether the starting point is a clean surface or it is modified with some functionality. This is where aromatic compounds can become especially valuable. On one hand, the stable aromatic groups possess sufficient reactivity to chemically interact with a very reactive clean silicon surface. On the other hand, they can act as relatively inert spacers if any other functional group is available on an aromatic compound. If the starting surface is functionalized, for example hydrogen terminated, the aromatic spacers prove to be exceptionally reliable inert geometric linkers between the desired functional groups: one needed to react with the surface and the other available for further modification.
With the intriguing possibilities of using molecules as potential components of molecular electronics, where conduction and switching is performed by means of a single molecule or small molecular agglomerates, the use of aromatic compounds on semiconductor surfaces received an additional boost, once the possibility of lateral conduction along the π-stacked aromatic rings was considered.
Although structurally diverse semiconductor surfaces are currently available, the very idea of creating a defect-free interface relies heavily on the ideally ordered starting surface. The easily accessible single crystalline elemental semiconductor surfaces make them an even more attractive playing field. Both fundamental studies and practical applications make a full use of single crystalline semiconductors and the surface chemistry of single crystalline silicon is by far the most studied. The next section will introduce the most commonly studied silicon surfaces and they will indeed be the focus of this chapter. Occasionally, where appropriate, the surfaces of other elemental semiconductors, particularly germanium, will also be considered.
6.1.2 Structure of Most Common Elemental Semiconductor Surfaces: Comparison of Silicon with Germanium and Carbon
One of the main reasons why silicon single crystals became the focus of fundamental scientific research in 1980s and 1990s was that they could provide an idealized starting point with exact positions of the surface atoms being known. The two major reviews, one by Waltenburg and Yates [2] and the other by Yoshinobu et al. [3], have indicated that silicon surface chemistry has reached the potential of a separate and highly regarded scientific field. Detailed reviews in nearly every aspect of this field started appearing regularly in the scientific literature and this chapter will not attempt to summarize all the enormous amount of work in this field. What we will try to achieve is to place the most recent work involving aromatic compounds, mostly on the traditionally best understood silicon single crystal (100) and (111) surfaces, in context of general rules and guidelines to design an appropriate pathway for semiconductor surface functionalization. The clean silicon surfaces of the (100) and (111) orientations can exhibit several different surface reconstructions, and the specifics of surface chemistry of aromatic compounds can definitely depend on the specific reconstruction of the same surface. Chapter 3 of this book focuses on the structures and origins of reactivity of elemental semiconductor surfaces. We will only highlight a few key features of the two most commonly studied reconstructions of clean silicon surfaces, Si(100)-(2×1) and Si(111)-(7×7) that will be used to illustrate the basic rules governing interactions of multifunctional aromatic molecules with silicon.
The structure of the clean Si(100)-(2×1) surface has been a subject of more than a decade-long debate, which is mostlyre solved now. The tetracoordinate atoms of the bulk silicon are lacking saturation on the freshly cleaved (100) surface. This causes them to pair up and form Si–Si dimers. These dimers form long-range rows, with the orientation of these rows switching to perpendicular at the single atomic height step edges. Despite this reconstruction, each atom of the silicon surface still has formally one unpaired electron, making this surface very reactive. Interestingly enough, each of these silicon surface dimers is slightly buckled, with electrons redistributed between the two participating atoms. This buckling is in a dynamic equilibrium at room temperature but can be “frozen” at cryogenic conditions. The low barrier for a dimer inversion and the fact that the overall potential for the process is rather flat, as discussed by Jung et al. [4], indeed caused substantial debate, since the specific temperature of this “freeze” was difficult to determine; but for the purposes of this chapter it is of less importance, since most surface modification chemistry is performed at room temperature or above. Figure 6.1 shows the STM studies of the formation of the silicon surface dimers and their buckling at cryogenic temperatures.
As was noted above, this reconstructed surface still has one unpaired electron per surface silicon atom, which can definitely cause a set of very interesting relations between its structure and reactivity [6]. In fact, the redistribution of the electronic density between the two silicon atoms of the surface dimer draws parallels with the chemistry of biradical species, zwitterionic compounds, and even the carbon–carbon double bond in alkenes [7]. The arguments in favor of one of these descriptions over another sometimes hinge on the specifics of the proposed surface reaction mechanisms, which are still often debated. However, most of the time, the final species, the results of the proposed surface transformations, are very stable at room temperature and can be undoubtedly characterized by common spectroscopic and microscopic techniques.
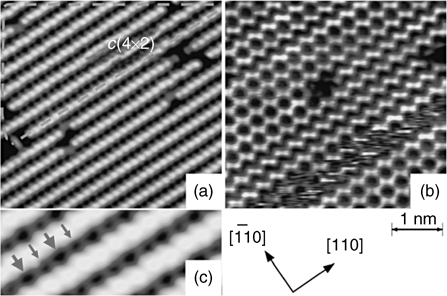
FIGURE 6.1 STM images taken below 10K with a sample bias voltage of −2.2 V (a) and + 1.6 V (b). Tunneling current is set at 38 and 55 pA, respectively. The filled-state image (a) apparently shows symmetric dimers, while the empty-state image (b) shows buckled dimers. In an area surrounded by a dashed green line in image (a) (zoomed in (c)) a faint trace of the c(4×2) structure, alternative array of dark spots between the dimer rows, is observed, as pointed with arrows, indicating an existence of the c(4×2) periodicity and the buckled dimers there. Figure reprinted with permission from Ref. 5. Copyright 2009 The American Physical Society.
In case of elemental group IV semiconductors, there is also an additional axis for comparison of the electronic properties for the (100) surfaces. Ge(100) and C(100) can be prepared and characterized as well. They also both exhibit a stable 2×1 reconstruction. However, in case of Ge(100)-(2×1), the surface germanium dimers are permanently buckled even at room temperature, while for the diamond surface, these dimers are symmetric even at cryogenic temperatures. These subtle differences add to the fundamentals of the comparison of all three semiconductors, as their dimers of the (100)-(2×1) surfaces illustrated in Fig. 6.2 can contribute to the differences in reactivity with respect to similar test reactions by altering the mechanisms of these transformations.
Unlike the Si(100)-(2×1) surface, Si(111) has a more complex reconstruction, which has no known direct analogues for other group IV semiconductors. The well-studied 7×7 reconstruction of the Si(111) surface is shown schematically in Fig. 6.3.
The unit cell of this complex structure has 12 adatoms with available dangling bonds, which are the obvious origin of chemical reactivity, 6 rest atoms, and 1 corner atom. Very useful information about the electronic properties of this surface can be obtained with the help of scanning tunneling microscopy with typical images shown in Fig. 6.4. Interestingly, despite a variety of nonequivalent surface reactive sites and unusual electronic properties, the chemistry exhibited by the Si(111)-(7×7) surface is largely determined by the reactivity of an adatom–rest atom pair, which nicely mimics the structure and reactivity of a silicon dimer of the Si(100)-(2×1) surface. In the case of Si(111)-(2×1), of course, the two atoms involved remain in fixed positions determined by the overall reconstruction. There are several different adatom–rest atom pairs on the Si(111)-(7×7) surface, which can result in very large clusters for its modeling; however, most often simplified cluster models can be used, as suggested in Fig. 6.5 [8].
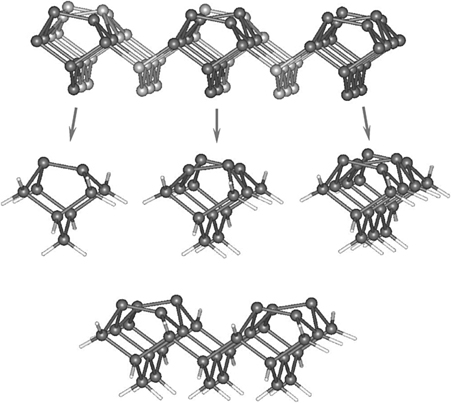
FIGURE 6.2 A representative view of a Si(100)-(2×1) surface reconstruction and representative clusters used in density functional theory (DFT) calculations to model the silicon dimers of this surface. On Ge(100)-(2×1) these dimers are permanently buckled and on C (100)-(2×1) they are symmetric at room temperature.
As some of the studies summarized in this chapter will involve hydrogen-covered Si(100) and Si(111) surfaces, it should be noted that the simplest form of hydrogenation yields the structures shown in Fig. 6.6, where hydrogen atoms are simply added to the two dangling bonds of the silicon dimer of the Si(100)-(2×1) surface or to every dangling bond of the unreconstructed Si(111).
Since the procedure to produce a well-defined flat Si(111) surface terminated with hydrogen that is stable in ambient for several hours was suggested by Chabal et al. [10], it has undergone several improvements [11,12] and currently serves as the main way to prepare this substrate. There are also a number of alternative methods to prepare both H–Si(100) and H–Si(111) in ambient and in vacuum, as summarized in an excellent review by Wayner and Wolkow [13], a report by Baluch et al. [14], and a recent report by Mayne et al. [15].
The H-terminated silicon surfaces are stable in air for up to several hours but longer exposure leads to the formation of oxide layers. In vacuum these surfaces are very stable for extended periods of time. It should be pointed out that unlike ultrahigh vacuum preparation, the traditional etching-based methods for preparing hydrogen-terminated Si(100) surface produce a mixture of surface reactive sites with various chemical reactivity; however, it is indeed possible to prepare the nearly ideal monohydride surface termination shown in Fig. 6.6 (left) in ultrahigh vacuum [16].
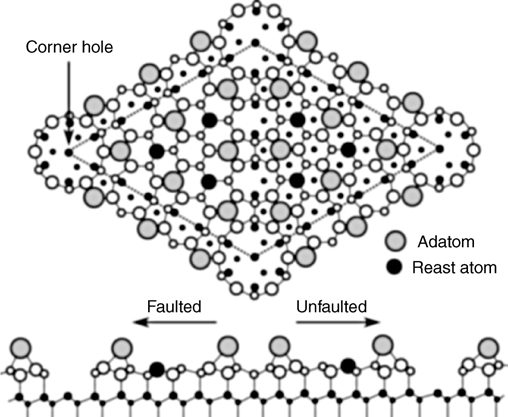
FIGURE 6.3 A representative view of the Si(111)-(7×7) surface with corner, adatoms and rest atoms marked on the reconstructed view, faulted and unfaulted halves of the unit cell shown as a cross section.

FIGURE 6.4 STM overviews comparing the clean 7×7 and the hydrogen-exposed surfaces both filled- and empty-state images. (a) Empty-state STM image of the Si(111)-(7×7) surface recorded with a tip voltage of –2 V. (b) Filled-state STM image of the Si(111)-(7×7) surface recorded with a tip voltage of +1.4 V. The size of images is 180 Å× 180 Å. The tunneling current was 0.3 nA. Figure reprinted with permission from Ref. 9. Copyright 2009 The American Physical Society.
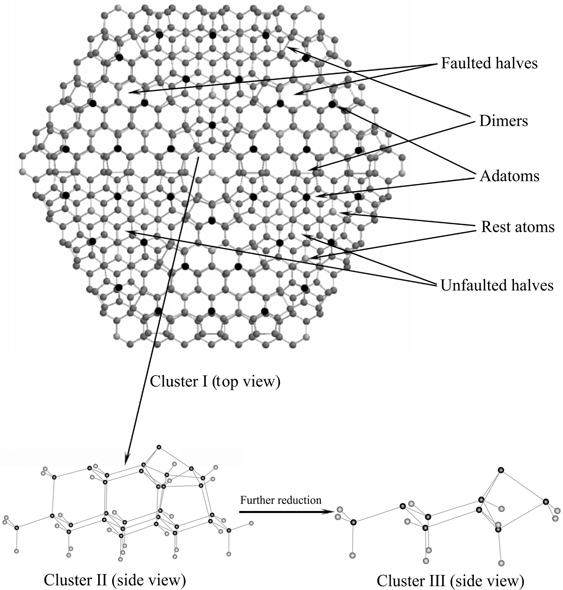
FIGURE 6.5 A representative view of a Si(111)-(7×7) surface and representative clusters used in DFT calculations to model this surface. Cluster models II and III are enhanced for clarity. Figure reprinted with permission from Ref. 8. Copyright 2009 American Chemical Society.
6.1.3 Brief Overview of the Types of Chemical Reactions Relevant for Aromatic Surface Modification of Clean Semiconductor Surfaces
The intrinsic multifunctionality of most aromatic compounds makes the world of reactions of aromatic compounds on semiconductor surfaces very complex. In fact, they often undergo a combination of several reactions simultaneously. While below we will consider multiple specific examples in detail, it is important to place the possible reactions considered in a context of several major classes of surface transformations. In order to understand the multifunctional reactivity on semiconductor surfaces, we should remember three classes of major reactions that are known to occur on the clean semiconductor surfaces: (a) formation of a dative bond (e.g., for substituted amines; this class of surface reaction may also lead to nonlocal surface effects following the initial adsorption); (b) dissociative addition (e.g., for alcohols); and (c) cycloaddition. The most common and probably the best understood type of cycloaddition is the [2+2] addition, where two electrons from the silicon atoms forming the surface dimer interact with the two electrons of the adsorbate, such as ethylene. This would be a symmetry forbidden reaction that should only occur with an assistance of external excitation, such as UV. However, these reactions readily occur on many semiconductor surfaces at room temperature. The exact mechanism of this reaction may be rather complex but the final product is always characterized by the formation of two σ-bonds between silicon surface atoms and the two atoms that are initially connected by an unsaturated bond, such as in ethylene, and if at least one of the atoms of the adsorbate involved is different from carbon (such as in carbonyls), this process could be characterized as heterocyclo [2+2] addition. The second type of cycloaddition was discovered relatively recently and its closest analogue is a common Diels–Alder reaction, such as the one between ethylene and 1,3-butadiene. If this process involves two electrons of the Si(100)-(2×1) surface dimer (instead of ethylene) and four electrons of the diene entity, then the observed process can be best described as a [4+2] cycloaddition and is observed for many dienes on Si(100)-(2×1), Ge(100)-(2×1), and C(100)-(2×1), as well as for the adatom–rest atom combination of the Si(111)-(7×7) surface. Finally, dipolar cycloadditions (or 1,3-cycloaddition processes) were observed for multiple groups that can be represented by a resonance structure with a 1,3-dipole, such as −NO2 or −N3. There are also special types of reactions that can follow some of the more common types, such as group-transfer that can sometimes follow dative bonding. Interestingly, the structure of aromatic compounds is such that all of these processes could be potentially observed for them to occur simultaneously. One can imagine a datively bonded benzene molecule that can dissociate its C–H bond, react using one –C= C– part of the aromatic ring, participate in a Diels–Alder [4+2] process with its –C= C–C= C– diene-like entity, or even to undergo some type of 1,3-cycloaddition process, especially if one of the carbon atoms of the ring is replaced with a heteroatom. That is why it is important to understand the ground rules that govern relative reactivity of the aromatic compounds with respect to the elemental group IV semiconductor surfaces.
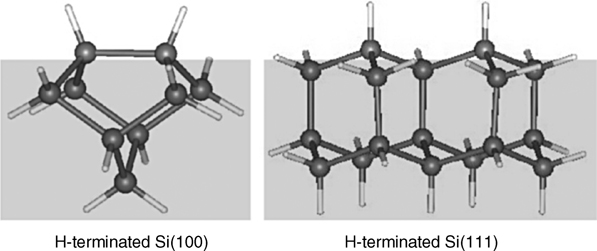
FIGURE 6.6 Clusters used in DFT calculations to model monohydride-terminated Si(100) surface and H-terminated Si(111) surface.
Two recent reviews have addressed multifunctionality in reactions on semiconductors. Filler and Bent [17] compared the reactivity of Si(100)-(2×1) and Ge(100)-(2×1) pointing out that on average the more stable products and more closely spaced activation barriers are typically observed for multifunctional compounds on Si(100)-(2×1) as compared to the same reagents on Ge(100)-(2×1), which can explain higher selectivity in some processes on germanium. However, the work of Leftwich and Teplyakov [7] also examined the cases on Si(100)-(2×1) where kinetics (the barrier height) rules in favor of the much less stable product, meaning that the temperature of the experiment can dictate the product distribution. The detailed discussion below will address both approaches.
If the semiconductor surface is fully or partially hydrogenated, there could be potentially other types of reactions observed for aromatic compounds. The brief summary of the examples below will include the mechanism of radical chain reaction of alkenes with a H-terminated Si(111) surface, where the reaction starts at a defect, such as a dangling bond and the cyclocondensation reaction that does not require the defect presence but is rather thermally driven [18]. These two types of reactions will be considered for specific cases only as discussed below.
6.2 MULTIFUNCTIONAL AROMATIC REACTIONS ON CLEAN SILICON SURFACES
6.2.1 Homoaromatic Compounds Without Additional Functional Groups
The next chapter will focus specifically on the reactions of polycyclic aromatic hydrocarbons on semiconductors. However, in order to understand and predict the reactions of multifunctional aromatic compounds, the reactivity of the simplest aromatic substituents will be considered here. It is most important to realize that within the framework of this chapter, aromatic groups are not just the relatively inert “linkers” but fully functional chemical groups. We start with a simple generic energy diagram shown in Fig. 6.7. It is in fact rather characteristic of many multichannel chemical processes on clean silicon surfaces. Reactant R may form one or more types of surface precursors, SP1 and SP2, which then can be converted into products of different stability, P1 and P2 via transition states TS1 and TS2. The energy gain in the formation of surface-bound precursors is generally in the order of 10–40kJ/mol; however there are examples of reactions without known precursor states and also with extremely strongly bound precursors, well outside this approximate range. Since the precursor states have been reported both for the more or less obvious functionalities, such as ketones and amines, and for the compounds that were less expected to form these states, such as ethylene, the formation of such a bound state should be considered as a prerequisite for most transformations on clean silicon surfaces. It will also be described later that even for the reactions shown to proceed straight downhill in computational studies, experimental barriers were found on the way to the formation of such precursor states.
A brief examination of this generic reaction diagram suggests the possibility of kinetic control, where the chemical reaction proceeds preferentially via the pathway with the lower kinetic barrier, or thermodynamic control, where all the states of the system can be sampled and the most thermodynamically stable product is formed. On silicon, these two control regimes often give very different results and since the products for all the reactions described above are rather stable, their formation is essentially irreversible.
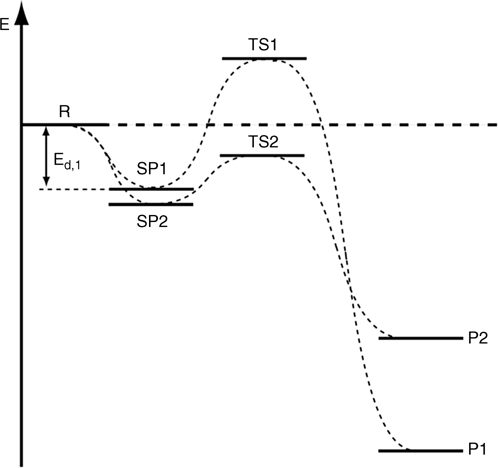
FIGURE 6.7 Comparison of the potential energy profiles for competing chemical reactions on silicon. Most of the time the difference in energy between TS1 and TS2 is substantially smaller than the difference in energy between P1 and P2. The thermodynamic stability of the products is normally hundreds of kJ/mol, while the differences between TS1 and TS2 are substantially smaller, so the scheme is not to scale.
Of course, the case presented in Fig. 6.7 is very simplistic. Often, a combination of two different functionalities on the same molecule can produce very stable surface species or the reaction of one may trigger the reaction of the other. Reactions with metal-containing aromatic compounds may proceed by an entirely different scheme. However, for the purposes of this chapter, it is important to illustrate the fact that even the simplest aromatic substituent alters the energy landscape of the surface reaction and should be considered a functional group. Thus, the reactivity of the simplest aromatic homonuclear compounds will be considered first, with the most emphasis on the recent developments.
The reactivity of a phenyl group should generally follow the trends previously discovered for this type of chemistry and delivering this group on silicon may serve as a starting point for further modification [19,20]. Thus, we will briefly review the reactions of benzene on silicon and germanium. Despite its seeming simplicity, benzene is one of those compounds that could potentially undergo all the classes of reactions listed above. Its chemical transformations on clean silicon surfaces have eluded scientists for well over a decade. A detailed review by Wolkow highlights the most important results of the benzene surface transformations of a clean Si(100)-(2×1) surface [21]. After the chemisorption of benzene was demonstrated by Taguchi et al. [22], the work from several different research groups involved microscopy, spectroscopy, and DFT studies to discern the initial formation of the metastable [4+2], or Diels–Alder, cycloaddition product between benzene and a single silicon dimer of the Si(100)-(2×1) surface. This state slowly converts to the asymmetric tight state bridging two neighboring silicon dimers via the activation energy barrier of 0.94 eV and with a rate constant of (1.5 ± 0.05)× 103 s−1 [23–30]. Some recent studies suggest that at high coverage the initial [4+2] adduct is stabilized and in fact forms the most stable structure [31–36]. There are two major conclusions that can be drawn from this research: (1) benzene (and by extension phenyl group) are indeed reactive entities and should be treated as such when considering reactions on clean silicon; and (2) if phenyl group in a substituted compound is found to react with a clean silicon surface, multiple reaction pathways and kinetic studies should be undertaken. These observations can be generalized for polyaromatic compounds. Naphthalene [37], pentacene [38–46], tetracene [47,48], perylene [48,49], and coronene derivatives [50–52] were all found to chemisorb on a clean Si(100)-(2×1) surface in a rather complex fashion that sometimes can even involve silicon dimers from different dimer rows. Pentacene was suggested to cleave a C–H bond leading to the irreversible chemisorption [38,53–55] and similar reactions have been explored computationally for benzene [56] but no experimental evidence for the C–H dissociation in benzene has been reported so far. Interestingly, on Ge(100), only weakly bound benzene was reported to desorb at 234 and 252K, the desorption regimes corresponding to binding to terraces and steps [57]. The very weak Ge–C bond, compared to the strong Si–C bond driving the addition processes on silicon, is the most likely explanation for such different reactivities, as was examined in a computational study [58].
Although the amount of work on a clean Si(111)-(7×7) surface is much less extensive compared to the studies of the Si(100)-(2×1) surface, there are certainly parallels that can be drawn between these two surfaces. There are several recent reviews highlighting the chemical reactivity of Si(111)-(7×7) and comparing it to Si(100)-(2×1) [7,8,59,60]. Benzene reaction could serve as a segue into this comparison. It is important to realize that the dynamic dipole within the dimer of the Si(100)-(2×1) has very similar zwitterionic properties if compared to the stationary charge distribution between the adatom and the rest atom of the Si(111)-(7×7) surface. Since the distances between these atoms differ on these two substrates and also since the dipole nature is not identical, one should be careful, however, transferring the chemistry discovered on Si(100)-(2×1) directly to Si(111)-(7×7). For example, benzene attachment to the adatom–rest atom pair of the Si(111)-(7×7) does result in a [4+2] Diels–Alder-type product; however this reaction was suggested to proceed via a very different, diradical mechanism, on Si(111)-(7×7) and to form the majority stable species [61]. A theoretical study by Petsalakis and Polanyi followed this process and also compared the calculations with the radiation-induced attachment of benzene on this surface [62]. Similarly, STM and DFT studies of naphthalene suggested that this compound forms nearly exclusively the [4+2] adduct involving the opposite carbon atoms of the same ring of naphthalene but this highly selective chemistry exhibits complex coverage dependence and preference for selected surface adatom–rest atom pairs [63]. A recent study by Guan et al. explored the adsorption of a large polyaromatic tetracene on Si(111)-(7×7) by ultraviolet photoelectron spectroscopy (UPS) and DFT and proposed a plausible model for the most stable adsorption configuration [64] near the top of a center adatom with the longer molecular axis along the (110) direction. Previous tetracene studies [65,66] seem to suggest that the tetracene acts as an acceptor of electrons with respect to the Si(111)-(7×7) surface and that it does participate in covalent bonding with the several combination of adatom–rest atom pairs on this substrate. Because of the larger size, pentacene actually loses all its aromatic character when reacted with Si(111)-(7×7) leading to the formation of multiple Si–C bonds [53,67].
Despite the rich chemistry of benzene and polyaromatic compounds, biphenyl will perhaps show more similar reactivity to substituted benzene. Biphenyl was suggested to adsorb across two adjacent dimers within the same row [68,69]. Several recent studies have addressed different possible adsorption configurations [70–72]; however, an interesting aspect of the biphenyl behavior is the observation that at 5K, biphenyl could be trapped in a bistable configuration pivoting about a rotational axis, and that the STM could be used to switch the pivot reproducibly [73]. Interesting electronic properties [74,75] coupled with the most recent STM-induced chemistry studies [76] summarized in Fig. 6.8 keep fueling the scientific interest toward biphenyl. In fact, the STM manipulation shown in Fig. 6.8 could serve as a stepping stone to developing molecular switches for practical applications because of such a rich set of electronic properties of biphenyl on Si(100)-(2×1). Even more intriguing behavior was recorded for para-hexaphenyl molecules, where six phenyl rings are coupled to one another sequentially in para-positions, forming a line of aromatic structures on Si(100)-(2×1) [77]. Two different states are observed by STM at room temperature and a very strong angular dependence is found between the axis of this molecule and the direction of propagation of the dimer lines of the Si(100)-(2×1) surface.
6.2.2 Functionalized Aromatics
6.2.2.1 Dissociative Addition
With all the reactivity exhibited by the benzene and phenyl groups described above, it may seem impossible to deliver an unreacted phenyl substituent to the silicon substrate. There are, however, at least two general approaches that help address this problem. One is based on decreasing the reactivity of the silicon surface, for example, by saturating the available surface silicon dangling bonds with hydrogen. This approach will be addressed later. The other is based on selecting appropriate chemical reactivity of a polyfunctional aromatic molecule. This can be illustrated by following the previously discussed energy diagram in Fig. 6.7. Since the formation of any covalent bonds with the silicon surface is very often irreversible, the preferential selective attachment schemes should either be based on the thermodynamically controlled regime where the target surface species is the most stable, or on a kinetically controlled approach to reach the less stable species over the lower energy barrier. Fortunately, both approaches can work on silicon. The attachment chemistries described below involve halogens (X), oxygen, nitrogen, or sulfur and often lead to the formation of two bonds at the expense of one, meaning that the processes leading to the formation of Si–O, Si–N, or Si–X bonds and accompanied by the formation of Si–H surface sites should be thermodynamically preferred compared to the reaction of the phenyl group. In addition, most of these processes do have lower reaction barriers compared to the phenyl reactions on silicon. There are plenty of exceptions from these rules, as will be outlined below.
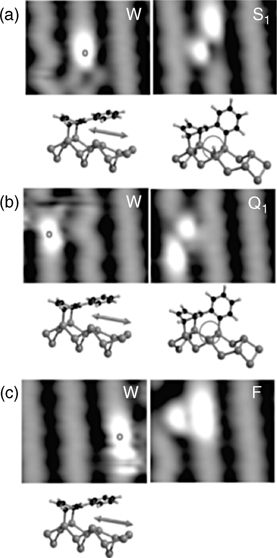
FIGURE 6.8 Changes of adsorption configuration by STM manipulation with positive surface voltage pulses. All 2.2×1.8 nm2 STM topographies are recorded at VS = – 2 Vand I = 240 pA. Left-hand and right-hand side STM topographies are recorded before and after STM manipulation, respectively. The dots indicate the location of the STM tip during the surface voltage pulse at VS = + 4 V. (a) Change from the weakly chemisorbed (W) to the strongly chemisorbed bistable (S1) configuration. (b) Change from the weakly chemisorbed (W) to the strongly chemisorbed quadristable (Q1) configuration. (c) Change from the weakly chemisorbed (W) to the strongly chemisorbed fixed (F) configuration. The adsorption configuration of the biphenyl molecule is shown below each STM topography. For the S1 configuration, the hydrogen atom dissociated from the fixed phenyl ring is circled. In the Q1 configuration, the hydrogen atom has been desorbed. Figure reprinted with permission from Ref. 76. Copyright 2009 American Chemical Society.
A substantial number of experimental and theoretical studies do suggest that the reactivity of phenyl group in singly substituted aromatic molecules is lower than the reactivity of almost any functional group. In fact even in toluene and xylenes, where phenyl group competes in reactivity with very stable methyl groups, there has been reported methyl C–H dissociation on Si(100)-(2×1) [78], where isotopically substituted compounds served as a marker to confirm that the source of surface hydrogen is the methyl group and not the aromatic ring. The first principles calculation studies by Costanzo et al. [79,80] were in complete agreement with the experiment. In a spectroscopic and DFT investigation of para-xylene (1,4-dimethylbenzene), it was suggested that some para-xylene molecules desorb intact at 400 and 470K after chemisorption pathways that were proposed to be similar to those for benzene; however, most surface species chemisorb via the formation of the Si–C bond following the loss of hydrogen from methyl groups [81]. The behavior of toluene on a clean Si(111)-(7×7) surface is somewhat different, as a photoemission study suggested the formation of the π-bond by the aromatic electrons [82] and STM studies followed the adsorption of both benzene and toluene at cryogenic temperature [83,84]; however, studies of further thermal transformations of toluene on this substrate are also consistent with methyl group dissociation [85].
Among the functional groups relevant to this chapter, the types of reactions with a clean silicon surface, either Si(100) or Si(111), can be generally divided into two major categories: Ph–Y–H dissociation and cycloaddition. Reactions of phenol, benzenethiol, aniline, 1,4-phenylenediamine, and benzoic (and 4-aminobenzoic) acid, can cleanly deliver an intact phenyl ring to the Si(100)-(2×1) surface by Ph–Y–H dissociation forming –O–[86], –S– [87], –NH– [88–91], and –O–CO– [90,92,93] linkages, respectively, and the corresponding Si–H species. A detailed review by Mui et al. analyzes the parallels between silicon and germanium chemistry with respect to these reactions [94]. For the case of aniline, multiple bonding configurations following dissociation of one and both N–H bonds have been considered and there are several possibilities for the exact long-range structures formed [88–91,95]; however, both these studies and the investigation of aniline adsorption on the Si(5 5 12) surface [96] suggest that the nitrogen atom is connected to silicon and that the aromatic ring structure is retained. If needed, the formation of an extra surface hydrogen atom evidenced in the dissociative addition processes can be avoided by employing the reaction of, for example, disulfide compounds, as was shown for diphenyl disulfide (C6H5S–SC6H5) [87]), where two C6H5S– surface groups were cleanly formed. Most of these reactions are relatively easy to follow spectroscopically, as the phenyl ring, in addition to having its own set of spectroscopic signatures (such as vibrational characteristics) helps to identify the chemical state of the elements in the adjacent functional groups. For example, upon dissociation of phenol on a clean Si(100)-(2×1), this reaction can be confirmed by following the photoemission spectra, as illustrated in Fig. 6.9 for the C 1s core-level spectrum of the submonolayer coverage of phenol [86]. Here, the carbon atom bound to oxygen can be easily distinguished from the lower binding energy component of carbon atoms in the phenyl ring [86]. The quantitative analysis confirms this assessment. A detailed computational study by Carbone et al. [97,98] explored the energy landscape for the phenol adsorption on Si(100)-(2×1) and also found the dissociation channel to lead to the most thermodynamically stable structure. The propensity of these dissociated molecules to form molecular lines on Si(100)-(2×1) has also been explored computationally [98]. Similar chemical behavior was observed for phenol on a clean Si(111)-(7×7) surface. It was shown to dissociate at temperatures as low as 50K [99]. Near-edge X-ray absorption fine structure (NEXAFS) studies of phenol, 4-fluorophenol, and 2,3,4-trifluorophenol on Si(111)-(7×7) confirm that this reaction is rather general [100]. Benzenethiol and 1,4-benzenethiol also dissociate one or both –SH functional groups, respectively, on Si(111)-(7×7) [101].
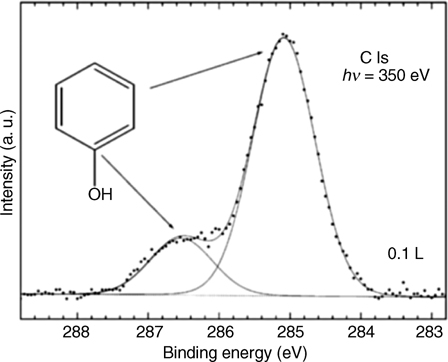
FIGURE 6.9 Line-shape analysis of the C 1s core-level spectrum after exposure to 0.1 L phenol, recorded at a photon energy of 350 eV. Figure reprinted with permission from Ref. 86. Copyright 2009 Elsevier B.V.
In some cases, the specific silicon surface reaction, and as a result the chemical nature of the surface-bound group of the aromatic compound can be altered by using a metal promoter. The Richardson group reported an investigation of the interaction of benzoic acid with a Si(100)-(2×1) surface covered with 0.5 monolayer of Na atoms, adsorbed on cave sites between silicon dimers [92,102]. Promoting interaction of benzoic acid with sodium caused the formation of a bidentate ligand instead of the Si–O–(C=O)–C linker observed for the reaction on an unpromoted surface. The observation of the Si–H stretching mode at 2090 cm–1 and the benzoate functionality characterized by v(OCO)=1425 cm–1 in HREELS studies confirmed this assessment and was supplemented by other analytical techniques. In this specific example, the selectivity of the bifunctional 4-aminobenzoic acid in a reaction with the Na-promoted Si(100)-(2×1) surface does not change [93], as the carboxylic group reacts predominantly with silicon regardless of the presence of metal promoter; however, the formation of the benzoate bidentate linker leads to the formation of a much more stable adduct, which is stable up to 300°C [92].
The dissociative attachment of the Ph–X compounds to the clean silicon surfaces can also deliver the intact aromatic group; however, depending on the reactivity of the C–X bond, this reaction may not be as selective as the dissociative reactions described above. For example, the low reactivity of the C–Cl bond on a Si(100)-(2×1) surface leaves the door open for the reaction of the phenyl group with this surface. The combination of DFT and STM studies by Polanyi's group [103,104] suggests that at room temperature both chlorobenzene and 1,2-dichlorobenzene yield 45–50% of [4+2] adduct and only approximately 20% surface products consistent with C–Cl bond scission and the deposition of the intact phenyl ring. However, the same study showed that doubly substituted 1,4-dichlorobenzene selectively dissociates both C–Cl bonds and forms a 1,4-adduct across the neighboring silicon dimer rows. Spectroscopic and computational study by Zhou and Leung also suggested that chlorobenzene and 1,2-dichlorobenzene, as well as 1,3-dichlorobenzene adsorb at room temperature on Si(100)-(2×1) mostly via the benzene ring [105]. The comparison within the same column of the periodic table followed 1,2-difluoro-, 1,2-dichloro-, and 1,2-dibromobenzenes [106]. The adsorption of 1,2-difluorobenzene was found to be completely reversible, as it desorbed intact in a molecular state at elevated temperatures, 25% of 1,2-dichlorobenzene was found to undergo dechlorination and 1,2-dibromobenzene was shown to undergo irreversible double debromination forming 1,2-phenylene species on Si(100)-(2×1) [106].
It seems that in a reaction of the phenyl ring with the adatom–rest atom pair of the clean Si(111)-(7×7) surface, the formation of the [4+2] surface adducts should be even more preferential as compared to the Si(100)-(2×1). Consistent with this assessment, no direct dissociation of the C–Cl bond in chlorobenzene was observed on Si(111)-(7×7) [107–110]. The physisorbed chlorobenzene and toluene were proposed as reversible molecular switches [111]. In fact, only bromo derivatives were shown to dissociate on this surface at room temperature, overpowering the reactivity of the phenyl ring [112] and more recently a detailed computational study of 1,2-dibromobenzene on Si(111)-(7×7) helped explain the experimental results [113–115]. Interestingly, the reactivity of the 1,2- and 1,4-dibromoxylenes was found to be lower than that for the corresponding substituted benzenes [116].
6.2.2.2 Cycloaddition
The cycloaddition processes on silicon can be generally grouped into 1,2-, 1,3-, and 1,4-cycloaddition classes of reactions. The 1,2- (or [2+2]) and 1,4- (or [4+2]) processes were mentioned above for the reactions of the phenyl ring. However, similar attachment chemistry can be used to deliver an intact phenyl group to the silicon surface.
The reactions of ethylene and acetylene described earlier in this book proceed by [2+2] pathways. Despite continuous studies of these seemingly simple hydrocarbons on clean silicon surfaces, there is still some debate over the exact mechanism of the attachment process. However, for the purposes of this chapter, the result of the attachment reaction, the Si–C–C–Si cycle, is fully described and agreed upon in the surface science community. Substituting one of the hydrogen atoms in ethylene or acetylene by a phenyl group should provide a convenient vehicle to deliver the aromatic functionality onto a silicon surface by this 1,2-cycloaddition chemistry. In fact, both styrene (Ph–CH=CH2) and phenylacetylene (Ph–CCH) react with the Si(100)-(2×1) surface by [2+2] cycloaddition. Considering all the possibilities for styrene, one could suggest that interaction of the vinyl group with the phenyl functionality could potentially lead to complex reactions including [4+2] addition; however, all the current studies suggest that a simple [2+2] process across a single silicon dimer dominates the reactions of this compound with a clean Si(100)-(2×1) surface [117–120]. As was done previously many times, reactions on the silicon surface can be paralleled with the test reactions with disilenes, and the reaction of styrene with Si(100)-(2×1) is a perfect example. As shown schematically in Fig. 6.10, the structure of the proposed attachment product for styrene on Si (100)-(2×1) is the same as for a variety of disilenes [121–123], suggesting the possible parallels in electronic structure of a Si–Si surface dimer of this substrate and a Si=Si double bond. The same addition reaction of styrene seems to take place on the permanently buckled dimer of the Ge(100)-(2×1) surface [124] or on Ge-covered Si(100) [125].
Since the need to deliver intact phenyl groups to a clean silicon surface was in part dictated by the possible electronic and switchable behavior of the aromatic compounds, it is important to highlight that the switching between two rotational states in a chemisorbed styrene molecule on a clean Si(100)-(2×1) surface could be followed experimentally by STM, however, this motion could be suppressed by chemical modification and by using 5-vinyl-2,3-dihydrobenzofuran instead of styrene, as summarized by schematics and STM images in Figs. 6.11 and 6.12 [126].
Despite such a high selectivity on Si(100)-(2×1), the studies of the isotope-substituted styrene on Si(111)-(7×7) [127] by HREELS clearly show rehybridization of the phenyl ring carbon atoms from sp2 to sp3, which would support the Diels–Alder-like chemistry involving the phenyl ring. Since the reactivity of the Si(111)-(7×7) seems to favor the [4+2] additions whenever possible, it is likely that the potential for conjugation between the vinyl group and the phenyl mentioned above for the reaction with Si(100)-(2×1) is actually a reality in a reaction with Si(111)-(2×1).
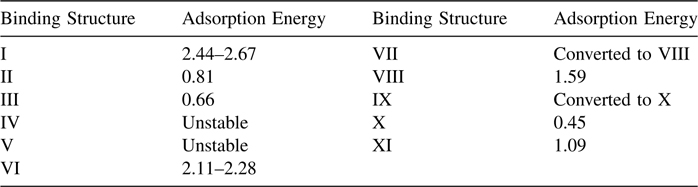
The geometrical restrictions of a C ≡ C bond make surface chemistry of phenylacetylene on silicon more complex compared to that of styrene. A simple [2+2] addition similar to that of styrene was proposed by Tao et al. [128] to dominate the reaction of phenylacetylene with Si(100)-(2×1), while possible dissociation of a weak ≡ C–H bond was not observed. STM and DFT investigation reported by Kim et al. [129] used phenylacetylene and 1-phenyl-1-propyne (a molecule with an extra methyl marker group to help distinguish structural differences in STM images) to observe two different surface structures. One of these structures was proposed to be the same as the one reported by Tao et al. [128], the result of [2+2] addition. The other was proposed to involve both functionalities, with phenyl group bound to one silicon dimer row and acetylene entity interacting with the other silicon dimer row, as shown in Fig. 6.13. Table 6.1 summarizes the predicted stabilities of these structures, with the [2+2] structure I indeed being the most stable; however, several other bridging structures were also observed experimentally. A more recent STM study combined with Monte Carlo simulation also confirmed the presence of two different adsorption modes for phenylacetylene on Si(100)-(2×1), one occupying two surface dangling bonds and one occupying three, and offered an accurate kinetic model for the adsorption process [130].
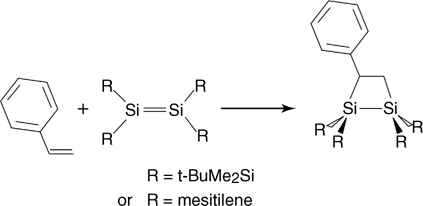
FIGURE 6.10 Reaction of styrene with tetramesityldisilene and tetradisilylene. Based on a schematic from Ref. 117.

FIGURE 6.11 (a) Schematic of [2+2] cycloaddition of a 4-methoxystyrene molecule on a dimer on the clean Si(100) surface. The methoxy group is expected to have the rotational degree of freedom indicated in the figure; (b) UHV STM image of 4-methoxystyrene molecules on the clean Si(100) surface following a dose of 0.7 L. Several of the molecules (indicated by white arrows) exhibit multiple switching events. Imaging conditions are sample bias of –2.0 V and tunneling current of 0.1 nA. Figure reprinted with permission from Ref. 126. Copyright 2009 American Vacuum Society.
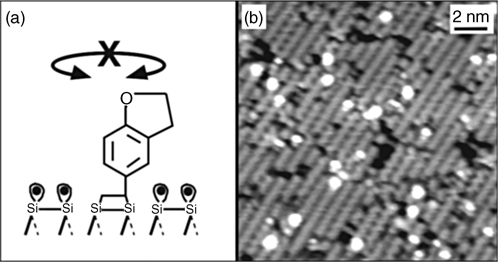
FIGURE 6.12 (a) Schematic of a 5-vinyl-2,3-dihydrobenzofuran bound to Si(100). This molecule is analogous to 4-methoxystyrene except that its methoxy group is covalently linked to the aromatic ring in order to inhibit its rotational degree of freedom; (b) UHV STM image of a submonolayer coverage of 5-vinyl-2,3-dihydrobenzofuran on the clean Si(100) surface following a dose of 0.4 L. No switching events are observed in this case. Imaging conditions are sample bias of –2.0 V and tunneling current of 0.1 nA. Figure reprinted with permission from Ref. 126. Copyright 2009 American Vacuum Society.
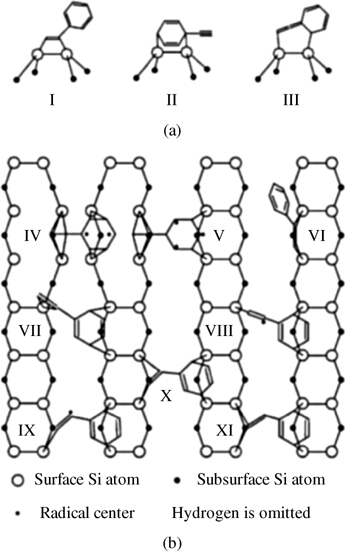
FIGURE 6.13 Schematic presentation of the proposed binding structures of phenylacetylene on Si(100)-(2×1). (a) Side view for structures I–III. (b) Top view for structures IV–XI. The structure of phenylacetylene is drawn only for the carbon skeleton without showing hydrogen atoms. Figure reprinted with permission from Ref. 129. Copyright 2009 American Chemical Society.
TABLE 6.1 Adsorption Energy of the Proposed Binding Structures in Fig. 6.13 (in eV/molecule) for Phenylacetylene on Si(100) Calculated from DFT (slab-GGA) for a Coverage of 1/8 ML [129]
The [2+2] cycloaddition processes on Si(100)-(2×1) are not restricted to only ethylene and acetylene derivatives. Benzaldehyde (Ph–COH), a heteroanalogue of styrene, also reacts via the same pathway despite the fact that it could also potentially dissociate the C–H bond of the aldehyde functionality [131]. This reaction was very conveniently followed by HREELS, as the very intense stretching mode vC=O = 1713 cm−1 disappeared upon adsorption. Additionally, bright protrusions assigned to the phenyl rings on top of the silicon dimers in low-coverage STM investigation corresponded to a single product and confirmed the absence of the inter-row reactions [131]. The preference with respect to the [2+2] addition could not even be hindered by substituting α-hydrogen for a methyl group in acetophenone (C6H5–CO–CH3). According to the HREELS and DFT investigation, this compound still selectively undergoes [2+2] heterocycloaddition via its C=O bond [132].
Another heteroanalogue of styrene, nitrosobenzene (Ph–NO), has both carbon atoms substituted but this does not seem to affect its propensity to [2+2] cycloaddition on Si(100)-(2×1). Perrine et al. used a combination of infrared spectroscopy and XPS with computational analysis to confirm the reactivity of the –N=O group [133]. The initial [2+2] cycloaddition was followed by the oxygen migration subsurface, as expected, and this process could be accelerated by thermal annealing. One of the interesting aspects of this example is the observation of the datively bonded Ph–NO at cryogenic temperatures, as suggested by the infrared studies summarized in Fig. 6.14 and substantiated by computational investigation.
A nitrogen-substituted heteroanalogue of phenylacetylene, benzonitrile (C6H5–C≡N), has also been investigated on Si(100)-(2×1) and it was originally suggested to react exclusively by [2+2] addition via the nitrile functionality [134]. The easily distinguishable VC≡N absorption band at 2247 cm−1 disappeared upon chemisorption and another intense VC=N absorption band at 1629 cm−1 appeared when reaction was carried out at 110K [134] and followed by HREELS. A combination of XPS analysis and DFT investigation agreed with these results. It should be pointed out that in the case of benzonitrile, the reversibility of the adsorption process was confirmed by thermal desorption as the molecular desorption was recorded at ~490K [134]. The [2+2] addition of benzonitrile found even more support in a computational study by Qu and Han [135] but careful investigation combining computational studies, XPS, and NEXAFS by Rangan et al. [136] suggested that in addition to the [2+2] addition product, another adduct bonded via the phenyl ring is observed at room temperature.
On Si(111)-(7×7) the unsaturated phenylacetylene [127] and benzonitrile [137] keep their phenyl groups intact. The HREELS studies observe the formation of styrene-like surface adducts. The high selectivity on this surface is likely aided both by high reactivities of both C≡C and C≡N in electrophilic addition reactions and by the fact that the geometry of the triple bond forces the phenyl ring to point away from the surface upon reaction with the adatom–rest atom pair, making it virtually impossible for any subsequent surface transformations to occur.
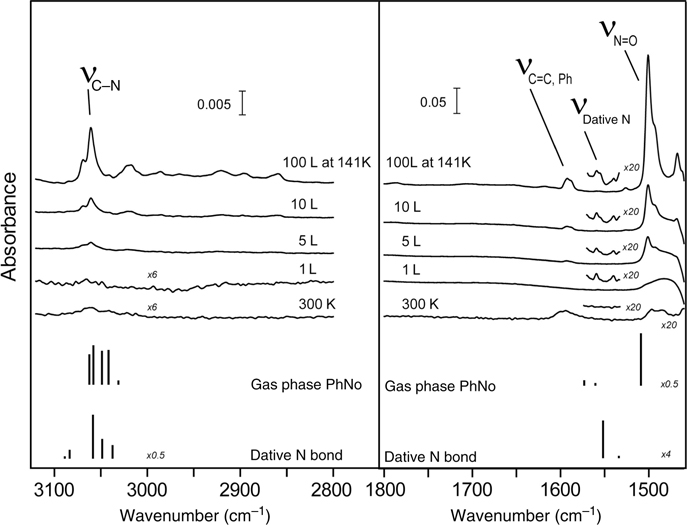
FIGURE 6.14 MIR-FTIR coverage profile of nitrosobenzene reacted on Si(100)-(2×1) at cryogenic and room temperatures. Left panel: C–H stretch region. Right panel: C=C and N=O stretch region. The exposure of the nitrosobenzene on Si(100)-(2×1) at 300K results in saturation of surface sites, based on IR signal. Below the experimental spectra are shown the computationally predicted infrared absorption bands corresponding to structures of a single molecule of nitrosobenzene in the gas phase and to the datively bonded nitrosobenzene on a Si (100)-(2×1) surface. The computationally predicted frequencies are scaled by 0.9564. Figure reprinted with permission from Ref. 133. Copyright 2009 American Chemical Society.
Finally, larger aromatic systems can also be delivered onto a silicon surface via [2+2] cycloaddition reaction. The investigation reported by Schwarts et al. focused on a possibility to deliver a stackable derivative of naphthalene, acenaphthalene, to react selectively with the silicon dimers of the Si(100)-(2×1) surface, as summarized in Fig. 6.15 [138]. A detailed set of STM and infrared spectroscopic studies of selectively deuterated acenaphthalene drew on a comparison with silylation chemistry to produce the acenaphthalene [2+2] product over a dimer row with selectivity over 90%, thus creating a sharp interface between silicon and assembled polycyclic aromatic molecules. This work was also complemented by computational studies by Nunzi et al. that confirmed the stability and preferential formation of the [2+2] cycloaddition product [139].
Within the general class of cycloaddition reactions on silicon, reactions of 1,3-cycloaddition have received a very substantial attention over the last decade. The reaction initially considered for nitromethane and nitroethane was proposed to lead to the formation of the Si–O–C–O–Si with subsequent oxygen migration into the silicon back-bond [141–144]. A detailed set of experimental and computational studies suggested that nitrobenzene (PhNO2) would also react with a clean Si(100)-(2×1) surface via 1,3-dipolar cycloaddition [145,146], as summarized in Fig. 6.16. Similar to other nitro compounds, nitrobenzene was shown to react with its –NO2 group exclusively, as no remaining nitrofunctionality was observed by MIR–FTIR following nitrobenzene reaction at room temperature and the binding between two different silicon dimers was prohibitively expensive as confirmed in the computational studies. Interestingly, a barrier was observed experimentally for this cycloaddition reaction despite the fact that the entire transformation was shown theoretically to proceed downhill.
FIGURE 6.15 Selected reaction products for acenaphthalene on Si(100)-(2×1) from Ref. 138 and a parallel with the silylation process from Ref. 140.
As a sidenote, the initial five-membered ring adduct was suggested computationally to have a potential to work as a molecular switch. The geometry of the nitrogen center could be switched between two positions, as shown in Fig. 6.17. The phenyl ring could change its position from being nearly parallel to the surface (IB) to almost exactly perpendicular (IA or IC) [146]. This potentially useful process could benefit from low-temperature STM studies. The cryogenic experiments would be required since the barriers found for this “flip” are rather low; however, experimentally found barrier for the attachment (40.8kJ/mol) [145] may make these studies difficult.
In many molecules amenable to 1,3-dipolar cycloaddition, there also exists a possibility for 1,2-cycloaddition reactions for the same functional group. For example, phenyl isothiocyanate (Ph–N=C=S) could potentially react with Si (100)-(2×1) via [2+2] cycloaddition either to the N=C bond or to the C=S bond, as well as by 1,3-dipolar cycloaddition to form a five-membered ring (Si–N–C–S–Si), as was predicted by Barriocanal and Doren for nitrile sulfide [142]. A very thorough investigation reported by Ellison and Hamers [147] used FTIR, XPS, STM, and DFT to prove that the major surface reaction is the 1,2-cycloaddition via the N=C group, as shown in Fig. 6.16b above. This reaction would also be expected based on the previous studies of solution silylation chemistry [148,149]. The reaction of the isoelectronic functional group was tested in a study on Ge(100), where Kim et al. used ethylenediamine coupling with 1,4-phenylene diisocyanate (O=C=N–C6H4–N=C=O) to initiate a layer-by-layer growth [150]. This investigation employed infrared spectroscopy to prove that the reaction occurs via 1,2-cycloaddition to form Si–N and Si–C bonds, very similarly to the reaction of phenyl isothiocyanate on Si(100)-(2×1).
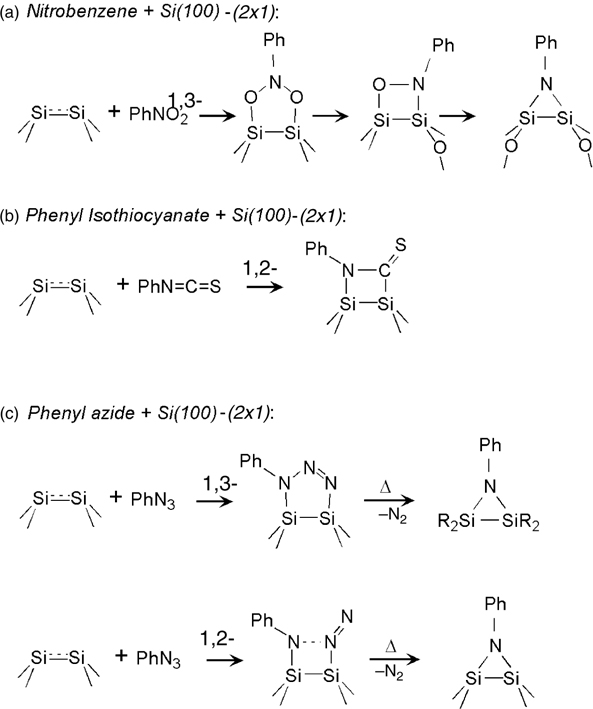
FIGURE 6.16 Schematic representation of selected approaches to introduce a phenyl ring onto a Si(100)-(2×1) substrate: (a) reaction with nitrobenzene; (b) reaction with isothiocyanate; (c) possible reactions with phenyl azide.
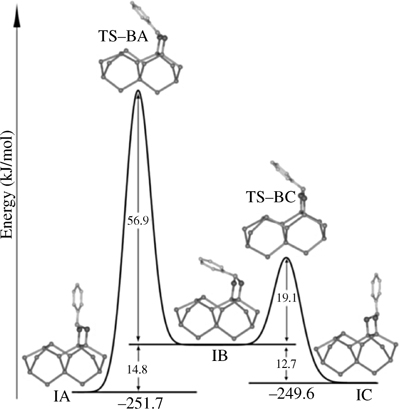
FIGURE 6.17 Proposed reaction pathway leading from structure IB (with the phenyl group nearly parallel to the surface) to either structure IA or structure IC with the phenyl group nearly perpendicular to the surface. Figure reprinted with permission from Ref. 146. Copyright 2009 American Chemical Society.
Despite great reaction selectivity and exceptional stability of the products of interaction of both nitrobenzene [145,146] and phenyl isothiocyanate [147], the nature of the chemical reactions delivering the aromatic functionality to the silicon surface in both cases implies that there is a potential for further transformations of the initial adducts in both cases. Specifically, the presence of sulfur or oxygen near the silicon surface can alter the electronic properties of the produced interface tremendously. Thus, it would be desirable to have the high selectivity of this cycloaddition chemistry without the risk of interface contamination. One of the ways around this problem was offered by Barriocanal and Doren [142]. The general trends in 1,3-cycloaddition chemistry on Si(100)-(2×1) were considered for azido groups, –N3, which could undergo this process to form a five-membered ring of the structure Si–N–N=N–Si, as shown in Fig. 6.16c for phenylazide. Interestingly, regardless of the exact surface reaction, 1,2- or 1,3-cycloaddition, if nitrogen could be eliminated in a thermal reaction, both pathways would lead to the formation of identical stable three-membered ring structures Si–N–Si. Despite the fact that phenylazide is explosive under certain experimental conditions, a successful study of this compound and its comparison with another azide homologue, benzylazide (Ph–CH2–N3), which is more stable, suggest that both 1,2- and 1,3- cycloadditions likely occur for these compounds [151,152]. Leftwich and Teplyakov compared the chemistry of phenylazide and benzylazide using a combination of thermal desorption, MIR-FTIR, and DFT and found that 1,3-cycloaddition is favored in both cases but 1,2-addition was observed spectroscopically for benzylazide and not for phenylazide. The infrared spectroscopy experiments summarized in Fig. 6.18 clearly follow the computationally predicted frequencies for 1,2-cycloaddition processes; however, computational investigation suggested that this process is much less likely in case of phenylazide. Also consistent with the computational predictions, thermal release of nitrogen from benzylazide, presumably from the 1,3-adduct, was found to occur at higher temperature compared to the same process for phenylazide [152]. A recent computational study of reaction of nitrones, including N-phenyl nitrone (Ph–N(OH)–CH–CH2) explored the propensity of the 1,3-cycloaddition on the C(100)-(2×1) surface and found that it is indeed the preferred cycloaddition pathway because of very substantial kinetic barriers for the cycloaddition processes [153].
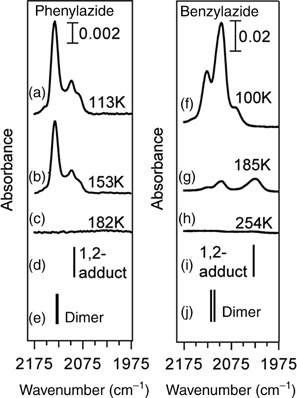
FIGURE 6.18 Infrared studies of phenylazide, left, and benzylazide, right, on Si(100)-(2×1): (a) 5L submonolayer coverage of phenylazide condensed on a Si(100)-(2×1) surface at 113K; (b) same as (a) but briefly annealed to 153K; (c) same as (a) but briefly annealed to 182K; (d) predicted infrared spectrum for a 1,2-adduct of phenylazide on a Si(100)-(2×1) surface; (e) predicted infrared spectrum for a dimer of phenylazide; (f) 10 L submonolayer coverage of benzylazide condensed on a Si(100)-(2×1) surface at 100K; (g) same as (f) but briefly annealed to 185K; (h) same as (f) but briefly annealed to 254K; (i) predicted infrared spectrum for a 1,2-adduct of benzylazide on a Si(100)-(2×1) surface; (j) predicted infrared spectrum for a dimer of benzylazide. The frequencies of all predicted spectra are scaled by 0.95. The spectra corresponding to the adsorbed benzylazide and phenylazide are reported with a deuterium-substituted silicon cluster to avoid interference from Si–H vibrations. Figure reprinted with permission from Ref. 152. Copyright 2009 American Chemical Society.
Although involving multiple chemical functionalities in a formation of a single cyclic product seems to possibly lead to multiple surface binding configurations, sometimes a clever design can produce very well characterized and expected product with exceptional selectivity. The Hamers group used 9,10-phenanthrenequinone on Si(100)-(2×1) [154,155] in hopes of selectively inducing a [4+2] hetero–Diels–Alder reaction leading to the formation of two Si–O–C linkages, and retaining an intact aromatic system. This very selective reaction forms a cyclic ring as shown in Fig. 6.19. Stretching vibrations within the 3000–3200 cm−1 spectral range observed with FTIR and detailed investigations of O 1s and C 1s core levels confirmed the presence of an intact aromatic ring and the Si–O–C bridges [154]. The STM studies of this system summarized in Fig. 6.20 show clear nodal structure corresponding to a single molecule of 9,10-phenanthrenequinone chemisorbed on Si (100)-(2×1) [154].
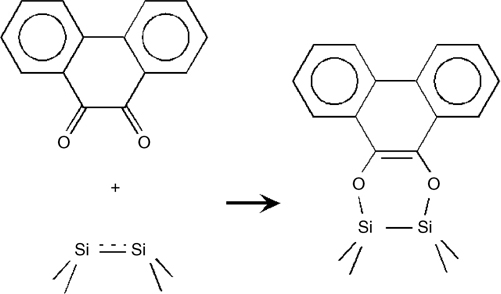
FIGURE 6.19 Reaction of 9,10-phenanthrenequinone with Si(100)-(2×1).
6.2.3 Heteroaromatics: Aromaticity as a Driving Force in Surface Processes
In most cases, the reactions of heteroaromatic compounds on clean semiconductor surfaces are rather complex and understandably so, as multiple reaction pathways are generally available. This is why there is a very limited effort to use these reactions for attaching target groups to semiconductors. However, aromaticity is a very important factor to consider while designing surface modification schemes because very often transformations that require breaking up an aromatic system are kinetically slow and at the same time if a stable aromatic system is created as a result of surface transformation, this could be used as a driving force in increasing selectivity of surface processes. Chapter 5 in this book details the binding of five- and six-membered aromatic molecules on semiconductors and this section will only briefly consider the influence of aromaticity on the reactions that either start with an aromatic system or whose resulting product is an aromatic system. Thus, the effect of aromaticity on the selectivity of the surface processes will be considered from the opposite points of view. Most of the compounds discussed in this section are shown in Fig. 6.21.
A recent review by Tao et al. [156] has considered electronic and structural factors determining the reactions of various heteroaromatics on silicon and germanium. One of the most studied classes of heteroaromatic compounds is derivatives of pyrrole. The rich multiple functionalities of pyrrole imply the variety of possible surface reactions on Si(100)-(2×1). However, as illustrated in Fig. 6.22, the dominant pathway for the chemical reaction of this molecule (a) with the silicon dimers of the Si(100)-(2×1) surface is the one that retains the aromatic stabilization, leading to the N–H dissociation and the formation of product (b) [89,157–160]. Further reaction following N–H dissociation involves C–H dissociation shown as (c), as was confirmed by the studies of deuterium-substituted pyrrole [89]. Thus, the retention of aromaticity dictates the selectivity of the reaction. At the same time, the reaction of a nonaromatic compound, 3-pyrroline (d) with the same surface is driven toward attaining aromatic character by the surface product. Consistent with the computational studies [161], a mixture of possible adducts is formed initially in this case. Most notably, N–H dissociation (e) competes with [2+2] cycloaddition of the C=C bond (g) [89]. However, if the product of the [2+2] pathway was suggested to lose extra hydrogen even at room temperature to form species (h), surface annealing past 550K leads to the formation of species (f), which is exactly the same as the result of pyrrole reaction with the same surface at room temperature [89].
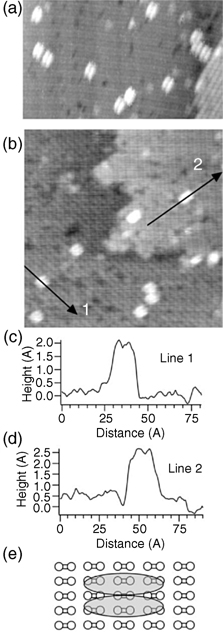
FIGURE 6.20 (a) STM image of Si(001) surface after it was exposed to 0.03 L 9,10-phenanthrenequinone. Molecular features are observed on one surface domain. (b) STM image obtained from another Si(001) surface exposed to 0.03 L 9,10-phenanthrenequinone. Molecular features are observed on two adjacent surface domains. (c) Height profile along line labeled “1” in (b). (d) Height profile along line labeled “2” in (b). (e) Schematic illustration showing the bonding location of the observed features with respect to the underlying surface reconstruction, based upon high-resolution images. Figure reprinted with permission from Ref. 154. Copyright 2009 Elsevier B.V.
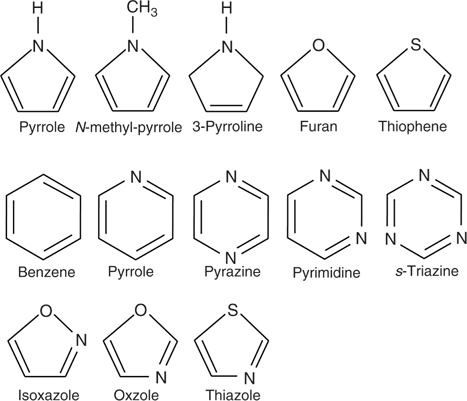
FIGURE 6.21 Compounds used in this section to illustrate the effect of aromaticity as a driving force for reactions of heteroaromatic compounds with silicon.
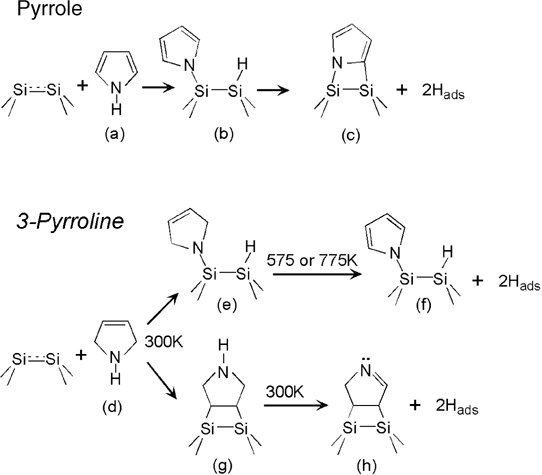
FIGURE 6.22 Reactions of pyrrole and 3-pyrroline with Si(100)-(2×1).
Similarly to the reaction on Si(100)-(2×1), pyrrole also dissociates on Si(111)-(2×1), as was confirmed by following the disappearance of the N–H stretching at 3384 cm–1 upon adsorption at room temperature in a set of vibrational spectroscopy studies [162]. This process occurred concurrently with the appearance of the vibrational signatures corresponding to Si–N and Si–H. However, if N–H functionality was replaced by C–CH3 in N-methylpyrrole, [2+2] and [4+2] cycloaddition products dominated the surface reaction both on Si(100)-(2×1) and on Si(111)-(7×7) [163], as the C–N bond has a very high dissociation barrier compared to that for N–H.
The parallel between pyrrole and N-methylpyrrole suggests that the suppression of the N–H dissociation channel should lead to the cycloaddition processes. Thus, in the absence of the possibility for the dissociative addition available for pyrrole and pyrroline, other heterocycles, such as furan and thiophene, predominantly react with the Si(100)-(2×1) via [4+2] cycloaddition as shown in Fig. 6.23.
A set of detailed studies by Qiao et al. used HREELS to detect the conversion of sp2 carbon into sp3 for both furan [164] and thiophene [165] on Si(100)-(2×1) and also observed peaks at 284.7 and 285.5 eV in the C 1s XPS spectra corresponding to α- and β-carbon atoms, respectively. The singular features at 532.3 eV for O 1s in furan [164] and at 227.4 eV for S 2s in thiophene [165] observed following the chemisorption of these compounds at room temperature agree with the structure proposed for the products of [4+2] cycloaddition reactions for both compounds. Thermal annealing causes further transformation of the adsorbed species including oxygen (or sulfur) migration to the neighboring silicon surface dimer [166–169]. Lee et al. confirmed the [4+2] addition of furan on Si(100)-(2×1) by NEXAFS investigation [170]. Recent STM studies combined with DFT analysis confirmed the presence of the di-σ-bonded structure for thiophene that further converts to more tightly bonded structures even at room temperature [171]. It should be pointed out that on Si(111)-(7×7), where there is a higher propensity for the [4+2] cycloadditions, the aromaticity of furan is also broken. However, in addition to the expected cycloaddition reactions, thermal desorption and HREELS studies also showed that at high coverages, there is a formation of mono–σ-bonded species resting on two adjacent adatoms, that can dimerize via α-carbon coupling [172]. This type of chemical transformation is impossible on Si(100)-(2×1).
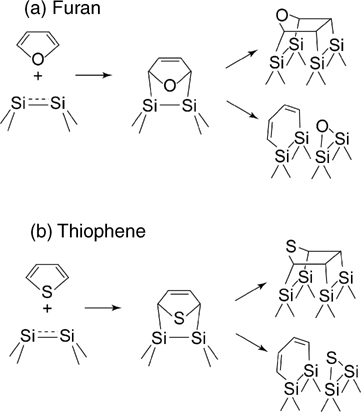
FIGURE 6.23 General reaction pathways of (a) furan and (b) thiophene with Si(100)-(2×1).
It can be mentioned here that the reaction of maleic anhydride could potentially produce an aromatic surface adduct structurally similar to furan; however, a set of detailed studies by the Richardson group employed HREELS, NEXAFS, photoluminescence, STM, and computational investigations to show that this seemingly complex compound reacts exclusively by its C=C function [173–176]. [2+2] cycloaddition pathways dominate at low coverage and involve either one surface silicon dimer or two dimers from the neighboring rows, while the high coverage provides an opportunity for C–H dissociation. Both regimes retain the –CO–O–CO–entity. Nor does cyclopentadiene form a potential aromatic adduct on Si(100)-(2×1), as it initially undergoes almost exclusively a [4+2] cycloaddition [177] similarly to furan and thiophene.
Although the formation of a datively bonded surface intermediate between heteroaromatic compounds and the silicon surface is often considered to be the starting point for many computational investigations, it is sometimes difficult to confirm this state experimentally. However, in case of nitrogen-containing aromatics, it is indeed the case. As was mentioned above, the existence of the N-datively bonded surface intermediate was shown experimentally for nitrozobenzene with the help of infrared spectroscopy and DFT calculations [133]. For pyridine, extensive studies both on Si(100)-(2×1) [178–180] and on Si(111)-(7×7) [181] confirm the existence of the intermediate datively bonded through the nitrogen atom, along with the [4+2] cycloaddition product. The datively bonded adduct undergoes cycloaddition or desorbs if the surface temperature is increased. The [4+2] cycloaddition products were shown theoretically to further convert to more stable configurations involving neighboring silicon dimers on Si(100)-(2×1) [180]. This suggestion was confirmed experimentally by surface differential reflectance spectroscopy studies [182,183] and by a detailed temperature dependent molecularly resolved STM investigation by Miwa et al. [179]. Further heating above room temperature causes pyridine to undergo several complex reactions including dissociation, fragmentation, and dissociative desorption [184].
The analysis by Lu and Lin [169] compared the behavior of pyridine, pyrazine, and s-triazine on Si(100)-(2×1) and placed the chemistry observed in a context of general understanding of chemical transformations of different heterocycles on this surface. Selected surface transformations available for these compounds are shown in Fig. 6.24. The DFT investigation suggests that all three molecules could form datively bonded intermediates via Si–N linkage [185]. In the case of pyridine, as suggested above, [4+2] cycloaddition follows the formation of the datively bonded intermediate. Because of the symmetry of the molecule, this cycloaddition process can either involve only the formation of Si–C bonds or nitrogen atom can participate in the [4+2] scheme, which seems to be the dominant process, as would be expected considering that the reaction starts with the datively bonded intermediate. Compared to pyridine, pyrazine has two nitrogen atoms within a six-membered aromatic structure and its [4+2] cycloaddition that follows the formation of the datively bonded adduct could form either a product involving a single silicon dimer or neighboring silicon dimers (as shown in Fig. 6.24) of the Si(100)-(2×1) surface [185–187] or involve the adatom–rest atom pair of the Si(111)-(7×7) surface [188]. Theoretical studies initially suggested that the [4+2] adduct that forms two Si–C σ bonds would be favorable [169,185]; however, experimental investigations involving HREELS and XPS confirmed binding via para-nitrogens [189]. If instead of para-heteroatoms, two nitrogen atoms are located at meta-positions, as in pyrimi-dine, [4+2] cycloaddition on Si(111)-(7×7) involves a carbon atom and a nitrogen atom in a para-position with respect to it, both reacting with the adatom–rest atom pair [190]. Out of the two possibilities for this [4+2] cycloaddition processes, N bonded to the adatom, and N bonded to the rest atom, the product with the N–Siadatom bond is more stable by more than 7.5 eV and is indeed the most stable of all the products considered. It is likely that the kinetics also favors this cycloaddition pathway since its starting point is the original surface adduct datively bonded via nitrogen to the adatom of the Si(111)-(7×7) surface. Neither pyrimidine nor s-triazine were confirmed to form the datively bonded adducts as intermediates on the way to further transformations; however, for these compounds the dative bonding is weaker than that for pyridine on Si(111)-(7×7) by 4.0 and 21.6 kcal/mol, respectively [190]. Finally, only one possible [4+2] bonding configuration is available for s-triazine because of the symmetry of this molecule. In a set of studies by Bu et al. [191], the behavior of s-triazine on Si(100)-(2×1) was investigated by a combination of HREELS, UPS, XPS, and thermal desorption. Since the asymmetric ring-breathing mode at 145 meV in the HREELS spectra above 200K was not observed, it was suggested that at this temperature s-pyrazine molecules lie flat on this surface. A weakly bound species was found to desorb at 235K from the monolayer, while decomposition was observed by 550K [191]. A more recent study of substituted triazines also combined several analytical techniques and suggested that a saturation coverage of one molecule per two surface silicon dimers of the Si(100)-(2×1) sample can be reached [192]. A derivative of s-triazine, 2,4,6-triphenyl-1,3,5-triazine was also studied on Si(111)-(7×7) by STM and suggested to exhibit nondissociative adsorption [193]. Despite the fact that the specific nature of the bound configuration was disputed by Lu and Lin [169], this microscopy study seems to be consistent with general behavior of triazines on silicon.
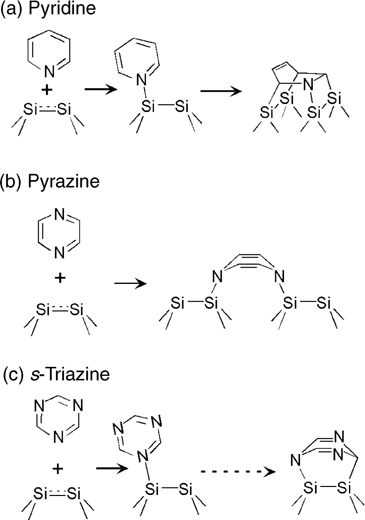
FIGURE 6.24 Reactions of (a) pyridine, (b) pyrazine, and (c) s-triazine with Si(100)-(2×1).
Thus, in six-membered N-containing aromatic heterocycles the aromaticity is broken in a thermodynamic regime, where the most stable surface structures are formed; however, kinetic trapping at low temperatures can be used to deliver a datively bonded aromatic ring to the silicon surface. The comparison of the computational investigations suggests that this kinetic trapping could work best for pyridine [185]. The appearance of the vertically oriented aromatic rings in an STM study of pyridine on a high-index silicon surface, Si(5 5 12)-(2×1) [194,195], confirms the existence of the datively bonded species in this reaction and also suggests that geometric restriction may play a role in stabilizing aromatic systems delivered to the semiconductor surfaces. Finally, it should be noted that Ge(100)-(2×1) substrate that has similar structure and often follows the same reactions with the hydrocarbons as Si(100)-(2×1), reacts with pyridine very differently. In this case the datively bonded state is formed almost exclusively leading to a stable c(4 × 2) pattern observed in STM [196] and consistent with the instability of the two-dimer bridge structures on Ge(100)-(2×1) predicted by DFT [180]. Thus, in the case of germanium, the aromaticity of the pyridine ring is retained at higher temperature compared to that on silicon. In general, datively bound surface adducts are stable on the Ge(100) surface and often determine the outcome of surface reactions, especially for the thermodynamic regime. These reactions are described in much more detail in Chapters 5 and 8 of this book.
Recent computational exploration of the potential use of pyridine on the selectively hydrogen-covered Si(100) surface shows that this approach also stabilizes the datively bonded configuration and pyridine molecules may form continuous lines of aromatic functionalities on this substrate [197]. More detailed description of the organized delivery of aromatic systems onto the hydrogen-precovered surface is presented below.
A recent review by Tao et al. [156] has analyzed the reaction of one more class of aromatic heterocyclic compounds on silicon. If a five-membered aromatic ring contains two different heteroatoms, the effect of the electronic structure on surface adsorption and reactions can be explored systematically. The complete analysis of the electronic effects in the three molecules considered in this review [156] and in the original paper by Tao and Bernasek [59] is based on specific distribution of the molecular electronic density, polarity, geometry, electronegativity of the heteroatoms, and their contribution to the driving of the resulting surface adduct toward the stable “formal” (4n+2) π-electron aromatic systems. The reactions of thiazole, oxazole, and isoxazole were followed by HREELS as a function of temperature and compared to those known for other 5- and 6-membered aromatic systems [59]. The most relevant to the material covered in this chapter is the fact that although isoxazole and oxazole form a mixture of surface products even at 100K, thiazole initially forms a stable datively bonded adduct, which later converts into a [4+2]-type product, opening the possibility of kinetic trapping of an aromatic system similarly to that for pyridine.
6.2.4 Chemistry of Aromatic Compounds on Partially Hydrogen-Covered Silicon Surfaces
As was briefly mentioned in the previous section, surface hydrogen may play a stabilizing role in delivering aromatic groups to semiconductor surfaces. The chemistry of fully hydrogenated surfaces is linked directly to this statement as the reactions on fully hydrogen-covered silicon surfaces most of the time start at the defect sites, which can be simply unoccupied silicon atoms. While the chemistry of hydrogen-covered surfaces has been reviewed extensively before, this section will focus on the information that can be obtained from investigating the behavior of aromatic compounds on hydrogen-covered surfaces where selected hydrogen atoms are removed, creating very specific surface sites with well-defined (and often limited) reactivity.
Since the understanding of the reactivity of the specific silicon surface reactive sites “unblocked” from precovered hydrogen literally has to be performed on the atomic and molecular level, this section will rely on the single crystalline silicon surfaces carefully prepared and studied in vacuum by scanning tunneling microscopy. This technique allows both for the atomic resolution and for the removal of a selected hydrogen atom from the H-covered silicon. This idea, proposed in the late 1990s [198,199], was developed by multiple research groups to become an entirely novel approach to understanding chemical reactions by observing and manipulating single molecules and atoms [198,200–214]. The reactive features produced on silicon surfaces in an unbelievably controlled fashion provided the opportunities to template the reactive sites on the scale ranging from a single silicon dangling bond to lines of empty dimers on Si(100) to objects of nearly macroscopic sizes [15,215], and to ask fundamental questions never posed before.
This atomic template approach is illustrated in Fig. 6.25, where the STM tip was used to selectively remove 1, 2, 3, or 4 surface hydrogen atoms to create the reactive sites of a truly atomic pattern [216].

FIGURE 6.25 Shows the detailed configurations of typical elements of the array of dangling bonds fabricated on a H-terminated Si(100) with a current-voltage pulse of + 3.5 V/4.0 A, 10 ms. The corresponding sketches of each configuration are plotted inside of each figure; a filled circle denotes a hydrogen-terminated Si atom, an unfilled circle denotes a dangling bond. Figure reprinted with permission from Ref. 216. Copyright 2009 Elsevier B.V.
The atomic templating based on creating well-defined surface reactive sites by the removal of hydrogen atoms blocking specific dangling bonds on silicon has been explored to understand binding and electronic structures of many different molecules, including norbornadiene (NBE), copper phthalocyanine (CuPc), and C60 fullerene [217]. For example, in a molecule as complex as CuPc, molecular rotation and charge transfer from the semiconductor substrate have been investigated. Here, we will attempt to summarize some of the rules for manipulating chemical reactions involving aromatic compounds.
As was mentioned above, biphenyl is a very interesting compound with unusual adsorption configuration available on a clean Si(100)-(2×1) surface, with a structure pivoting around one of the phenyl groups at 5K [68,69,73]. This is a perfect structure to investigate on a hydrogen-terminated Si(100) surface, if selected hydrogen atoms can be removed with the help of the STM tip. To determine the size limitations of the adsorption site, the experiments were performed on a template with 1, 2, 3, 4 (two dimers), and more adjacent silicon atoms stripped of surface hydrogen. The room temperature adsorption required at least four surface atoms on two adjacent silicon dimers to be cleared and available for binding [218]. This template, described by the authors as a “molecular mold,” directed the biphenyl binding into a fixed configuration, on a clean Si(100)-(2×1) surface that was achieved with the help of the STM tip. The effects of the hydrogen-templated artificial surface site on the binding of biphenyl are illustrated in Fig. 6.26, where a chronological sequence of events illustrates binding of this molecule onto a template of three silicon surface dimers stripped of hydrogen protection.
The design of molecular wires relies on the templating and assembling of the appropriate compounds in a highly ordered fashion. Sometimes this can be achieved by chemical methods adapted to self-assembly, as described later in this chapter; however, the alternative approach is to design the wires based on the large molecular structures and then “staple” them to the semiconductor surface using appropriate chemistry or an STM tip. Similarly to the concept of using para-hexaphenyl molecules described above [77], other molecules can also be used. The main problem is that the more complex molecules tend to also have a very complex set of chemical interactions with clean semiconductor surfaces. For example, 1,4″-paratriphenyl-dimethylacetone, whose structure is presented in Fig. 6.27 adsorbs on a clean Si(100)-(2×1) in a rather well-defined direction. The adsorption of this molecule on a clean Si(100)-(2×1) surface requires five sequential silicon dimers [210,218]. However, the specific thermal or STM tip-induced chemistry may be very complex, as was suggested in a recent theoretical investigation [219]. Nevertheless, a combination of chemical design, microscopy studies, computational investigation, and possibly selective blocking of the potential reactive sites with hydrogen by creating appropriate “molecular molds” should ultimately lead to well-controlled electronic behavior. The applications may not just be limited to the molecular wires but could also extend to other fields, such as photonics, as a photochromic behavior of a complex cis-1,2-dicyano-1,2-bis(2,4,5-trimethyl-3-thienyl)ethene (CMTE) molecule was also demonstrated on Si(100) with the help of STM [219a].

FIGURE 6.26 Chronological sequence of four filled-state STM topographies (each 50 Å×50 Å, –1.5 V, and 0.5 nA) of the same area of the hydrogenated Si(100)-(2×1) surface. The left-hand column shows (a) the clean hydrogenated surface, (c) the surface after fabricating the molecular mold, (e) a moving biphenyl molecule adsorbed in the mold, and (g) the biphenyl molecule is fixed in the mold. These four stages are shown schematically in the right-hand column (b), (d), (f), and (h). Note in (f), only one of the bistable states of the molecule is shown. The structure of the molecule is shown at the bottom. Figure reprinted with permission from Ref. 218. Copyright 2009 American Institute of Physics.
FIGURE 6.27 Structure of 1,4″-paratriphenyl-dimethylacetone.
The examples above showed that some of the ensemble effect limitations can be studied and understood directly with the help of STM, especially when it is obvious that more than one surface dangling bond is needed for the bonding to occur. However, some reactions could potentially need only one available surface dangling bond. For example, as was discussed above for pyridine, there is only one surface dangling bond involved initially in the formation of the Si–N linkage and later this structure converts into more stable adducts on the Si(100)-(2×1) surface. Thus, it could be expected that the individual dangling bonds available as defects on H-covered Si(100) should readily bind pyridine as well as prevent its conversion by cycloaddition pathways, leaving an intact aromatic ring available for investigations or further modification of this substrate. Miwa et al. posed exactly this question in their STM studies [179]. However, as shown in Fig. 6.28, pyridine did not bind to readily available isolated dangling bonds on H-covered Si(100). Instead, it selectively formed the Si–N linkages with the surface sites where both hydrogen atoms of the same silicon surface dimer were removed. The authors explained this phenomena by the electron-accepting role of the “down” atom of the silicon surface dimer, while an individual surface dangling bond on a silicon surface could not possibly act in this manner [179]. On the other hand, the adsorption of an iron-based porphyrin heme(b) on the H-covered Si(111) studied computationally suggested only weak adsorption on the ideal surface; however, the removal of a single hydrogen atom to create a dangling bond increased the binding energy to 0.543 eVand a pronounced interaction between the dangling bond of the silicon substrate and the orbitals of the iron atom in the porphyrin frame was noted [220].
Starting with an individual dangling bond on otherwise ideally H-terminated silicon, the entire surface could be modified. The complete analysis of the STM manipulation of the molecular systems on silicon is described in detail in Chapter 10 and wet chemical methods to create organic layers on semiconductors are highlighted in Chapters 11 and 12, so the next few paragraphs will only focus on the specifics of the delivery and assembly of aromatic structures by these approaches.
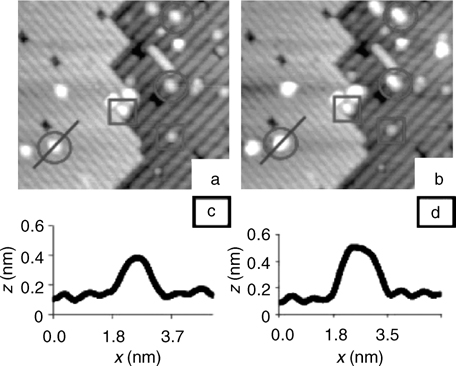
FIGURE 6.28 (a) STM image (17nm×17nm, −2.1 V, 26 pA) of a hydrogen-terminated Si(100) surface with a small number of reactive sites. Examples of clean dimers (circles) and single dangling bonds (squares) are indicated. (b) Shows the same area after dosing the surface with pyridine. The molecules only adsorb to clean dimers and appear as asymmetric protrusions across a single dimer. Cross sections through the clean dimer before (c) and after (d) the reaction are also shown. Figure reprinted with permission from Ref. 179. Copyright 2009 American Chemical Society.
On a well-defined H-terminated Si(111) surface that can be prepared by wet methods and on H-terminated Si(100) that can be prepared by controlled-temperature reaction of hydrogen molecules dissociated on a hot tungsten filament with a clean Si(100)-(2×1) surface, the predominant surface site is a monohydride Si–H. The modification approach suggested in the early 1990s by the Chidsey group used the chain reaction illustrated in Fig. 6.29 [221,222] to build self-assembled monolayers by chain reaction involving an alkene function.
This process can be initiated at a dangling bond that is either naturally available on the surface as a defect or induced by radical precursors or UV exposure [223,224]. The reaction scheme is similar to what is known for organosilane chemistry [225]. What is most relevant to the material covered in this chapter is whether aromatic functionality could be introduced onto a well-defined semiconductor by such an approach. It was indeed the fact, as a stable, cheap, and easy to manipulate and monitor styrene molecule reacted with H-terminated Si(100) or Si(111) as was followed by STM. The studies by Cicero et al. [226] presented in Fig. 6.30 follow the formation of exactly one styrene molecule high islands on a H-terminated Si(111) and confirmed that the growth of each of these islands started at an isolated silicon dangling bond.
The functionality of the aromatic compounds used to grow these islands can be altered proving the robustness of this surface functionalization approach. For example, a monolayer of 4-bromostyrene was also grown and characterized on Si(111) [227,228].
FIGURE 6.29 The mechanism of radical chain reaction of alkenes with a H-terminated Si(111) surface. Figure based on Fig. 7 from Ref. [13].

FIGURE 6.30 Occupied state STM images (215 Å× 130 Å, –2.1V, 44 pA) of an H-terminated Si(111) surface with isolated dangling bonds created by desorption activated with the STM tip (a) before dosing with styrene and (b) after exposure to 12 Langmuirs of styrene. The black dots in (b) mark the positions of the initial dangling bonds, showing that these sites serve to nucleate the growth of styrene islands. The gray scale ranges in the two images are different; the maximum height range in (a) is ~5 Å and ~10 Å in (b). Figure reprinted with permission from Ref. 226. Copyright 2009 American Chemical Society.

FIGURE 6.31 Constant current filled-state STM imaging of a methylstyrene/styrene heterostructure on the H:Si(100) surface. (a) Growth of the first line segment of methylstyrene (8L exposure). The chemically reactive silicon radical that initiated the line growth (not shown) is now located at the end of the CH3-styrene segment (indicated by white arrow). This silicon radical will serve as the initiation site for the growth of the second line segment. (b) Following a 78 L exposure of styrene, the methylstyrene segment in panel (a) has been extended to form a molecular heterostructure. The white wedge indicates the location of the heterointerface. At Vs = –3.0 V, the methylstyrene line segment images higher than the styrene segment. (c) Vs = –2.4 V, the methylstyrene and styrene line segments image with similar height. (d) Vs = –1.8 V, the methylstyrene and styrene line segments reveal different molecular contrast. Images were acquired using a constant tunnel current of 60 pA. Image areas: ~9 × 9 nm2 [229]. Figure reprinted with permission from Ref. 229. Copyright 2009 The American Physical Society.
A brief examination of the chemistry of this growth process suggests that as the styrene molecule starts the reaction at an individual dangling bond, it can only create the required surface-bound Ph–CH2–CH2– group by plucking a hydrogen atom from a neighboring silicon site. However, on Si(111) there are six directions of propagation for such a process so the shapes of the islands created lack specific directionality. On Si(100) covered with monohydride, the directionality of the silicon surface dimer rows implies the possibility of growing molecular lines along the direction of these rows, which is exactly the case. The STM image of a molecular line and a junction produced when the dosing compound is switched from styrene to methylstyrene is presented in Fig. 6.31 and the molecular model is given in Fig. 6.32 [229]. The fact that the compound delivered onto the silicon surface as a molecular line is aromatic triggered a very substantial interest in this approach as the overlap of aromatic π-systems gives this molecular line a possible attribute of a conducting molecular wire [226,229–242]. Different methods of initiation of this reaction, and various measurements relevant to the potential applications of these molecular lines as electron conductors on different substrates have been reported [126,227,238,243–256]. One of the possible initiators, a stable radical species TEMPO, could actually serve both as an initiator of this reaction by removing a single hydrogen atom from H-precovered silicon, and as an inhibitor by passivating available dangling bonds [237,257].
FIGURE 6.32 Model structure of a heterojunction between methylstyrene and styrene chains (each of four molecules) on a hydrogen-passivated silicon cluster representing a dimerized H-terminated Si(001) surface. Carbon atoms are black, silicon is gray, hydrogen is white. Inset: Another view of the molecules and nearby substrate atoms. Figure reprinted with permission from Ref. 229. Copyright 2009 The American Physical Society.
In the context of potential future applications it should be pointed out that the specific electronic structure of the wires produced may be complex and the aromatic rings, especially at the end of the lines could be fluxional [258]. In addition, the silicon substrate plays a very important role in the electron transport phenomena using molecular ethylbenzene wires [259]. One of the plausible pathways to decouple the properties of the silicon substrate from the electronic conductance through the π-stacked aromatics was offered in a theoretical study by Rochefort and Beausoleil [260], who considered π-stacking in a molecular line of 1-hexyl-naphthalene molecules on Si(100)-(2×1). Here, the stack of aromatic molecules is separated from the silicon surface by a hexyl linker; however, experimental study of this specific system may be difficult. Considering all these factors in a combination with designing more functional molecular lines should ultimately lead to the design of a functional device.
The mechanism of the surface chain reaction proposed for styrene proved to be quite robust. In addition to adding a functionalized junction with methylstyrene [229] and building the self-aligned structures from bromostyrene [227], dimethylstyrene was also shown to react with the dangling bond on the Si(100)-(2×1)-H surface similarly to styrene [233]. However, some structurally similar molecules, including the aromatic 4-vinylpyridine did not show any observable line growth [233]. On the other hand, functionalized aromatics such as benzonitrile [248] and benzaldehyde [252] follow the same overall chemistry as styrene on an H-terminated Si(100)-(2×1) surface. In the case of benzaldehyde [252], the chemical functionality actually provides two alternative possibilities for the reaction leading to the formation of the surface species of a general formula Ph–O–CH2–. As illustrated in Fig. 6.33 for an aldehyde of a general formula R–C=O, the attachment to an individual dangling bond A on an ideally H-terminated Si(100)-(2×1) surface leads to the formation of intermediate C, which in turn can abstract a hydrogen atom from the same dimer (F-G) or from the neighboring dimer (E-D), potentially causing two different mechanisms for the growth of the molecular chain, as depicted in Fig. 6.34 [252]. The STM studies of benzaldehyde presented in Fig. 6.35 show the existence of both single and double lines, including the transformation of a single line into a double line, confirming the possibility of these mechanisms [252].
FIGURE 6.33 Reaction mechanisms for the self-directed growth of aldehyde lines on H–Si (100)-(2×1). For benzaldehyde, R=C6H5, and for acetaldehyde, R=CH3. The calculated C···H distances in the transition-state structures are 1.65–1.70 Å. Figure reprinted with permission from Ref. 252. Copyright 2009 American Chemical Society.
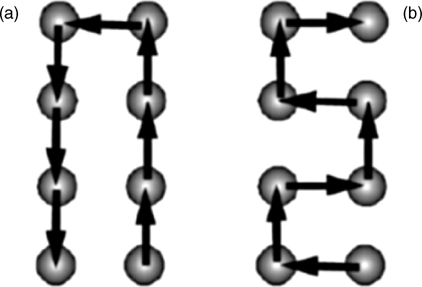
FIGURE 6.34 Cartoon representing (a) antiparallel and (b) square wave growth mechanisms for the formation of double lines. Horizontal pairs of circles represent a silicon dimer on the H–Si(100)-(2×1) surface. The tail of the arrow indicates the adsorption site for the aldehyde, while the head of the arrow indicates the site of hydrogen abstraction. Figure reprinted with permission from Ref. 252. Copyright 2009 American Chemical Society.

FIGURE 6.35 STM images demonstrating the mechanisms for aldehyde line growth. (a) Benzaldehyde double line that has a dangling bond at its end extends its growth (b) via the square wave mechanism upon additional exposure to benzaldehyde. (c) Nanostructure composed of double and single line portions with a dangling bond at the end of the single line. Further exposure to benzaldehyde leads to the formation of a complete double line through the antiparallel growth mechanism (d). Wedges indicate the location of the dangling bonds. STM images: 12 nm × 12 nm, –3.5 V, 0.1 nA. Figure reprinted with permission from Ref. 252. Copyright 2009 American Chemical Society.
The continuing STM studies proved that the chain reaction of H-terminated Si(100) with styrene can be reversed at elevated temperatures [261] and also explored the possibility of cross–linking neighboring silicon dimer rows with chemical functionalities [233,262,263]; however, the linking with aromatic compounds to attempt to bridge the neighboring rows of silicon dimers electronically has not been attempted thus far. To conclude this section, it can be summarized that despite a very rich chemistry for the aromatic molecules on semiconductor surfaces, decreasing the ensemble of surface sites available for the interaction of the aromatics with clean semiconductor surfaces helps to retain aromaticity in surface functionalization processes, providing the means to use the aromatic groups both as stable linkers and as building blocks for molecular electronic structures and future devices.
6.2.5 Delivery of Aromatic Groups onto a Fully Hydrogen Covered Silicon Surface
6.2.5.1 Hydrosilylation
Chapters 11 and 12 will describe all the subtle points of using wet chemistry methods for hydrosilylation of H-terminated silicon and germanium surfaces and for chemical modification of the Cl-terminated surfaces that can be prepared starting with H-terminated substrates. Because of stability, wide variety of produced functionalities, cheapness and attractive electronic properties, aromatic linkers are often used in these reactions. Other chapters in this book will delineate the mechanistic details of these general processes. Here it will be sufficient to say that in all those reactions aryl groups are delivered to the surface intact and do not participate in surface bonding. They are mostly used as stable linkers. The potential to use these linkers for applications other than just the para-position functionality does exist. However, it should be pointed out that most of the time the monolayers created by these methods, despite making the silicon surface very resistive to oxidation in ambient conditions, do not form dense monolayers. Coverages of only about 20–30% are found to be reached at full saturation, meaning that there are remaining Si–H or Si–Cl species available for further reactions. Nevertheless, recent advances in alkylation procedures can safely deliver phenyl- [264] and benzyl- [265] groups by chlorination/alkylation protocol, para-functionalized aryls in diazonium reactions starting with tetrafluoroborates [266], and sterically hindered aromatics also by diazonium chemistry [267]. A photoinduced reaction of Cl-terminated layers on Si(111) could be performed with benzonitrile [268]. However, in addition to the fact that not every reactive surface site can be modified with a desired group, it should also be pointed out that the need to use solvents in the wet chemistry preparation schemes often plays a very substantial role in determining surface modification reaction. For example, silicon hydrosilylation of both H-terminated Si(111) and Si(100) with the use of Spier catalyst, H2[PtCl6], leads exclusively to the alcohol solvent grafting instead of the intended attachment of the complex “indene ligand” (3-[2′ (1H-inden-3″-yl)ethyl]-5-(4″′-vinylphenyl)-1H-indene) containing aromatic moieties. Only UV light initiation and only on the H-terminated Si(111) surface produced the target result [269].
6.2.5.2 Cyclocondensation
Despite being commonly used in homogenous chemistry, cyclocondensation processes in the chemistry of H-terminated silicon surfaces, where the surface provides the hydrogen for the elimination process, have only been discovered very recently. As shown in Fig. 6.36, dehydrative cyclocondensation can deliver a nitroso–fragment onto an H-terminated silicon surface. A detailed set of studies compared the reaction of the nitrozobenzene with H-terminated Si(100) and Si(111) and also with the reaction of the nitrozobenzene on a clean Si(100)-(2×1) in vacuum (used as a spectroscopic marker) to confirm that this reaction occurs with a retention of the aromatic ring. A set of infrared spectroscopy experiments proved the formation of the aromatic adduct on a surface following the reaction and were also used to follow the reduction of the intensity of the Si–H stretching vibration around 2100 cm–1 [18]. In addition, as shown in Fig. 6.37, XPS spectra for a variety of possible surface reactions were analyzed with the help of computational methods [18,270]. Despite a variety of possible reactions with different reactive sites on Si(100) and Si(111) surfaces terminated with hydrogen, these studies suggest that the primary product of the dehydrative cyclocondensation process is nitrosoadduct (E, H, or K in Fig. 6.37) [18]. Further heating of the silicon substrate past the surface modification temperature may drive the oxygen of the nitroso-group subsurface [133] to form a phenylnitrene adduct (C, F, I, L in Fig. 6.37).
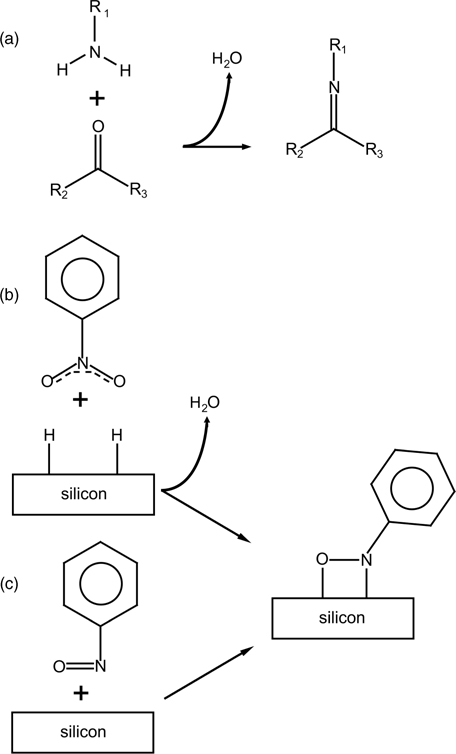
FIGURE 6.36 Schematic representation of (a) a condensation reaction; (b) the cyclocondensation reaction of nitrobenzene with hydrogen-terminated silicon, and (c) the cycloaddition reaction of nitrosobenzene with Si(100)-(2×1). Figure reprinted with permission from Ref. 18. Copyright © 2009 American Chemical Society.
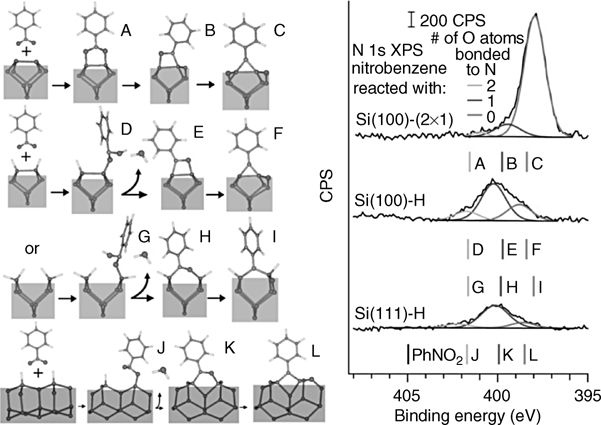
FIGURE 6.37 Comparison of the 1,3-cycloaddition reaction of nitrobenzene with a Si(100)-(2×1) surface with the condensation reactions of nitrobenzene with an ideal monohydride-terminated Si(100) surface, a dihydride-terminated Si(100) surface, and an ideal monohydride-terminated Si(111) surface, descending order on the left. The ex situ high-resolution N 1s XPS spectra, with the predicted N 1s binding energy of the corresponding products, after these reactions occur, and nitrobenzene itself, are presented on the right of the respective reaction. Computations were preformed at the B3LYP/6–311+G(d,p) level of theory. Organic fragments are represented as follows: black, oxygen; dark gray, nitrogen; light gray, carbon; white, hydrogen. The nitrobenzene reaction with the Si(100)-(2×1) surface was performed in ultrahigh vacuum as described previously and the XPS spectrum was collected ex situ. The condensation reaction was performed under nitrogen ex situ. On the schematic, hydrogen atoms terminating the silicon clusters are omitted for clarity. Figure reprinted with permission from Ref. 18. Copyright © 2009 American Chemical Society.
Thermal control is one of the most attractive aspects of this reaction. It is slow at room temperature but can be completed within hours at 200°C. A computational investigation was used to follow the possible reaction pathways for the dehydrative cyclocondensation scheme, and discovered a substantial barrier for the first step of surface modification. The opportunity to use thermal control for surface coverage dependence is illustrated in Fig. 6.38, where the decrease of the intensity of the infrared absorption band corresponding to the Si–H stretch shows that partial coverage can be obtained by varying reaction time and temperature [18].
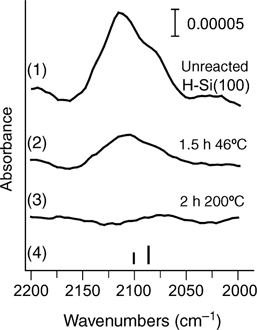
FIGURE 6.38 (1) Infrared spectrum of the Si–H stretch region of a hydrogen-terminated Si(100) surface. (2) Infrared spectrum of the Si–H stretch region of a hydrogen-terminated Si(100) surface after a 1.5 h reaction with nitrobenzene at 46°C. (3) Infrared spectrum of the Si–H stretch region of a hydrogen-terminated Si(100) surface after 2 h reaction with nitrobenzene at 200°C. (4) Predicted infrared spectrum of the Si–H stretch region of structure H. Figure reprinted with permission from Ref. 18. Copyright © 2009 American Chemical Society.
The use of this dehydrative cyclocondensation technique is amenable for silicon surface functionalization in a controlled manner but the current model also delivers an additional surface oxygen atom. To combat this process, which can be detrimental to the interface formed during the reaction, we have also attempted to use nitrosobenzene as a starting point; however, for the moment, alternative reaction pathways seem to preclude us from creating an oxygen-free interface [271].
6.2.6 Delivery of Aromatic Compounds onto Protected Silicon Substrates
In some cases, covalent binding of aromatic molecules delivered to the semiconductor surface may be undesired. In fact, a chemically inert layer can be employed to prevent this chemisorption. For example, hydrogen passivation may provide this buffer zone with limited chemical interaction, as was suggested for the iron-based porphyrin described above [220]. Of course, in that case the presence of a surface dangling bond at a defect site was found to increase the binding energy substantially. In fact, even an ideally H-terminated silicon surface may bind aromatic compounds very strongly via dispersion interactions, as was found for pentacene [272]. The covalent bonding might also occur by an unintended catalysis, as was proposed in a computational study of phenylacetylene reaction with hydrogenated silicon surfaces in the presence of Lewis acids [273].
In order to avoid these interactions, the silicon surface could be premodified before introducing the aromatic functionality. For example, Wakatsuchi et al. used the reaction of 1,4-cyclohexadiene with a clean Si(100)-(2×1) surface to produce a monolayer that served as a buffer zone to prevent the chemisorption of the subsequently used pentacene, which was allowed to self-assemble in the target surface structures as a result of this modification [274]. Metal passivation of the silicon surface has been demonstrated to promote the diffusion of perylene tetracarboxylic dianhydride (PTCDA) and tetraaminobenzene (TAB) on the Ag/Si (111)-(√3 × √3) R30° that form stable hydrogen-bonded structures [275] suggesting that the choice of metal may not only promote the chemical interaction between aromatic molecules and silicon, as was the case for Na-promoted adsorption of benzoic acid [92,93,102], but also block the chemisorption entirely.
Alternatively, the strong and well-defined bonding on a premodified silicon surface may be desired as the building of molecular structures extends into the 3rd dimension. Some of the building blocks used in this modification approach are based on aromatic systems. For example, Bitzer and Richardson [276,277] suggested to use the initial surface reaction of 1,4-phenylene diamine followed by imide coupling to build a 3D network in a controllable layer-by-layer method [276,278]. This approach, bearing close resemblance to the atomic layer deposition chemistry, can be termed molecular layer deposition and is currently being developed for multiple types of reactions. The coupling of amino–terminated monolayers on Si(111) with benzaldehyde was recently used by Eves et al. [279]. Azide reactions with acetylene-terminated monolayers on Si(100) [280] and the attachment of branched substituents to the organic layer on an ester-terminated Si(111) surface [281] have also been performed. More elaborate growth strategies involve covalently linked multiporphyrin-imide architectures on Si(100), as reported by Jiao et al. [282]. One could hope that inclusion of aromatic functionalities into the 3D building approaches will introduce something more than just relatively inert linker into these novel structures, but also bring the electronic structure of these functionalities into play for designing materials with unusual electronic, photochemical, and photochromic properties.
6.3 SUMMARY
This chapter focused on the role of aromaticity as a factor in determining surface reactions on group IV semiconductor surfaces. The retention of aromaticity and the drive toward the formation of an aromatic adduct as a result of chemical reactions leading to surface functionalization most of the time govern the chemical processes in these systems. Thus, the chemistry that has been previously discovered for other organic compounds on silicon can often be applied (although with some degree of caution) to compounds containing aromatic functionality. The chemistry of silicon, germanium, and diamond surfaces can be utilized for these purposes, but while some reactions can be directly replicated on all these substrates, many chemical transformations will depend drastically on the specific surface and specific element. The aromatic groups attached to the semiconductor surface may further serve as the molecular electronic components, structures, and devices, which makes the in-depth study of this field ever so important.
ACKNOWLEDGMENTS
AVT acknowledges financial support from the National Science Foundation (CHE-313803, CHE-0415979, and CHE-1057374) and from the Donors of the American Chemical Society Petroleum Research Fund for making this work possible. I greatly appreciate the help of the following researchers who graciously provided the permission to use their figures in this review: Maria Pia Casaletto, Gérald Dujardin, Göran Hansson, Yukio Hasegawa, Robert J. Hamers, Mark Hersam, Heon Kang, Gregory P. Lopinski, Andrew J. Mayne, Robert A. Wolkow, Qin Xu.
REFERENCES
1. Schleyer, P. v. R. Aromaticity (Editorial), Chem. Rev. 2001, 101, 1115.
2. Waltenburg, H. N.; Yates, J. T. Jr., Chem. Rev. 1995, 95, 1589.
3. Yoshinobu, J.; Tanaka, S.; Nishijima, M. Jpn. J. Appl. Phys. 1993, 32, 1171.
4. Jung, Y.; Shao, Y.; Gordon, M. S.; Doren, D. J.; Head-Gordon, M. J. Chem. Phys. 2003, 119, 10917.
5. Ono, M.; Kamoshida, A.; Matsuura, N.; Ishikawa, E.; Eguchi, T.; Hasegawa, Y. Phys. Rev. B 2003, 67, 201306.
6. Yoshinobu, J. Prog. Surf. Sci. 2004, 77, 37.
7. Leftwich, T. R.; Teplyakov, A. V. Surf. Sci. Rep. 2008, 63, 1.
8. Tao, F.; Xu, G. Q. Acc. Chem. Res. 2004, 37, 882.
9. Razado, I. C.; Zhang, H. M.; Uhrberg, R. I. G.; Hansson, G. V. Phys. Rev. B 2005, 71, 235411.
10. Higashi, G. S.; Chabal, Y. J.; Trucks, G. W.; Raghavachari, K. Appl. Phys. Lett. 1990, 56, 656.
11. Wade, C. P.; Chidsey, C. E. D. Appl. Phys. Lett. 1998, 71, 1679.
12. Allongue, P.; de Villeneuve, C. H.; Morin, S.; Boukerroub, R.; Wayner, D. D. M. Electrochim. Acta 2000, 45, 4591.
13. Wayner, D. D. M.; Wolkow, R. A. J. Chem. Soc., Perkin Trans. 2002, 2, 23.
14. Baluch, A. S.; Guisinger, N. P.; Basu, R.; Foley, E. T.; Hersam, M. C. J. Vac. Sci. Technol. A 2004, 22, L1.
15. Mayne, A. J.; Riedel, D.; Comtet, G.; Dujardin, G. Prog. Surf. Sci. 2006, 81, 1.
16. Faggin, M. F.; Green, S. K.; Clark, I. T.; Queeney, K. T.; Hines, M. A. J. Am. Chem. Soc. 2006, 128, 11455.
17. Filler, M. A.; Bent, S. F. Prog. Surf. Sci. 2003, 73, 1.
18. Leftwich, T. R.; Madachik, M. R.; Teplyakov, A. V. J. Am. Chem. Soc. 2008, 130, 16216.
19. Morrison, R. T.; Boyd, R. N. Organic Chemistry. Boston, Allyn and Bacon, 1959.
20. Wade, L. G., Jr., Organic Chemistry. Prentice-Hall, Inc., NJ, 1995.
21. Wolkow, R. A. Annu. Rev. Phys. Chem. 1999, 50, 413.
22. Taguchi, Y.; Fujisawa, M.; Takaoka, T.; Okada, T.; Nishijima, M. J. Chem. Phys. 1991, 95, 6870.
23. Borovsky, B.; Krueger, M.; Ganz, E. Phys. Rev. B 1998, 57, R4269.
24. Jeong, H. D.; Ryu, S.; Lee, Y. S.; Kim, S. Surf. Sci. 1995, 344, L1226.
25. Lopinski, G. P.; Fortier, T. M.; Moffatt, D. J.; Wolkow, R. A. J. Vac. Sci. Technol. A 1998, 16, 1037.
26. Lopinski, G. P.; Moffatt, D. J.; Wolkow, R. A. Chem. Phys. Lett. 1998, 282, 305.
27. Wolkow, R. A.; Lopinski, G. P.; Moffatt, D. J. Surf. Sci. 1998, 416, L1107.
28. Gokhale, S.; Trischberger, P.; Menzel, D.; Widdra, W.; Dröge, H.; Steinrück, H.-P.; Birkenheuer, U.; Gutdeutsch, U.; Rösch, N. J. Chem. Phys. 1998, 108, 5554.
29. Birkenheuer, U.; Gutdeutsch, U.; Rösch, N. Surf. Sci. 1998, 409, 213.
30. Kong, M. J.; Teplyakov, A. V.; Lyubovitsky, J. G.; Bent, S. F. Surf. Sci. 1998, 411, 286.
31. Silvestrelli, P. L.; Ancilotto, F.; Toigo, F. Phys. Rev. B 2000, 62, 1596.
32. Witkowski, N.; Hennies, F.; Pietzsch, A.; Mattsson, S.; Fiohlisch, A.; Wurth, W. Phys. Rev. B 2003, 68, 115408.
33. Shimomura, M.; Munakata, M.; Honma, K.; Widstrand, S. M.; Johansson, L.; Abukawa, T.; Kono, S. Surf. Rev. Lett. 2003, 10, 499.
34. Witkowski, N.; Pluchery, O.; Borensztein, Y. Phys. Rev. B 2005, 72, 075354.
35. Jung, Y.; Gordon, M. S. J. Am. Chem. Soc. 2005, 127, 3131.
36. Besley, N. A.; Blundy, A. J. J. Phys. Chem. B 2006, 110, 1701.
37. Okamura, K.; Ishii, H.; Kimura, Y.; Niwano, M. Surf. Sci. 2005, 576, 45.
38. Kasaya, M.; Tabata, H.; Kawai, T. Surf. Sci. 1998, 400, 367.
39. Kasaya, M.; Tabata, H.; Kawai, T. Surf. Sci. 1998, 406, 302.
40. Hughes, G.; Roche, J.; Carty, D.; Cafolla, T.; Smith, K. E. J. Vac. Sci. Technol. B 2002, 20, 1620.
41. Weidkamp, K. P.; Hacker, C. A.; Schwartz, M. P.; Cao, X.; Tromp, R. M.; Hamers, R. J. J. Phys. Chem. B 2003, 107, 11142.
42. Tejima, M.; Kita, K.; Kyuno, K.; Toriumi, A. Appl. Phys. Lett. 2004, 85, 3746.
43. Okamura, K.; Hosoi, Y.; Kimura, Y.; Ishii, H.; Niwano, M. Appl. Surf. Sci. 2004, 237, 439.
44. Choudhary, D.; Clancy, P.; Bowler, D. R. Surf. Sci. 2005, 578, 20.
45. Suzuki, T.; Sorescu, D. C.; Yates, J. T., Jr., Surf. Sci. 2006, 600, 5092.
46. Lee, H.-K.; Han, J.-H.; Kim, K.-J.; Kang, T.-H.; Kim, B. Surf. Sci. 2007, 601, 1456.
47. Rada, T.; Chen, Q.; Richardson, N. V. J. Phys.: Condens. Matter 2003, 15, S2749.
48. Rada, T.; Chen, Q.; Richardson, N. Phys. Status Solidi B 2004, 241, 2353.
49. Preuss, M.; Miotto, R.; Bechstedt, F.; Rada, T.; Richardson, N. V.; Schmidt, W. G. Phys. Rev. B 2006, 74, 115402/1.
50. Suzuki, T.; Sorescu, D. C.; Jordan, K. D.; Yates, J. T., Jr., J. Chem. Phys. 2006, 124, 224708/1.
51. Suzuki, T.; Sorescu, D. C.; Jordan, K. D.; Levy, J.; Yates, J. T., Jr., J. Chem. Phys. 2006, 124, 054701/1.
52. Suzuki, T.; Levy, J.; Yates, J. T., Jr., Nano Lett. 2006, 6, 138.
53. Hughes, G.; Carty, D.; Cafolla, A. A. Surf. Sci. 2005, 582, 90.
54. Weidkamp, K. P.; Tromp, R. M.; Hamers, R. J. J. Phys. Chem. C 2007, 111, 16489.
55. Ruiz, R.; Choudhary, D.; Nickel, B.; Toccoli, T.; Chang, K.-C.; Mayer, A. C.; Clancy, P.; Blakely, J. M.; Headrick, R. L.; Iannotta, S.; Malliaras, G. G. Chem. Mater. 2004, 16, 4497.
56. Nunzi, F.; Sgamellotti, A.; Re, N. J. Phys. Chem. C 2007, 111, 1392.
57. Fink, A.; Menzel, D.; Widdra, W. J. Phys. Chem. B 2001, 105, 3828.
58. Cho, J. H.; Kim, K. S.; Morikawa, Y. J. Chem. Phys. 2006, 24, 024716.
59. Tao, F.; Bernasek, S. L. Am. Chem. Soc. 2007, 129, 4815.
60. Ma, Z.; Zaera, F. Surf. Sci. Rep. 2006, 61, 229.
61. Lu, X.; Wang, X. L.; Yuan, Q. H.; Zhang, Q. J. Am. Chem. Soc. 2003, 125, 7923.
62. Petsalakis, I. D.; Polanyi, J. C.; Theodorakopoulos, G. Surf. Sci. 2003, 544, 162.
63. Yong, K. S.; Zhang, Y. P.; Yang, S.-W.; Xu, G. Q. Surf. Sci. 2008, 602, 1921.
64. Guan, D. D.; Mao, H. Y.; Chen, M. L.; Dou, W. D.; Song, F.; Zhang, H. J.; Li, H. Y.; He, P.; Chen, H. Z.; Bao, S. N. J. Chem. Phys. 2009, 130, 174712.
65. Schedel, T.; Frank, K.-H.; Karlsson, U.; Koch, E. E. Vacuum 1990, 41, 652.
66. Yong, K. S.; Zhang, Y. P.; Yang, S.-W.; Wu, P.; Xu, G. Q. J. Phys. Chem. A 2007, 111, 12266.
67. Yong, K. S.; Zhang, Y. P.; Yang, S.-W.; Wu, P.; Xu, G. Q. J. Phys. Chem. C 2007, 111, 4285.
68. Mayne, A. J.; Lastapis, M.; Baffou, G.; Soukiassian, L.; Comtet, G.; Hellner, L.; Dujardin, G. Phys. Rev. B 2004, 69, 045409.
69. Cranney, M.; Mayne, A. J.; Comtet, G.; Dujardin, G. Surf. Sci. 2005, 593, 139.
70. Mamatkulov, M.; Stauffer, L.; Minot, C.; Sonnet, P. Phys. Rev. B 2006, 73, 035321.
71. Dubois, M.; Delerue, C.; Rubio, A. Phys. Rev. B 2007, 75, 041302(R).
72. Martin, M.; Lastapis, M.; Riedel, D.; Dujardin, G.; Mamatkulov, M.; Stauffer, L.; Sonnet, P. Phys. Rev. Lett. 2006, 97, 216103.
73. Lastapis, M.; Martin, M.; Riedel, D.; Comtet, G.; Hellner, L.; Dujardin, G. Science 2005 308, 1000.
74. Cranney, M.; Comtet, G.; Dujardin, G.; Kim, J. W.; Kampen, T. U.; Horn, K. Phys. Rev. B 2007, 76, 075324.
75. Lima, C. P.; Miwa, R. H.; Schmidt, T. M. Surf. Sci. 2008, 602, 2634.
76. Riedel, D.; Bocquet, M.-L.; Lesnard, H.; Lastapis, M.; Lorente, N.; Sonnet, P.; Dujardin, G. J. Am. Chem. Soc. 2009, 131, 7344.
77. Cranney, M.; Chalopin, Y.; Mayne, A. J.; Dujardin, G. Appl. Phys. A 2009, 94, 767.
78. Coulter, S. K.; Hovis, J. S.; Ellison, M. D.; Hamers, R. J. J. Vac. Sci. Technol. A 2000, 18, 1965.
79. Costanzo, F.; Sbraccia, C.; Silvestrelli, P. L.; Ancilotto, F. J. Phys. Chem. B 2003, 107, 10209.
80. Costanzo, F.; Sbraccia, C.; Silvestrelli, P. L.; Ancilotto, F. Surf. Sci. 2004, 566, 971.
81. Li, Q.; He, Z. H.; Zhou, X. J.; Yang, X.; Leung, K. T. Surf. Sci. 2004, 560, 191.
82. Carbone, M.; Piancastelli, M. N.; Casaletto, M. P.; Zanoni, R.; Comtet, G.; Dujardin, G.; Hellner, L. Surf. Sci. 2002, 498, 186.
83. Tomimoto, H.; Takehara, T.; Fukawa, K.; Sumii, R.; Sekitani, T.; Tanaka, K. Surf. Sci. 2003, 526, 341.
84. Tomimoto, H.; Sekitani, T.; Sumii, R.; Sako, E. O.; Wada, S.-I.; Tanaka, K. Surf. Sci. 2004, 566, 664.
85. Macpherson, C. D.; Leung, K. T. Surf. Sci. 1995, 326, 141.
86. Casaletto, M. P.; Carbone, M.; Piancastelli, M. N.; Horn, K.; Weiss, K.; Zanoni, R. Surf. Sci. 2005, 582, 42.
87. Coulter, S. K.; Schwartz, M. P.; Hamers, R. J. J. Phys. Chem. B 2001, 105, 3079.
88. Kugler, T.; Thibaut, U.; Abraham, M.; Folkers, G.; Göpel, W. Surf. Sci. 1992, 260, 64.
89. Cao, X.; Coulter, S. K.; Ellison, M. D.; Liu, H.; Liu, J.; Hamers, R. J. J. Phys. Chem. B 2001, 105, 3759.
90. Bitzer, T.; Alkunshalie, T.; Richardson, N. V. Surf. Sci. 1996, 368, 202.
91. Rummel, R. M.; Ziegler, C. Surf. Sci. 1998, 418, 303.
92. Bitzer, T.; Richardson, N. V. Surf. Sci. 1999, 427–428, 369.
93. Lopez, A.; Bitzer, T.; Heller, T.; Richardson, N. V. Surf. Sci. 2001, 480, 65.
94. Mui, C.; Han, J. H.; Wang, G. T.; Musgrave, C. B.; Bent, S. F. J. Am. Chem. Soc. 2002, 124, 4027.
95. Kugler, T.; Ziegler, C.; Göpel, W. Mater. Sci. Eng. B 1996, 37, 112.
96. Jang, S. H.; Jeong, S.; Hahn, J. R. J. Chem. Phys. 2009, 130, 234703.
97. Carbone, M.; Meloni, S.; Caminiti, R. Phys. Rev. B 2007, 76, 085332.
98. Carbone, M.; Cazzato, P.; Caminiti, R. Surf. Sci. 2009, 603, 611.
99. Carbone, M.; Piancastelli, M. N.; Zanoni, R.; Comtet, G.; Dujardin, G.; Hellner, L. Surf. Sci. 1999, 419, 114.
100. Zheng, F.; McChesney, J. L.; Liu, X.; Himpsel, F. J. Phys. Rev. B 2006, 73, 205315.
101. Baik, J.; Park, J.; Kim, M.; Ahn, J. R.; Park, C.-Y.; An, K.-S.; Hwang, C.-C.; Hwang, H.-N.; Kim, B. J. Korean Phys. Soc. 2007, 50, 690.
102. Bitzer, T.; Richardson, N. V.; Reiss, S.; Wuhn, M.; Woll, C. Surf. Sci. 2000, 458, 173.
103. Naumkin, F. Y.; Polanyi, J. C.; Rogers, D. Surf. Sci. 2003, 547, 335.
104. Naumkin, F. Y.; Polanyi, J. C.; Rogers, D.; Hofer, W.; Fisher, A. Surf. Sci. 2003, 547, 324.
105. Zhou, X. J.; Leung, K. T. J. Phys. Chem. B 2006, 110, 9601.
106. Zhou, X. J.; Leung, K. T. Surf. Sci. 2006, 600, 3285.
107. Jiang, G. P.; Polanyi, J. C.; Rogers, D. Surf. Sci. 2003, 544, 147.
108. Matta, C. F.; Polanyi, J. C. Phil. Trans. Royal Soc. A 2004, 362, 1185.
109. Li, Z. H.; Li, Y. C.; Wang, W. N.; Cao, Y.; Fan, K. N. J. Phys. Chem. B 2006, 108, 14049.
110. Sloan, P. A.; Palmer, R. E. J. Phys.: Condens. Matter 2006, 18, S1873.
111. Lu, X.; Polanyi, J. C.; Yang, J. Nano Lett. 2006, 6, 809.
112. Dobrin, S.; Harikumar, K. R.; Polanyi, J. C. Surf. Sci. 2004, 561, 11.
113. Harikumar, K. R.; Petsalakis, I. D.; Polanyi, J. C.; Theodorakopoulos, G. Surf. Sci. 2004, 572, 162.
114. Dobrin, S. J. Phys. Chem. B 2005, 109, 22976.
115. Petsalakis, I. D.; Polanyi, J. C.; Theodorakopoulos, G. Israel J. Chem. 2005, 45, 111.
116. Dobrin, S.; Harikumar, K. R.; Matta, C. F.; Polanyi, J. C. Surf. Sci. 2005, 580, 39.
117. Schwartz, M. P.; Ellison, M. D.; Coulter, S. K.; Hovis, J. S.; Hamers, R. J. J. Am. Chem. Soc. 2000, 122, 8529.
118. Hamers, R. J.; Hovis, J. S.; Coulter, S. K.; Ellison, M. D.; Padowitz, D. F. Jpn. J. Appl. Phys. 2000, 39, 4366.
119. Calzolari, A.; Ruini, A.; Molinari, E.; Caldas, M. J. Phys. Rev. B 2006, 73, 125420.
120. Wang, Y.; Ma, J.; Inagaki, S.; Pei, Y. J. Phys. Chem. B 2005, 109, 5199.
121. Dixon, C. E.; Liu, H. W.; Vander Kant, C. M.; Baines, K. M. Organometallics 1996, 15, 5701.
122. Dixon, C. E.; Baines, K. M. Phosphorus, Sulfur Silicon Relat. Elem. 1997, 125, 123.
123. Iwamoto, T.; Sakurai, H.; Kira, M. Bull. Chem. Soc. Jpn. 1998, 71, 2741.
124. Hwang, Y. J.; Kim, A.; Hwang, E. Y.; Kim, S. J. Am. Chem. Soc. 2005, 127, 5016.
125. Zhang, Y. P.; Yang, L.; Lai, Y. H.; Xu, G. Q.; Wang, X. S. Appl. Phys. Lett. 2004, 84, 401.
126. Basu, R.; Tovar, J. D.; Hersam, M. C. J. Vac. Sci. Technol. B 2005, 23, 1785.
127. Tao, F.; Wang, Z. H.; Lai, Y. H.; Xu, G. Q. J. Am. Chem. Soc. 2003, 125, 6687.
128. Tao, F.; Qiao, M. H.; Li, Z. H.; Yang, L.; Dai, Y. J.; Huang, H. G.; Xu, G. Q. Phys. Rev. B 2003, 67, 115334.
129. Kim, K.-Y.; Song, B.-K.; Jeong, S.; Kang, H. J. Phys. Chem. B 2003, 107, 11987.
130. Pluchery, O.; Coustel, R.; Witkowski, N.; Borensztein, Y. J. Phys. Chem. B 2006, 110, 22635.
131. Huang, H. G.; Zhang, Y. P.; Cai, Y. H.; Huang, J. Y.; Yong, K. S.; Xu, G. Q. J. Chem. Phys. 2005, 123, 104702-1- 104702–6.
132. Huang, H. G.; Huang, J. Y.; Cai, Y. H.; Xu, G. Q. Chem. Phys. Lett. 2005, 414, 143.
133. Perrine, K. A.; Leftwich, T. R.; Weiland, C. R.; Madachik, M.; Opila, R. L.; Teplyakov, A. V. J. Phys. Chem. C 2009, 113, 6643.
134. Tao, F.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2002, 106, 3557.
135. Qu, Y.-Q.; Han, K.-L. J. Phys. Chem. B 2004, 108, 8305.
136. Rangan, S.; Bournel, F.; Gallet, J. J.; Kubsky, S.; Le Guen, K.; Dufour, G.; Rochet, F.; Sirotti, F.; Carniato, S.; Ilakovac, V. Phys. Rev. B 2005, 71, 165319.
137. Tao, F.; Wang, Z. H.; Chen, X. F.; Xu, G. Q. Phys. Rev. B 2002, 65, 115311.
138. Schwartz, M. P.; Halter, R. J.; McMahon, R. J.; Hamers, R. J. J. Phys. Chem. B 2003, 107, 224.
139. Nunzi, F.; Sgamellotti, A.; Re, N. J. Phys. Chem. B 2004, 108, 10881.
140. Laguerre, M.; Felix, G.; Dunogues, J.; Calas, R. J. Org. Chem. 1979, 44, 4275.
141. Barriocanal, J. A.; Doren, D. J. J. Phys. Chem. B 2000, 104, 12269.
142. Barriocanal, J. A.; Doren, D. J. J. Vac. Sci. Technol. A 2000, 18, 1959.
143. Bocharov, S.; Mathauser, A. T.; Teplyakov, A. V. J. Phys. Chem. B 2003, 107, 7776.
144. Eng, J., Jr.,; Hubner, I. A.; Barriocanal, J.; Opila, R. L.; Doren, D. J. J. Appl. Phys. 2004, 95, 1963.
145. Bocharov, S.; Teplyakov, A. V. Surf. Sci. 2004, 573, 403.
146. Méndez De Leo, L. P.; Teplyakov, A. V. J. Phys. Chem. B 2006, 110, 6899.
147. Ellison, M. D.; Hamers, R. J. J. Phys. Chem. B 1999, 103, 6243.
148. Schaumann, E.; Bauch, H.-G.; Sieveking, S.; Adiwidjaja, G. Chem. Ber. 1982, 115, 3340.
149. Reddy, C. K.; Reddy, P. S. N.; Ratnam, C. V. Ind. J. Chem. 1987, 26B, 882.
150. Kim, A.; Filler, M. A.; Kim, S.; Bent, S. F. J. Am. Chem. Soc. 2005, 127, 6123.
151. Bocharov, S.; Dmitrenko, O.; Méndez De Leo, L. P.; Teplyakov, A. V. J. Am. Chem. Soc. 2006, 128, 9300.
152. Leftwich, T.; Teplyakov, A. V. J. Phys. Chem. C 2008, 112, 4297.
153. Wang, Z.; Wang, H.; Liu, Y.; Suo, Y. J. Mol. Struct.: THEOCHEM 2008, 850, 72.
154. Fang, L.; Liu, J.; Coulter, S.; Cao, X.; Schwartz, M. P.; Hacker, C.; Hamers, R. J. Surf. Sci. 2002, 514, 362.
155. Hacker, C. A.; Hamers, R. J. J. Phys. Chem. B 2003, 107, 7689.
156. Tao, F.; Bernasek, S. L.; Xu, G.-Q. Chem. Rev. 2009, 109, 3991.
157. Qiao, M. H.; Cao, Y.; Deng, J. F.; Xu, G. Q. Chem. Phys. Lett. 2000, 325, 508.
158. Wang, G. T.; Mui, C.; Tannaci, J. F.; Filler, M. A.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 2003, 107, 4982.
159. Qiao, M. H.; Tao, F.; Cao, Y.; Xu, G. Q. Surf. Sci. 2003, 544, 285.
160. Kim, K.-J.; Han, J.; Kang, T.-H.;Ihm, K.; Jeon, C.; Moon, S.; Hwang, C.-C.; Hwang, H.-N.; Kim, B. J. Elect. Spectrosc. Rel. Phenom. 2005, 144–147, 429.
161. Romero, A. H.; Sbraccia, C.; Silvestrelli, P. L. J. Chem. Phys. 2004, 120, 9745.
162. Yuan, Z. L.; Chen, X. F.; Wang, Z. H.; Yong, K. S.; Cao, Y.; Xu, G. Q. J. Chem. Phys. 2003, 119, 10389.
163. Tao, F.; Yuan, Z. L.; Chen, X. F.; Qiao, M. H.; Wang, Z. H.; Dai, Y. J.; Huang, H. G.; Cao, Y.; Xu, G. Q. Phys. Rev. B 2003, 67, 245406.
164. Qiao, M. H.; Tao, F.; Cao, Y.; Li, Z. H.; Dai, W. L.; Deng, J. F.; Xu, G. Q. J. Chem. Phys. 2001, 114, 2766.
165. Qiao, M. H.; Cao, Y.; Tao, F.; Liu, Q.; Deng, J. F.; Xu, G. Q. J. Phys. Chem. B 2000, 104, 11211.
166. Lu, X.; Xu, X.; Wang, N. Q.; Zhang, Q.; Lin, M. C. J. Phys. Chem. B 2001, 105, 10069.
167. Lu, X.; Lin, M. C.; Xu, X.; Wang, N. Q.; Zhang, Q. N. Phys. Chem. Commun. 2001, 1.
168. Lu, X.; Lin, M. C.; Xu, X.; Wang, N. Q.; Zhang, Q. N. Sci. Chin. Ser. B 2001, 44, 473.
169. Lu, X.; Lin, M. C. Int. Rev. Phys. Chem. 2002, 21, 137.
170. Lee, H.-K.; Kim, K.-J.; Kang, T.-H.; Chung, J. W.; Kim, B. Surf. Sci. 2008, 602, 914.
171. Isobe, N.; Shibayama, T.; Mori, Y.; Shobatake, K.; Sawabe, K. Chem. Phys. Lett. 2007, 443, 347.
172. Cao, Y.; Wang, Z. H.; Deng, J. F.; Xu, G. Q. Angew. Chem. Int. Ed. 2000, 39, 2740.
173. Bitzer, T.; Dittrich, T.; Rada, T.; Richardson, N. V. Chem. Phys. Lett. 2000, 331, 433.
174. Lopez, A.; Bitzer, T.; Heller, T.; Richardson, N. V. Surf. Sci. 2001, 477, 219.
175. Bitzer, T.; Rada, T.; Richardson, N. V. J. Phys. Chem. B 2001, 105, 4535.
176. Hofer, W. A.; Fisher, A. J.; Bitzer, T.; Rada, T.; Richardson, N. V. Chem. Phys. Lett. 2002, 355, 347.
177. Wang, G. T.; Mui, C.; Musgrave, C. B.; Bent, S. F. J. Phys. Chem. B 1999, 103, 6803.
178. Tao, F.; Qiao, M. H.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2003, 107, 6384.
179. Miwa, J. A.; Eves, B. J.; Rosei, F.; Lopinski, G. P. Chem. B 2005, 109, 20055.
180. Kim, H.-J.; Cho, J.-H. J. Chem. Phys. 2004, 120, 8222.
181. Tao, F.; Lai, Y. H.; Xu, G. Q. Langmuir 2004, 20, 366.
182. Pluchery, O.; Witkowski, N.; Borensztein, Y. Phys. Status Solidi B 2005, 242, 2696.
183. Borensztein, Y.; Pluchery, O.; Witkowski, N. Phys. Rev. Lett. 2005, 95, 117402.
184. Li, Q.; Leung, K. T. Surf. Sci. 2003, 541, 113.
185. Lu, X.; Xu, X.; Wu, J.; Wang, N.; Zhang, Q. New J. Chem. 2002, 26, 160.
186. Shimomura, M.; Ichikawa, D.; Fukuda, Y.; Abukawa, T.; Aoyama, T.; Kono, S. Phys. Rev. B 2005, 72, 033303.
187. Shimomura, M.; Kono, S. Hyomen Kagaku 2006, 27, 298.
188. Huang, H. G.; Wang, Z. H.; Xu, G. Q. J. Phys. Chem. B 2004, 108, 12560.
189. Huang, H. G.; Huang, J. Y.; Ning, Y. S.; Xu, G. Q. J. Chem. Phys. 2004, 121, 4820.
190. Huang, H. G.; Huang, J. Y.; Wang, Z. H.; Ning, Y. S.; Tao, F.; Zhang, Y. P.; Cai, Y. H.; Tang, H. H.; Xu, G. Q. Surf. Sci. 2007, 601, 1184.
191. Bu, Y.; Lin, M. C. J. Phys. Chem. 1994, 98, 7871.
192. Casey, S. M.; LeMond, L. M. Silicon Surface Chemistry of Substituted Triazines. Logan, UT, United States, June 6–9, 2004.
193. Giinster, J.; Dieckhoff, S.; Souda, R. Thin Solid Films 1998, 325, 24.
194. Hahn, J. R.; Jeong, H.; Jeong, S.; Jang, S. H. Jpn. J. Appl. Phys. 2006, 45, 2175.
195. Jeong, H.; Jeong, S.; Jang, S. H.; Seo, J. M.; Hahn, J. R. J. Phys. Chem. B 2006, 110, 15912.
196. Cho, Y. E.; Maeng, J. Y.; Kim, S. J. Am. Chem. Soc. 2003, 125, 7514.
197. Choi, J.-H.; Cho, J.-H. J. Am. Chem. Soc. 2006, 128, 3890.
198. Avouris, P.; Walkup, R. E.; Rossi, A. R.; Akpati, H. C.; Nordlander, P.; Shen, T. C.; Abeln, G. C.; Lyding, J. W. Surf. Sci. 1996, 363, 368.
199. Abeln, G. C.; Hersam, M. C.; Thompson, D. S.; Hwang, S.-T.; Choi, H.; Moore, J. S.; Lyding, J. W. J. Vac. Sci. Technol. B 1998, 16, 3874.
200. Avouris, P.; Walkup, R. E.; Rossi, A. R.; Shen, T. C.; Abeln, G. C.; Tucker, J. R.; Lyding, J. W. Chem. Phys. Lett. 1996, 257, 148.
201. Foley, E. T.; Kam, A. F.; Lyding, J. W.; Avouris, P. Phys. Rev. Lett. 1998, 80, 1336.
202. Guyot-Sionnest, P.; Dumas, P.; Chabal, Y. J.; Higashi, G. S. Phys. Rev. Lett. 1990, 64, 2156.
203. Guyotsionnest, P.; Lin, P. H.; Hiller, E. M. J. Chem. Phys. 1995, 102, 4269.
204. Hashizume, T.; Heike, S.; Lutwyche, M. I.; Watanabe, S.; Nakajima, K.; Nishi, T.; Wada, Y. Jpn. J. Appl. Phys. Part 2-Lett. 1996, 35, L1085.
205. Hitosugi, T.; Hashizume, T.; Heike, S.; Kajiyama, H.; Wada, Y.; Watanabe, S.; Hasegawa, T.; Kitazawa, K. Appl. Surf. Sci. 1998, 132, 340.
206. Lyding, J. W.; Shen, T. C.; Hubacek, J. S.; Tucker, J. R.; Abeln, G. C. Appl. Phys. Lett. 1994, 64, 2010.
207. Sakurai, M.; Thirstrup, C.; Nakayama, T.; Aono, M. Appl. Surf. Sci. 1997, 121, 107.
208. Salam, G. P.; Persson, M.; Palmer, R. E. Phys. Rev. B 1994, 49, 10655.
209. Shen, T. C.; Wang, C.; Abeln, G. C.; Tucker, J. R.; Lyding, J. W.; Avouris, P.; Walkup, R. E. Science 1995, 268, 1590.
210. Soukiassian, L.; Mayne, A. J.; Comtet, G.; Hellner, L.; Dujardin, G.; Gourdon, A. J. Chem. Phys. 2005, 122, 134704.
211. Stokbro, K.; Hu, B. Y. K.; Thirstrup, C.; Xie, X. C. Phys. Rev. B 1998, 58, 8038.
212. Stokbro, K.; Thirstrup, C.; Sakurai, M.; Quaade, U.; Hu, B. Y. K.; Perez-Murano, F.; Grey, F. Phys. Rev. Lett. 1998, 80, 2618.
213. Thirstrup, C.; Sakurai, M.; Nakayama, T.; Stokbro, K. Surf. Sci. 1999, 424, L329.
214. Hersam, M. C.; Guisinger, N. P.; Lee, J.; Cheng, K.; Lyding, J. W. Appl. Phys. Lett. 2002, 80, 201.
215. Mayne, A. J.; Dujardin, G.; Comtet, G.; Riedel, D. Chem. Rev. 2006, 106, 4355.
216. Tong, X.; Wolkow, R. A. Surf. Sci. 2006, 600, L199.
217. Hersam, M. C.; Guisinger, N. P.; Lyding, J. W. Nanotechnology 2000, 11, 70.
218. Mayne, A. J.; Soukiassian, L.; Commaux, N.; Comtet, G.; Dujardin, G. Appl. Phys. Lett. 2004, 85, 5379.
219. Mamatkulov, M.; Stauffer, L.; Sonnet, P.; Mayne, A. J.; Comtet, G.; Dujardin, G. J. Chem. Phys. 2008, 128, 244710.
219a. Bellec, A.; Cranney, M.; Chalopin, Y.; Mayne, A. J., Comtet, G.; Dujardin, G. J. Phys. Chem. C 2007, 111, 14818.
220. Sena, A. M. P.; Brázdová, V.; Bowler, D. R. Phys. Rev. B 2009, 79, 245404.
221. Linford, M. R.; Chidsey, C. E. D. J. Am. Chem. Soc. 1993, 115, 12631.
222. Linford, M. R.; Fenter, P.; Eisenberger, P. M.; Chidsey, C. E. D. J. Am. Chem. Soc. 1995, 117, 3145.
223. Terry, J.; Mo, R.; Wigren, C.; Cao, R.; Mount, G.; Pianetta, P.; Linford, M. R.; Chidsey, C. E. D. Phys. Res., Sect. B 1997, 133, 94.
224. Terry, J.; Linford, M. R.; Wigren, C.; Cao, R.-Y.; Pianetta, P.; Chidsey, C. E. D. Appl. Phys. Lett. 1997, 71, 1056.
225. Chatgilialoglu, C. Acc. Chem. Res. 1992, 25, 188.
226. Cicero, R. L.; Chidsey, C. E. D.; Lopinski, G. P.; Wayner, D. D. M.; Wolkow, R. A. Langmuir, 2002, 18, 305.
227. Basu, R.; Kinser, C. R.; Tovar, J. D.; Hersam, M. C. Chem. Phys. 2006, 326, 144.
228. Basu, R.; Lin, J.-C.; Kim, C.-Y.; Schmitz, M. J.; Yoder, N. L.; Kellar, J. A.; Bedzyk, M. J.; Hersam, M. C. Langmuir 2007 23, 1905.
229. Kirczenow, G.; Piva, P. G.; Wolkow, R. A. Phys. Rev. B 2005, 72, 245306.
230. Basu, R.; Guisinger, N. P.; Greene, M. E.; Hersam, M. C. Appl. Phys. Lett. 2004, 85, 2619.
231. Cho, J. H.; Oh, D. H.; Kleinman, L. Phys. Rev. B 2002, 65.
232. Guisinger, N. P.; Basu, R.; Baluch, A. S.; Hersam, M. C. Ann. NY Acad. Sci 2003, 1006, 227.
233. Hossain, M. Z.; Kato, H. S.; Kawai, M. J. Phys. Chem. B 2005, 109, 23129.
234. Kang, J. K.; Musgrave, C. B. J. Chem. Phys. 2002, 116, 9907.
235. Lopinski, G. P.; Wayner, D. D. M.; Wolkow, R. A. Nature 2000, 406, 48.
236. Pei, Y.; Ma, J. Langmuir 2006, 22, 3040.
237. Pitters, J. L.; Piva, P. G.; Tong, X.; Wolkow, R. A. Nano Lett. 2003, 3, 1431.
238. Pitters, J. L.; Wolkow, R. A. J. Am. Chem. Soc. 2005, 127, 48.
239. Takeuchi, N.; Kanai, Y.; Selloni, A. J. Am. Chem. Soc. 2004, 126, 15890.
240. Takeuchi, N.; Selloni, A. J. Phys. Chem. B 2005, 109, 11967.
241. Tong, X.; DiLabio, G. A.; Wolkow, R. A. Nano Lett. 2004, 4, 979.
242. Wang, Y.; Hwang, G. S. Appl. Phys. Lett. 2005, 86.
243. Alavi, S.; Rousseau, R.; Patitsas, S. N.; Lopinski, G. P.; Wolkow, R. A.; Seideman, T. Phys. Rev. Lett. 2000, 85, 5372.
244. de Smet, L.; Zuilhof, H.; Sudholter, E. J. R.; Lie, L. H.; Houlton, A.; Horrocks, B. R. J. Phys. Chem. B 2005, 109, 12020.
245. Guisinger, N. P.; Greene, M. E.; Basu, R.; Baluch, A. S.; Hersam, M. C. Nano Lett. 2004, 4, 55.
246. Hallback, A. S.; Poelsema, B.; Zandvliet, H. J. W. Appl. Surf. Sci. 2007, 253, 4066.
247. Hofer, W. A.; Fisher, A. J.; Lopinski, G. P.; Wolkow, R. A. Chem. Phys. Lett. 2002, 365, 129.
248. Lee, J. Y.; Cho, J. H. J. Chem. Phys. 2004, 121, 8010.
249. Li, Q.; Leung, K. T. J. Phys. Chem. B 2005, 109, 1420.
250. Patitsas, S. N.; Lopinski, G. P.; Hul'ko, O.; Moffatt, D. J.; Wolkow, R. A. Surf. Sci. 2000, 457, L425.
251. Pitters, J. L.; Wolkow, R. A. Nano Lett. 2006, 6, 390.
252. Pitters, J. L.; Dogel, I.; DiLabio, G. A.; Wolkow, R. A. J. Phys. Chem. B 2006, 110, 2159.
253. Sun, Q. Y.; de Smet, L.; van Lagen, B.; Giesbers, M.; Thune, P. C.; van Engelenburg, J.; de Wolf, F. A.; Zuilhof, H.; Sudholter, E. J. R. J. Am. Chem. Soc. 2005, 127, 2514.
254. DiLabio, G. A.; Piva, P. G.; Kruse, P.; Wolkow, R. A. J. Am. Chem. Soc. 2004, 126, 16048.
255. Piva, P. G.; DiLabio, G. A.; Pitters, J. L.; Zikovsky, J.; Rezeq, M. D.; Dogel, S.; Hofer, W. A.; Wolkow, R. A. Nature, 2005, 435, 658.
256. Ratner, M. Nature, 2005, 435, 575.
257. Greene, M. E.; Guisinger, N. P.; Basu, R.; Baluch, A. S.; Hersam, M. C. Surf. Sci. 2004, 559, 16.
258. Yang, L.; Doren, D. J. J. Phys. Chem. C 2008, 112, 781.
259. Smeu, M.; Wolkow, R. A.; Guo, H. J. Am. Chem. Soc. 2009, 131, 11019.
260. Rochefort, A.; Beausoleil, A. Chem. Phys. Lett. 2004, 400, 347.
261. Hossain, M. Z.; Kato, H. S.; Kawai, M. J. Am. Chem. Soc. 2007, 129, 3328.
262. Tong, X.; DiLabio, G. A.; Clarkin, O. J.; Wolkow, R. A. Nano Lett. 2004, 4, 357.
263. Dogel, S. A.; DiLabio, G. A.; Zikovsky, J.; Pitters, J. L.; Wolkow, R. A. J. Phys. Chem. C, 2007, 111, 11965.
264. Nemanick, E. J.; Hurley, P. T.; Webb, L. J.; Knapp, D. W.; Michalak, D. J.; Brunschwig, B. S.; Lewis, N. S. J. Phys. Chem. B 2006, 110, 14770.
265. Nemanick, E. J.; Hurley, P. T.; Brunschwig, B. S.; Lewis, N. S. J. Phys. Chem. B 2006, 110, 14800.
266. Pandey, D.; Zemlyanov, D. Y.; Bevan, K.; Reifenberger, R. G.; Dirk, S. M.; Howell, S. W.; Wheeler, D. R. Langmuir 2007, 23, 4700.
267. Combellas, C.; Kanoufi, F.; Pinson, J.; Podvorica, F. I. J. Am. Chem. Soc. 2008, 130, 8576.
268. Cai, Y. H.; Shao, Y. X.; Xu, G. Q. J. Am. Chem. Soc. 2007, 129, 8404.
269. Langner, A.; Panarello, A.; Rivillon, S.; Vassylyev, O.; Khinast, J. G.; Chabal, Y. J. J. Am. Chem. Soc. 2005, 127, 12798.
270. Leftwich, T. R.; Teplyakov, A. V. J. Electron. Spectrosc. Relat. Phenom. 2009, 175, 31.
271. Leftwich, T. R.; Teplyakov, A. V. Unpublished data, 2009.
272. DiLabio, G. A.; Johnson, E. R.; Pitters, J. J. Phys. Chem. C 2009, 113, 9969.
273. Zipoli, F.; Bernasconi, M. J. Phys. Chem. B 2006, 110, 23403.
274. Wakatsuchi, M.; Kato, H. S.; Yamada, T.; Kawai, M. Jpn. J. Appl. Phys. 2005, 44, 514.
275. Ma, J.; Rogers, B. L.; Humphry, M. J.; Ring, D. J.; Goretzki, G.; Champness, N. R.; Beton, P. H. J. Phys. Chem. B 2006, 110, 12207.
276. Bitzer, T.; Richardson, N. V. Appl. Phys. Lett. 1997, 71, 1890.
277. Bitzer, T.; Richardson, N. V. Appl. Surf. Sci. 1999, 144–145, 339.
278. Bitzer, T.; Richardson, N. V. Appl. Phys. Lett. 1997, 71, 662.
279. Eves, B. J.; Fan, C.; Lopinski, G. P. Small 2006, 2, 1379.
280. Ciampi, S.; Böcking, T.; Kilian, K. A.; James, M.; Harper, J. B.; Gooding, J. J. Langmuir 2007, 23, 9320.
281. Bin, X.; Mischki, T. K.; Fan, C.; Lopinski, G. P.; Wayner, D. D. M. J. Phys. Chem. C 2007, 111, 13547.
282. Jiao, J.; Anariba, F.; Tiznado, H.; Schmidt, I.; Lindsey, J. S.; Zaera, F.; Bocian, D. F. J. Am. Chem. Soc. 2006, 128, 6965.
Functionalization of Semiconductor Surfaces, First Edition
Edited by Franklin (Feng) Tao and Steven L. Bernasek.
© 2012 John Wiley & Sons, Inc. Published 2012 by John Wiley & Sons, Inc.