
4
Application of Minimal Anticipated Biological Effect Level (MABEL) in Human Starting Dose Selection for Immunomodulatory Protein Therapeutics – Principles and Case Studies
Haiqing Wang1, Zheng Yang1 and Rong Shi2
1Bristol‐Myers Squibb Co., Department of Metabolism and Pharmacokinetics, Pharmaceutical Candidate Optimization, 3551 Lawrenceville Princeton, Lawrence Township, NJ, 08648, USA
2Bristol‐Myers Squibb Co., Department of Clinical Pharmacology and Pharmacometrics, 3551 Lawrenceville Princeton, Lawrence Township, NJ, 08648, USA
4.1 Introduction
Immunomodulatory protein therapeutics (IMPTs) are designed to interact with one or more components of the immune system to either enhance or suppress immune responses. These protein therapeutics have revolutionized the treatment of cancer and immune‐related diseases in the past two decades. One of the most notable examples is the immune checkpoint inhibitors, such as anti‐cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4, Yervoy®) and anti‐programmed cell death protein 1 (PD‐1) (Opdivo® and Keytruda®) monoclonal antibodies (mAbs), which have demonstrated remarkable anti‐tumor efficacy by effectively enhancing patient immune responses against cancer cells [1]. Likewise, immune suppressive therapeutic proteins such as anti‐tumor necrosis factor α (TNFα) antibody (e.g. Humira®) and CTLA‐4 Fc fusion protein (Oriencia®) have also improved the treatment of various inflammatory and autoimmune diseases [2].
A key challenge in early clinical investigation of novel IMPTs is their inherent risk of adverse immune‐mediated effects, including inflammation, cytokine storm, tumor lysis syndrome, infection, hypersensitivity, cell and tissue injuries, and others [3–5]. Although some of these adverse effects may be acceptable based on the risk/benefit ratio to the patients, these adverse effects should be avoided at the starting dose of a first‐in‐human (FIH) study, particularly when normal healthy volunteers are the study subjects. As a result, determining a safe starting dose is an important first step in preparing new IMPTs for FIH studies. The starting dose needs to be safe so that it will not cause any harm to human subjects. In the case of the FIH study of anti‐cancer agents, where cancer patients are enrolled, the starting dose is also expected to be not far from the efficacious dose, so the number of patients exposed to ineffective doses can be reduced. In the past decade, the methods of the FIH starting dose selection for IMPTs have evolved from mostly toxicology‐based approaches to a more integrated pharmacology‐ and mechanism‐based method, such as determining a minimal anticipated biological effect level (MABEL) as the FIH stating dose [6]. The transformation triggered by the TGN1412 incident in year 2006, reflects an endeavor of thorough understanding of immunopharmacology of these protein therapeutics in humans and in animals, and safety risks mitigation in early clinical development [7–9].
This chapter intends to capture the evolution of FIH starting dose selection approaches of IMPTs over the past decade and to review the principles and procedures of more integrated pharmacology‐ and mechanism‐based methods for the MABEL determination, along with several case studies. The perspectives of further improvement of the starting dose selection approaches will also be discussed.
4.2 Safety and Immune‐Related Toxicities of Immunomodulatory Protein Therapeutics
Protein therapeutics, including mAbs, Fc fusion proteins, and polyethylene glycol (PEG)ylated proteins, are large size molecules that generally reside in the extracellular space and exert their biological activities by binding to cell surface targets or soluble antigens with a high affinity and specificity. In general, protein therapeutics are well tolerated in humans. Their high affinity and specificity toward their targets significantly minimize nonmechanism‐based toxicity, i.e. off‐target toxicity. In addition, the in vivo catabolites of mAbs and Fc fusion proteins are natural amino acids that are safe for humans and animals. In terms of proteins conjugated with high molecular weight (HMW) PEG, such as certolizumab pegol (anti‐TNFα Fab with 40 kDa branched PEG, Cimzia®), the HMW PEG moiety is demonstrated to be well tolerated even though there may be toxicity concerns over chronic treatment due to vacuolation in organs and tissues [10]. As a result, the primary safety risk of protein therapeutics, including those with immunomodulatory functions, in their early clinical development is on‐target toxicities or exaggerated pharmacology by blocking or enhancing the activities of their targets.
IMPTs are designed to bind directly to T cells, B cells, natural killer (NK) cells, antigen presenting cells (APCs, dendritic cells, and macrophages), cytokines, and chemokines to modulate immune functions. The immune suppressive protein therapeutics are intended to either deplete their targets, suppress their functions, prevent their homing to lymphoid organs and inflammatory sites, or induce energy in order to treat autoimmune diseases or prevent transplant rejection. Alternatively, the immune‐activating protein therapeutics for cancer treatment are able to activate immune cells through direct agonizing immune activation receptors or antagonizing immune inhibitory receptors or by depleting specific populations of Treg cells. Although the intended pharmacology is required for clinical efficacy, prolonged or exaggerated pharmacological effect, or, sometimes, irreversible changes of immune target cells/pathways, or any unintended pharmacological consequences may lead to immune toxicities [4]. For example, chronic treatment of immunosuppressive agents such as anti‐TNFα biologics (infliximab, adalimumab, or etanercept, etc.) in rheumatoid arthritis patients has shown increased risk for opportunistic bacterial, fungal, or parasitic infection, chronic viral infection, or virally induced cancers. In contrast, immune checkpoint inhibitors, such as anti‐CTLA4 antibody ipilimumab, have shown different levels of immune‐related adverse events (irAEs) in 90% treated patients, generally within three to six months of a treatment initiation. Most frequent Grade 3–5 irAEs of ipilimumab is gastrointestinal disorders including severe (Grade 3–4) diarrhea in 10% patients [11]. In comparison, immune agnostic agents developed for cancer indications, such as an anti‐CD28 superagonist TGN1412 and an anti‐CD40 agonistic antibody CP‐870893, exhibit more acute adverse effects associated with infusion and hypersensitivity reactions, and cytokine release syndromes (CRSs) [3]. In addition, antibodies such as rituximab and alemtuzumab exert their tumor cell cytotoxicity through Fc‐mediated cross‐linking between CD16A (FcγRIIIa) expressing cells, which lead to antibody‐dependent cell‐mediated cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC). The ADCC and CDC effects may also increase the risk of cytokine release, cytopenia, immune suppression, and tissue injuries [4,12,13].
The potential risk of immune toxicities of novel IMPTs must be carefully assessed in nonclinical studies to ensure the safety of normal healthy volunteers and patients enrolled in clinical trials. Generally speaking, toxicology studies following good laboratory practice (GLP) in nonclinical relevant species are conducted prior to the FIH study. The systemic exposure and toxicology data from the GLP toxicology study are then utilized to determine a FIH safe starting dose as described in the ICH S9 Guideline on Nonclinical Evaluation for Anticancer Pharmaceuticals or the Food Drug Administration Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers [14]. In order to assess on‐target toxicities of IMPTs arising from their exaggerated pharmacology in the nonclinical toxicology studies, the protein therapeutics must bind the same target in a toxicology animal species and in humans with a similar affinity. More importantly, the target function and its expression patterns in the toxicology animal species must be consistent with humans to ensure the same mechanism of action that is evaluated in the animal species. The similar target‐binding affinities can often be fulfilled by using monkey as the toxicology species because of genetic similarity between monkeys and humans. However, because subtle differences of the target expression level and functions in the whole immune systems between monkeys and humans, and our inadequate understanding of the pharmacology of IMPTs, the results from the GLP toxicology studies, in some cases, are not predictive of immune‐related adverse effects in humans [15]. Therefore, an integrated approach including all nonclinical pharmacology data (e.g. human in vitro data) together with the toxicology data is needed to provide a full assessment of the safety risk and provide a more mechanism‐based FIH safety starting dose. In the following sections, the limitations of the toxicology study in selecting FIH starting dose and a better and more integrated approach will be discussed in detail.
4.3 Uncertainties of Toxicology Approach in FIH Safe Starting Dose Selection for Immunomodulatory Protein Therapeutics
The GLP toxicology study in relevant animal species has been a conventional approach in selecting a safe starting dose of novel therapeutics in the FIH studies. Two industrial guidance issued by FDA, one for adult healthy volunteers and the other for cancer patients, provide detailed instructions on the starting dose calculation that have been well implemented by pharmaceutical industries [14,16]. Briefly, if a novel therapeutic is to be tested in adult healthy volunteers, the starting dose will be based on a no‐observed‐adverse‐effect‐levels (NOAELs) determined in most relevant animal species. If a novel therapeutic is for oncology indications, cancer patients are usually enrolled in the FIH study. In this case, treatment at the starting dose is expected not only to be safe but also to have pharmacologic effects that may bring some benefits to patients. Based on the FDA guidance for anticancer pharmaceuticals, if rodents are the most relevant toxicology species, the severely toxic dose in 10% of the tested animals (severely toxic dose, STD 10) needs to be determined from the GLP toxicology study. Alternatively, if nonrodents are the most appropriate species, the highest nonseverely toxic dose (HNSTD), defined as the highest dose that does not produce evidence of lethality, life‐threatening toxicities or irreversible findings, needs to be determined. The NOAEL, STD10, or HNSTD from the toxicology study is converted to human equivalent dose (HED) to which an empirical safety factor is applied to derive the maximum recommended starting dose (MRSD). One of the uncertainties in the toxicology‐based MRSD calculation for protein therapeutics is the method used in the HED calculation, and another uncertainty is the empirical nature of the safety factor, both of which are discussed below.
4.3.1 HED Calculation for Immunomodulatory Protein Therapeutics
The conversion of HED from NOAEL/STD10/HNSTD, as recommended by FDA, uses either body surface area or body weight as the scaling factor of the dose, depending on whether the drug molecular weight (MW) < 100 kDa or MW ≥ 100 kDa, respectively. For IMPTs, including antibodies, Fc‐fusion proteins, and PEGylated protein (PEG MW ≥ 40 kDa), the body weight‐based conversion appears to be more reasonable because of their high MW. However, in toxicology studies where immunogenicity of the protein therapeutics lead to much lower systemic exposure than expected, using body weight‐based dose conversion of animal NOAEL/HNSTD may overestimate the HED. To correct for the decreased systemic exposure due to immunogenicity, a systemic exposure‐based conversion using the predicted human clearance is preferred to estimate the HED. In addition, the systemic exposure‐based approach is also recommended when nonlinear pharmacokinetics (PKs) of the protein therapeutics is observed in animals and also expected in humans due to target‐mediated drug disposition (TMDD).
In order for the systemic exposure‐based conversion of NOAEL/STD10/HNSTD to HED to be conducted, the clearance (CL) and the steady‐state volume of distribution (V ss) of the protein therapeutics in humans need to be estimated from nonclinical animal species, mostly monkeys. Generally, if mAbs (including Fc‐fusion proteins) exhibit linear pharmacokinetics over a wide dose range in monkeys, simple allometry, with an exponent of 0.80–0.85 for the CL and 1 for the V ss, has been shown to reasonably predict the human PK parameters from monkey data [17–19]. However, if mAbs show nonlinear pharmacokinetics due to TMDD, simple allometry method is not suitable to estimate the clearance in humans. Rather, a TMDD or Michaelis–Menten PK model may be explored to separate the target‐ and nontarget‐mediated clearance in monkeys, from which target‐mediated clearance can be translated to humans based on the characteristics of a target (target expression, binding affinity, etc.) and the nontarget‐mediated clearance can be scaled‐up using simple allometry as described above [20]. Recently, physiologically based pharmacokinetic (PBPK) models have become available for antibodies (e.g. Simcyp v.12) and may be used to predict human pharmacokinetics and concentration–time profiles, as needed [21].
4.3.2 Determination of Safety Factor for Immunomodulatory Protein Therapeutics
The safety factor, often used as a denominator of the HED for the MRSD determination, is to account for uncertainties such as enhanced therapeutic potencies or unexpected sensitivity in humans vs. animals. It thereby serves as an additional assurance of safety in humans at the first dose in FIH studies. Generally, a safety factor of 10 is recommended by FDA to determine MRSD from the NOAEL‐derived HED, whereas, for anti‐cancer agents, a safety factor of 6 is recommended to determine the MRSD from the HNSTD‐derived HED [14,16]. In addition, FDA guidance also acknowledges that the safety factor should be raised or decreased depends on the confidence that investigators have on the safety at the first dose in FIH studies. A recent survey by Suh et al. on the starting dose of mAbs in FIH studies between 1990 and 2013 summarized the safety factors of 14 antibodies used to determine the NOAEL‐based MRSD [6]. The average of the safety factors was found to be 42 with a wide range of 3.2–1290, suggesting that the majority of the safety factors used by drug companies is greater than what the FDA recommends. Nevertheless, because of the empirical nature of the safety factor, a large safety factor does not necessarily guarantee the safety of the starting dose in FIH studies, as illustrated by the TGN1412 incident where a 160‐fold safety factor still led to a disastrous outcome at the starting dose.
4.3.3 TGN1412 Incident and Minimal Anticipated Biological Effect Level
The TGN1412 incident, happened in 2006, is a well‐known example of a failed toxicology‐based approach in the selection of a safe FIH starting dose. TGN1412 is an anti‐CD28 super agonistic monoclonal antibody that binds and activates T cell co‐stimulatory receptor CD28, allowing T cell activation without the need of a simultaneous T cell receptor (TCR) engagement. Nonclinical safety assessment of TGN1412 was conducted in monkeys because the extracellular loop of monkey CD28 shares 100% sequence homology with that of the human receptor, and TGN1412 demonstrated equivalent binding affinity and similar tissue staining patterns in both humans and monkeys. TGN1412 was well tolerated in monkeys with the NOAEL at the highest tested dose of 50 mg kg−1. The starting dose was then determined to be 0.1 mg kg−1, 160‐fold lower than the HED of 16 mg kg−1 [7]. Although the NOAEL level from the toxicology study and 160‐fold safety factor appear to guarantee the safety at 0.1 mg kg−1 starting dose, six healthy subjects who received the starting dose simultaneously experienced devastating side effects, such as cytokine storm, and life‐time high risk of developing cancers and autoimmune diseases [22,23].
Extensive research following the TGN1412 incident has revealed that the species difference in response to the CD28 superagonist is due to their subtle differences in the immune system. In humans, the immediate cytokine release is due to TGN1412‐mediated activation of CD28 receptors on CD4+ effector memory T (TEM) cells, which mostly reside in the tissues driven by multiple exposures to infectious agents. In laboratory rodents housed under clean conditions, there is little presence of CD4+ TEM cells. Consequently, administration of the CD28 superagonist to rodents lead to initial polyclonal T cell activation but was rapidly dominated by an expansion of immunosuppressive regulatory T (Treg) cells. In monkeys, the lack of the cytokine storm after the TGN1412 treatment in the toxicology study was because their CD4+ TEM does not express CD28 receptors, which, unfortunately, has gone unnoticed for many years of primate testing [24–26].
The TGN1412 lesson emphasizes the importance of a thorough understanding of target pharmacology between humans and animals during drug development. For novel IMPTs with limited pharmacology and toxicology understanding, a simplistic algorithm based on the toxicology data brings little assurance of the safety in a FIH study, even with a large and empirical margin of the starting dose relative to the NOAEL. Rather, a robust and well thought‐out process considering both NOAEL and all available pharmacological data is required for the selection of a safe FIH starting dose. As expected, after the TGN1412 incident, a MABEL approach was proposed by Sir Gordon Duff and was subsequently adopted into the EMA guideline for pharmaceutical industries in the determination of a safe starting dose for high‐risk IMPTs in FIH studies [7].
4.4 Incorporating Mabel Approach in FIH Starting Dose Selection for High‐Risk Immunomodulatory Protein Therapeutics
The rationale of the MABEL approach is that the toxicity of a novel therapeutic agent is due to its exaggerated or prolonged pharmacology. Therefore, if the starting dose is only to reach a minimal biologic effect level, the on‐target toxicity will be absent or at least minimal at that level as well. For example, a retrospective analysis on the TGN1412 starting dose suggests that, for an antibody with a superagonistic activity, a 10% receptor occupancy (RO) was appropriate for the starting dose, which is approximately 1.5 µg kg−1, assuming a K d of 1.88 nM and the total target expression of 0.65 nM. The retrospectively proposed starting dose of 1.5 µg kg−1 is 60‐fold lower than the actual Phase I starting dose (0.1 mg kg−1) that lead to >90% RO and a massive cytokine release. Recently, TGN1412 was renamed as TAB08 and returned in a healthy volunteers clinical study at very low doses (0.1–7 µg kg−1) demonstrated safety without a cytokine storm, further support the MABEL concept [27].
MABEL‐based approaches have been used for the starting dose selection of several high‐risk IMPTs [28–31] and have also been reviewed in a number of publications [8,32,33]. In particular, Muller et al. outlined an integrated MABEL and toxicology approach to mitigate the risk of both on‐ and off‐target toxicities for high‐risk protein therapeutics [33]. In this section, we modify the integrated starting dose selection scheme by Muller et al. incorporating the in vitro cytokine release assay (CRA) and other in vitro toxicity assessments as important components for the safety assessment in addition to the in vivo toxicology‐based approach. We also further delineate the approaches of pharmacological data integration for the MABEL calculation. As shown in Figure 4.1, the starting dose selection of high‐risk IMPTs are based on the MRSD from the in vivo toxicology study (Section 4.2), the in vitro cytokine release risk assessment, and the pharmacology‐based MABEL approach. The latter two elements are discussed below.
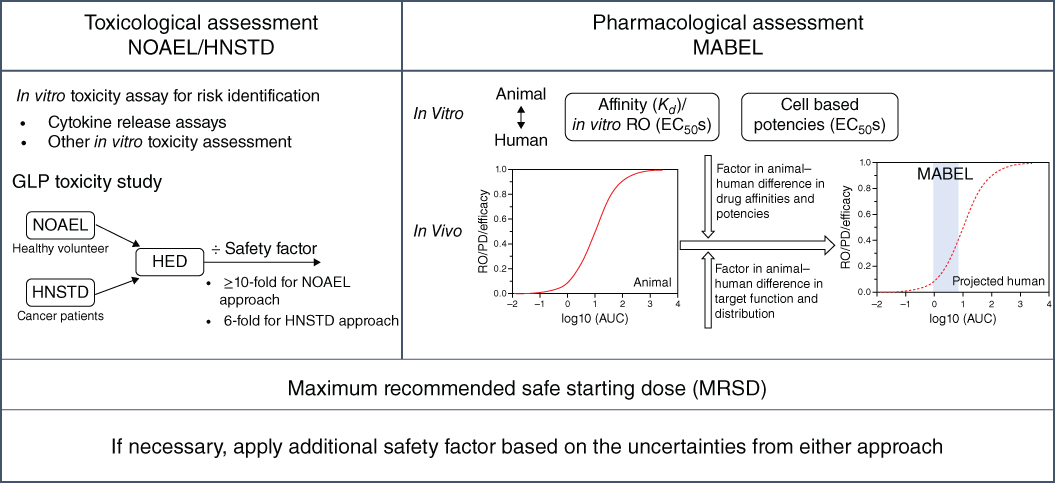
Figure 4.1 Approaches for FIH safe starting dose selection for novel immunomodulatory protein therapeutics.
4.4.1 In vitro Cytokine Release Assay and Other In vitro Assays as Toxicity Assessment for Immunomodulatory Protein Therapeutics
CRS (cytokine release syndrome) or its extreme case “cytokine storm” is one of the most dangerous side effects for IMPTs in the clinic. In addition to TGN1412 that leads to life‐threatening cytokine storm, other therapeutic antibodies, including muromonab (anti‐CD3), alemtuzumab (anti‐CD52), rituximab (anti‐CD20), CP‐870893 (anti‐CD40 agonist), also showed mild‐to‐severe CRS in the clinic [34]. The risk of CRS for new protein therapeutics usually cannot be assessed in animals, as shown in the TGN1412 case, but may be estimated by in vitro cytokine release assays (CRAs). Brennan et al. recently outlined a decision tree of novel protein therapeutics for CRA based on their target mechanism and their affinities with Fcγ receptors (FcγRs) [35]. They recommended CRAs for antibodies that bind to cell surface target or FcγRs on immune cells or other cell type capable of cytokine/chemokine production (endothelial, fibroblasts, etc.), and for the drug that has a strong capability to cross‐link FcγR even if the target cells do not produce cytokines. A recent FDA analysis of immune activating therapeutics for oncology indications suggests that 84% newly submitted investigative new drug (IND) applications conducted in vitro CRAs prior to FIH studies [3].
Several in vitro CRAs have been developed for preclinical risk assessment of new therapeutic proteins [35–37]. The solid‐phase and solution‐based assays are the two most widely used methods in which peripheral blood mononuclear cells (PBMCs) are incubated with the protein therapeutics either immobilized on a solid phase or in solution. The selection of a right assay format ties to at least two distinct mechanisms that govern the protein therapeutics‐induced systemic cytokine release: either through antibody Fab–antigen interaction (Type I) or through antibody Fc–Fcγ receptor interaction (Type II) or both. TGN1412 and muromonab‐CD3 are the examples of the Type I mechanism. Their antibody Fab regions cross‐link and cluster CD28 or CD3, respectively, on the CD4+ T cells resulted in cell activation and cytokine release (IL‐2, IFNγ, TNFα, IL‐10, IL‐6, etc.). Among these cytokines, IL‐2 is a signature of CD4+ T cell activation and often associated with a more severe cytokine release. Alemtuzumab and other antibodies follow the Type II mechanism by interacting with a nonactivatory cell surface marker (such as CD52 for alemtuzumab) and FcγRIII (CD16A) receptor on NK cells through their Fc region to trigger NK cell activation and release similar cytokines except IL‐2 [37]. It also appears that the Fc binding affinity to FcγRIII is correlated with the degree of NK cell‐mediated cytokine release. For example, the afucosylated anti‐CD20 and anti‐CD40 human IgG1 antibodies with improved affinity toward FcγRIII showed CRS in patients at much lower dose than the antibodies with wild‐type human IgG1 Fc. Vessillier et al. recently evaluated various CRAs under various solid phase and soluble incubation conditions with either whole blood or PBMC [37]. Their results suggested that PBMC solid‐phase CRA is most sensitive for the detection of TGN1412‐like cytokine release (Type I mechanism), whereas both soluble and solid‐phase assay seem suitable for alemtuzumab‐like NK cell‐mediated cytokine release (Type II mechanism).
Although the recent FDA survey suggests most sponsors conducted at least one or more of in vitro CRA as part of their IND packages of immune activating antibodies for cancer treatment, the CRAs mostly served as hazard identification, rather than projecting a safe starting dose with minimal cytokine release risk [3,35]. Translational research needs to be conducted by using antibodies with known cytokine release risk in the clinic to explore the possibility of establishing a quantitative exposure/response relationship in the cytokine‐released assays that can be used to project a safe dose/exposure with minimal cytokine release liability in the clinic.
Additional in vitro toxicological assessments include (i) Fc‐mediated direct activation of FcγR‐bearing cells that lead to cytokine release, ADCC by NK cells, and antibody‐dependent cellular phagocytosis (ADCP) by macrophages and neutrophils; (ii) Fc‐mediated complement activation that produces direct CDC of target cells [4,35]. The ADCC‐, ADCP‐, and CDC‐mediated cytotoxicities are important mechanisms of action (MOA) contributing to the tumor cell killing. The MOA, however, also leads to potential toxicities, such as cytokine release (including tumor lysis syndrome), inflammatory response by activating granulocytes, macrophages, mast cells, and basophils, and undesirable cytopenia and tissue injury due to the killing of normal cells expressing the target. The potencies of ADCC‐, ADCP‐, and CDC‐mediated cytotoxicity can be well tailored by selecting different Fc isotypes, i.e. the potencies of IgG1 > IgG2 > IgG4, or by modifications of the Fc through nonfucosylation, aglycosylation, and other mutations that lead to further enhanced or silenced Fc‐mediated effects [38]. Thus, selecting an optimal Fc format is an important consideration of novel IMPTs in order to balance the efficacy benefit and safety risk. If the ADCC‐, ADCP‐, and CDC‐mediated cytotoxicity, particularly with the modified Fc of enhanced activities, are required for the efficacy of novel IMPTs, the in vitro characterization of the toxicity risks is suggested to be based on the cells with different target densities that represent cell populations in patients. In addition, the use of a wide drug concentration range to delineate the concentration‐dependent Fc function for MABEL considerations is needed [35].
4.4.2 Integrate In vitro Pharmacology Data to Estimate MABEL for High‐risk Immunomodulatory Protein Therapeutics
The EMA guideline recommends all relevant in vitro and in vivo information from pharmacological and PK/pharmacodynamic (PD) studies to be utilized for MABEL calculation [39]. Discretion, however, needs to be made on which data is the most representative of the biological effect level. This is because the intention of MABEL is not to choose the most sensitive assay that leads to an extremely low starting dose and a prolonged dose escalation scheme. Rather, all the in vitro and in vivo pharmacological and PK/PD data need to be integrated in an aim to project human exposure/PD/efficacy profiles with reasonable confidence, from which the exposure at the minimal level can be estimated.
As shown in Figure 4.1, the nonclinical assessment of IMPTs for the MABEL determination focuses primarily on three aspects: target engagement, pharmacologic activity, and PK/PD/efficacy relationship. The in vitro target‐binding affinity, often measured by solid‐phase resonance (SPR) assays, and the cell‐based potency are evaluated under artificial experimental settings. Therefore, they may not directly reflect in vivo potency or activity. Nevertheless, comparing the in vitro data between animals and humans is important to evaluate the potency difference of the protein therapeutics between the species. The in vivo pharmacological assessment explores the relationships among the systemic exposure of protein therapeutics with whole blood RO (for target engagement), PD responses, and efficacy in relevant animal PD and disease models. Ideally, if the systemic exposure vs. RO and/or PD relationships can be established and well translated to the efficacy profiles in animal models, the human systemic RO/PD profiles, projected after factoring in the potency difference between the species, may be used as the predictive biomarkers for the efficacy in humans. As a result, the projected human exposure vs. RO or PD profiles may be suitable for the MABEL calculation. However, when target expression and measurable PD response lacks in nonclinical species, the relationship that best represents the pharmacology of the protein therapeutics to its target may be considered for the MABEL calculation.
It is worth noting that mechanistic PK/PD modeling often plays an important role in the MABEL determination by integrating nonclinical pharmacology data and incorporating the species difference in target load and biological function into the projection. The integration of pharmacological data for the MABEL calculation and the PK/PD model based MABEL approach is elucidated in the four case studies discussed below.
4.5 Case Studies of Mabel Calculation
4.5.1 Case Study I: MABEL Determination for Anti‐CD28 Antagonist Domain Antibody BMS‐931699
BMS‐931699 was the first protein therapeutics targeting human CD28 following the TGN1412 incident [28,40,41]. Unlike TGN1412 for oncology indications, BMS‐931699 was developed for auto‐immune diseases by blocking the CD28‐B7 (CD80/86) costimulation pathway to suppress T cell activities. In order to avoid the potential of immune activation through antibody bivalent binding cross‐link the CD28 receptor, BMS‐931699 is engineered as a 12 kDa domain antibody (dAb) conjugated with a 40 kDa branched PEG that binds monovalently to and antagonizes the CD28 receptor. All the available nonclinical data, including the CRA, support that the monovalent anti-human CD28 dAb (anti‐hCD28 dAb) is a T‐cell co‐stimulation inhibitors without any agonistic activity. Nevertheless, a MABEL‐based approach was used to select a safe starting dose for the FIH trial, given the fact the CD28 receptor is a high‐risk target.
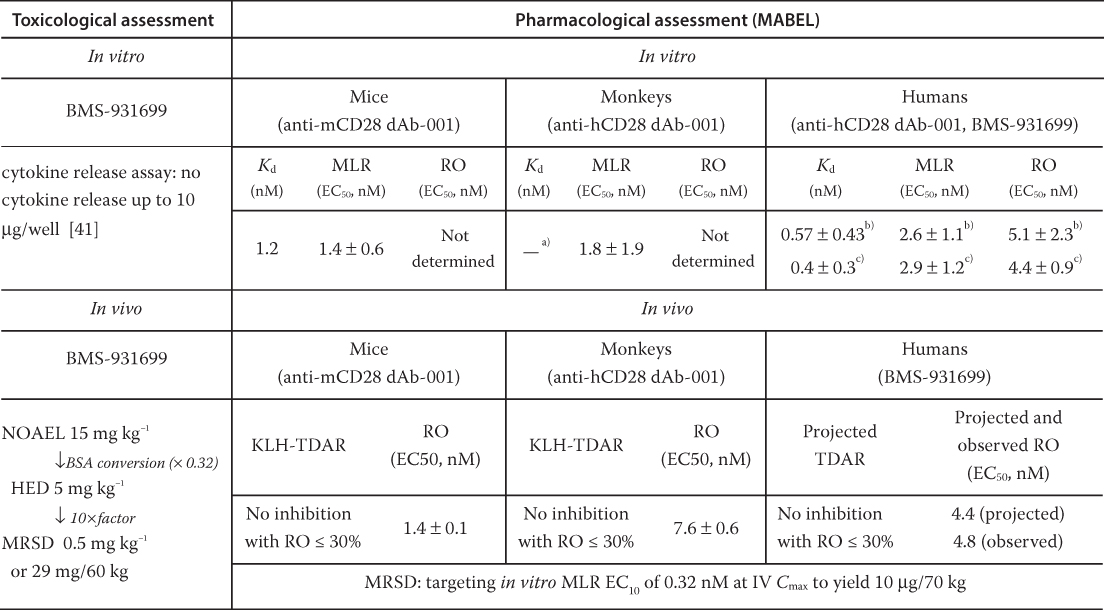
Yang et al. reported an integrated analysis using PK/PD modeling‐based approach for BMS‐931699 MABEL calculation [28]. In the article, the authors first compared the in vitro activities of BMS‐931699, anti‐hCD28‐001 (two extra amino acids at the N‐terminus of BMS‐931699), and a mouse surrogate (anti‐mCD28‐001) in mixed lymphocyte reaction (MLR) assay. As shown in Table 4.1, the MLR EC50 values, which are similar among the three dAbs, 1.8 ± 1.9 nM of anti‐mCD28‐001 in mice, 2.6 ± 1.1 nM of anti‐hCD28‐001 and 2.9 ± 1.2 nM of BMS‐931699, appears to be a sensitive and relevant measure reflecting the in vitro whole blood CD28 ROs. The authors subsequently determined the connectivity of in vivo and in vitro whole blood CD28 RO EC50. The PK/PD modeling of anti‐hCD28 dAb‐001 in cynomolgus monkeys estimated the in vivo RO EC50 (7.6 ± 0.6 nM), which is consistent with its human whole blood RO EC50 (5.1 ± 2.3 nM) measured in vitro as well as in vitro human whole blood RO EC50 (4.4 ± 0.9 nM) of BMS‐931699, indicating a strong agreement between in vitro and in vivo in the two species. Later, the human in vitro RO EC50 (4.4 ± 0.9 nM) of BMS‐931699 was subsequently proven to be in excellent agreement with its in vivo EC50 (4.8 ± 0.8 nM) generated from the FIH clinical studies [28]. More importantly, the authors further established the correlation of RO with the PD response as measured by the suppression of T cell‐dependent keyhole limpet hemocyanin antigen response (anti‐KLH TDAR) in the monkeys using PK/PD modeling based approach. In both mice and cynomolgus monkeys, a CD28 RO of ≤30% is unlikely to introduce a significant inhibition of the T‐cell–dependent keyhole limpet hemocyanin (KLH)‐induced IgG response or a significant immunosuppression activity.
Table 4.1 BMS‐931699 FIH starting dose selection using MABEL approach.
|
BSA, body surface area; TDAR, T cell‐dependent antibody response.
The extracellular domain of human and monkey CD28 is identical, thus the K d value in monkeys is expected to be the same as that in humans.
The data are from anti‐hCD28 dAb‐001, which has two extra amino acids in the N‐terminus compare to BMS‐931699.
The data are from BMS‐931699.
Taking all the above analyses into considerations, Yang et al. proposed an RO ≤ 10% to determine the MRSD from the MABEL approach. To estimate the 10% CD28 RO of BMS‐931699, the EC10 (0.32 nM) was obtained from the more sensitive in vitro human MLR assay (EC50 2.9 ± 1.2 nM) rather than the in vitro human whole blood RO EC50 (4.4 ± 0.9 nM). This is with the consideration that CD28 is a high‐risk target and both in vitro human MLR and RO EC50 are similar (1.5 fold), and the MLR assay is a sensitive way to reflect the CD28 blockade as demonstrated in nonclinical species. The MABEL‐based FIH starting dose was then calculated as the EC10 multiplied by the human plasma volume of 40 ml kg−1, which led to a dose of 10 µg for a 70‐kg human subject.
In the article, the Kd of 0.4 ± 0.28 nM from Biacore binding assay with human CD28 was also explored for the MABEL determination. Using the K d to calculate the CD28 RO resulting a starting dose (3 µg/70 kg that was threefold less than the dose based on the in vitro MLR assay. The authors further discussed the rationale of selecting the MLR assay approach over the Biacore K d method for the CD28 RO calculation. That is, the Biacore system is an artificial system with the CD28 receptors immobilized on a chip, and there is no natural ligand present to compete with binding. In comparison, the in vitro MLR assay may represent a closer approximation to the in vivo environment where cell–cell interactions were preserved.
The MRSD of BMS‐931699 from the GLP toxicology approach was also determined, as shown in Table 4.1. As expected, BMS‐931699 is well tolerated in cynomolgus monkeys, with the NOAEL determined to be 15 mg kg−1. Based on FDA guidance of estimating the MRSD in Initial Clinical Trials for therapeutics in adult healthy volunteers, the authors calculated HED based on the body surface area conversion to 5 mg kg−1. The MRSD of 0.5 mg kg−1 (or 29 mg/60 kg) is derived by applying a tenfold safety factor, which is 2900‐fold higher than the MABEL‐based starting dose of 10 µg [16]. In the FIH study with BMS‐931699, it was revealed that a single dose of 10 μg led to < 10% RO and a single dose of 25 mg led to ≥80% CD28 RO in healthy subjects [40].
4.5.2 Case Study II: MABEL Determination for Anti‐CD40L Receptor Antagonist BMS‐986004
The CD40 ligand (CD40L) and CD40 are members of the TNF and tumor necrosis factor receptor (TNFR) superfamily, respectively, and mediate the activation of innate and adaptive immune cells. CD40 is prominently expressed on APCs, whereas CD40L is induced on activated T cells and constitutively expressed on platelets. The CD40‐CD40L pathway has been an attractive therapeutic target for modulation of autoimmune disease and transplant rejection. Early clinical studies with two anti‐CD40L antibodies (hu5c8 IgG1 [BG9588] from Biogen; IDEC‐131 IgG1 from IDEC) showed efficacy in patients with immune thrombocytopenic purpura (ITP), and a positive clinical effect was noted with hu5c8 IgG1 in lupus nephritis patients [42–44]. However, further development of these molecules was halted due to drug‐related thromboembolism (TE) incidents occurred in treated patients [42,45]. The relationship of TE and the anti‐CD40L antibodies was carefully investigated. Xie et al. demonstrated that anti‐human CD40L antibodies with wild‐type human IgG1, such as hu5C8 and IDEC131, are able to cross‐link the platelets through binding of CD40L and FcγRIIa receptors (via their human IgG1 Fc portion) that are both expressed on the platelets [46]. The cross‐linking of the platelets leads to platelet activation and thromboembolism.
The revelation that human IgG1 Fc binding to FcγRIIa on platelets is responsible for the CD40L‐mediated platelet activation leads to the development of BMS‐986004 that is aimed to be efficacious for treating autoimmune diseases and transplant rejection but has a reduced TE risk [47]. BMS‐986004 is a dimeric, anti‐human CD40L antagonistic dAb fused to a mutated human IgG1 Fc with diminished FcγRIIa binding capability (EC50 > 3 µM vs. 240 nM of hu5c8 with wild type human IgG1 Fc). Nonclinical studies of BMS‐986004 and BMS‐986003 (differing from BMS‐986004 by one additional amino acid at the N terminus) showed the lack of platelet activation or TE in the in vitro and in vivo preclinical studies [47]. However, due to the TE incidents observed with previous two anti‐CD40L mAbs in previous nonclinical and clinical studies, a MABEL‐based approach was adopted to select a safe FIH starting dose of BMS‐986004 in order to ensure the safety of human healthy subjects.
Because the interaction of BMS‐986004 with CD40L, depending on its expression on platelets or activated T‐cells, may lead to distinct pharmacological responses, the MABEL of BMS‐986004 was determined to mitigate both the known risk of platelet activation/TE and the unknown risk of blocking T‐cell co‐stimulation.
The MABEL approach to mitigate the TE risk associated with the CD40L/antibody‐mediated platelet activation used hu5c8 as a benchmark. Hu5c8 exhibited a similar in vitro potency as BMS‐986004 in human and nonhuman primate primary B cell assays [47]. In cynomolgus monkeys, the suppressions of KLH‐induced IgG production were comparable between BMS‐986003 and hu5c8 (in vivo plasma EC50 of 74 ± 14 and 60 ± 18 nM, respectively, unpublished data). The in vitro and in vivo pharmacological effects between hu5C8 and BMS‐986004/BMS‐986003 provided a basis to leverage the nonclinical and clinical data of hu5c8 to determine the MABEL of BMS‐986004.
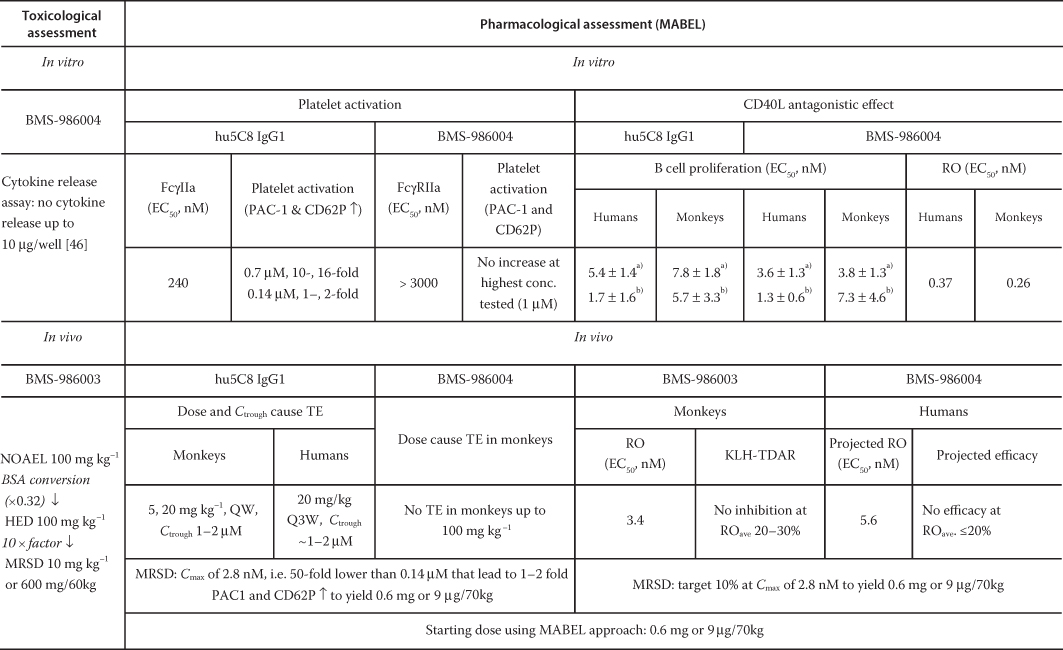
In the human whole blood platelet activation assay, BMS‐986004 did not activate platelets (no elevation of PAC‐1 and CD62P) at the concentration (1 µM) tested. Under the same testing condition, hu5c8 showed a robust platelet activation (>20‐fold elevation of PAC‐1 and CD62P) [47]. In a separate study, hu5c8 at 0.7 µM showed maximum platelet activation (10‐ and 16‐fold elevation of PAC‐1 and CD62P over the baseline, respectively), whereas at the lowest concentration (0.14 µM) tested, a ∼10% of the maximum response was observed (PAC‐1 and CD62P elevated by a respective one‐ and twofold over the baseline, unpublished data). In vivo assessment of BMS‐986004 TE risk was also evaluated in rhesus monkeys. No evidence of thrombosis or TE was observed in the rhesus monkeys treated with BMS‐986004 up to 100 mg kg−1 given weekly. In contrast, hu5c8 dosed weekly at 5 and 20 mg kg−1 led to a thrombosis incidence of 1/4 and 6/12 in rhesus monkeys at an exposure lower than or comparable to that of BMS‐986004 [48]. In humans, hu5c8 caused TE in lupus patients when given at 20 mg kg−1 once‐every‐three weeks [42]. The estimated trough concentrations of hu5c8 in rhesus monkeys and humans where TE was observed were ∼1–2 µM, which is in good agreement with the concentration (0.7 µM) that showed the maximum platelet activation in the in vitro settings, suggesting that the in vitro data may be predictive of the TE potential in vivo. Although no platelet activation was observed with BMS‐986004 either in vitro or in rhesus monkeys, the concentration at which hu5c8 resulted in no observable platelet activation in vitro would be a conservative way to determine the MABEL of BMS‐986004 for this high‐risk target. In this case, the MABEL was determined as a dose at which the C max of BMS‐986004 was below the concentration of hu5C8 that yielded no observable platelet activation in vitro. Because hu5c8 still exhibited 10–13% of maximal platelet activation at the lowest tested concentration (0.14 µM) in the in vitro study, a safety factor of 50‐fold was applied to the lowest concentration to yield a concentration of 2.8 nM (i.e. 0.14 µM/50 = 2.8 nM) that is considered no effect (Table 4.2). Consequently, the C max of 2.8 nM was converted to the corresponding human dose using a plasma volume of 0.04 l kg−1, which yielded the MABEL dose of approximately 9 µg kg−1. For a 70‐kg human subject, this corresponded to a dose of ∼0.6 mg.
Table 4.2 BMS‐986004 FIH starting dose selection using MABEL approach.
|
EC50 from CD40L trimer assay [47].
EC50 obtained from CHO‐CD40L assay [47].
The MABEL was also determined using a second approach based on the intended pharmacologic action of BMS‐986004. In this regard, the connectivity of the CD40L RO between in vitro and in vivo was evaluated. The in vivo CD40L RO on CD4+ T‐cells in whole blood was examined in cynomolgus monkeys, where the in vivo RO EC50 values, with a Hill factor of 3, was estimated to be 3.4 nM. These values were 15‐fold higher than the corresponding in vitro RO EC50 of 0.26 nM for cynomolgus, respectively. To estimate the extent of the CD40L RO in humans, the human in vivo RO EC50 of 5.6 nM was scaled from the in vitro RO EC50 of 0.37 nM by 15‐fold.
Furthermore, the relationship between the CD40L RO on CD4+ T‐cells in whole blood and the inhibition of KLH‐induced antigen response as the intended PD response was examined in monkeys. In cynomolgus monkeys, after a single subcutaneous (SC) dose of BMS‐986003, no inhibition of the KLH‐induced IgG response was observed at 0.2 mg kg−1, where the maximum RO was achieved at 99%, with an average RO of 20–30% over the study duration. On the other hand, 12% and 40% inhibition of the KLH‐induced IgG response (when estimated as the area under the average IgG response‐time curve up to Day 43) was observed at 2 and 20 mg kg−1 doses, with the average RO over the study duration of 40% and 75%, respectively. Collectively, these data indicate that an in vivo CD40L RO below 20% was unable to produce a significant inhibition of the KLH‐induced IgG response.
The relevance of the KLH‐induced IgG response in cynomolgus monkeys to the efficacy in patients with ITP was further examined with hu5c8 [43]. At the clinical efficacious dose of 20 mg kg−1, the estimated systemic exposure of hu5c8 in patients was ∼2 higher than that in cynomolgus monkeys at the same dose, where a 97% inhibition of the KLH‐induced IgG response was observed. This suggests that a complete inhibition of KLH‐induced IgG response may be required for clinical efficacy. To that extent, a dose with a maximum RO ≤ 20% is unlikely to achieve a meaningful efficacy in ITP patients.
To calculate the MABEL based on the intended pharmacologic effect, a CD40L RO of 10% was used. Based on the anticipated human in vivo RO EC50 (5.6 nM) along with a Hill factor (γ) of 3 derived from the PK/PD modeling of the in vivo RO data in cynomolgus and rhesus monkeys, the C
max value corresponding to a RO of 10% in humans was predicted to be 2.7 nM, using the equation ![]() . Accordingly, the C
max was then converted to the corresponding human dose using a plasma volume of 0.04 l kg−1, which yielded the MABEL dose of ∼9 µg kg−1. For a 70‐kg human subject, this also resulted in a MABEL dose of ∼0.6 mg that was in agreement with the earlier described approach.
. Accordingly, the C
max was then converted to the corresponding human dose using a plasma volume of 0.04 l kg−1, which yielded the MABEL dose of ∼9 µg kg−1. For a 70‐kg human subject, this also resulted in a MABEL dose of ∼0.6 mg that was in agreement with the earlier described approach.
Taken together, same MABEL dose of 0.6 mg was derived using two approaches to warrant no expected platelet activation and minimal intended pharmacological effect. At the MABEL dose, the predicted human exposure was more than 15 000‐fold below the NOAEL observed in a one month toxicology study in monkeys (100 mg kg−1). Therefore, the MABEL dose of 0.6 mg was recommended to be a safe FIH starting dose and used successfully in the FIH trial.
4.5.3 Case Study III: MABEL Determination for MOXR0916, an Agonistic Antibody Targeting OX40
MOXR0916 is a humanized agonistic IgG1 mAb targeting OX40, a costimulatory TNF super family receptor transiently expressed on CD4+ and CD8+ T cells during antigen‐specific priming [49]. MOXR0916 agonizes OX40 on activated CD8+ T cells to enhance effector T cell activation and proliferation, and inhibits or depletes inhibitory regulatory T cells, both of which are considered promising MOA in complementary to existing immune checkpoint inhibitors for cancer immunotherapies. MOXR0916 is being developed for the treatment of refractory solid tumors in early phase clinic studies. Because of the agonistic nature of MOXR0916, the FIH starting dose of MOXR0916 was determined after considering the totality of toxicology and pharmacology data [50].
Sukumaran presented the translational pharmacology and FIH starting dose projection of MOXR0916 in the National Biotechnology Conference (NBC) in 2015 [50]. In healthy cynomolgus monkeys, MOXR0916 was well tolerated up to the highest tested dose of 30 mg kg−1. The peripheral blood OX40 receptor occupancy was also measured. However, both the safety data and the peripheral blood RO were not used for the starting dose selection. Sukumaran explained that (i) the healthy monkeys were not predictive of MOXR0916 on‐target toxicity, because OX40 was transiently expressed on activated T cells and healthy monkeys enrolled in the study have negligible activated T cells over the study duration and (ii) without the T‐cell activation, there is a lack of connectivity of systemic RO with the safety and pharmacological effect. As a result, Sukumaran decided the starting dose of MOXR0916 based on the in vivo efficacy and MOA‐related PD responses of its mouse surrogate, PRO307205, an anti‐mouse OX40 with mIgG2a Fc in a mouse syngeneic breast tumor model (EMT6).
In the EMT6 tumor model, the mice treated with PRO307205 at 0.1, 1, and 10 mg kg−1 showed an average of 10%, 30%, and 40% complete response, respectively. Consistently, PRO307205 also exhibited dose‐dependent CD8+ T cell proliferation and Treg reduction in blood and tumors, with the lowest dose lead to the MOA‐related PD modulation in mice was 0.1 mg kg−1. Taking together, the minimal pharmacological active dose (MPAD) in the mouse tumor model was determined to be 0.1 mg kg−1. After adjusting mouse and projected human PK difference and factor in approximately eightfold higher affinity of MOXR0916 for human OX40 than PRO307205 for murine OX40, the starting dose of MOXR0916 was 2 µg kg−1 (∼200 µg flat dose).
It is worth to note that Sukumaran used the MPAD rather than the MABEL for the MOXR0916 FIH starting dose. Nevertheless, MABEL, which focuses on the biological effects takes into account of pharmacological activities of MPAD in the starting dose selection. Therefore, in this chapter, we consider both MPAD and MABEL to capture the on‐target pharmacological/biological effect as compare to the NOAEL/HNSTD to the toxicological effect.
4.5.4 Case Study IV: MABEL Determination for Bispecific Immunomodulatory P‐cadherin LP‐DART (PF‐06671008) in Immune‐oncology
Anti‐P‐cadherin human IgG1 Fc‐containing DART (dual‐affinity retargeting) (PF‐06671008) is a bispecific immunomodulatory biotherapeutic molecule that are designed to bind P‐cadherin overexpressed on breast tumors while simultaneously binding the CD3 receptors on T cells to form a tri‐molecular complex of drug, tumor cell, and T cell. The formation of the tri‐molecular complex results in killing of the target tumor cells either due to granzymes and perforin‐induced cell lysis or due to cytokine release caused by T‐cell activation [51]. The half‐life of PF‐06671008 was extended through incorporating an Fc fragment that does not bind to FcγRs but retains the binding with FcRn.
The mechanism of action of PF‐06671008 is similar to several clinical anti‐CD3 bispecific therapeutics, such as anti‐CD3/PSMA (prostate specific membrane antigen) and anti‐CD3/CEA (carcinoembryonic antigen) constructs, through cross‐linking T cells and cancer cells to activate T‐cell mediated cell killing. The dose limiting toxicities of these anti‐CD3 bispecific agents were mostly CRS and infusion‐related reaction (IRR) [3]. It is likely that PF‐06671008 may encounter similar immune‐related toxicity. As a result, MABEL was used for the FIH staring dose selection of PF‐06671008. Chen et al. recently reported an integrated mechanistic PK/PD modeling approach for the MABEL projection of PF‐06671008 [31]. The authors first evaluated in vitro time‐course of tumor cell killing and T‐cell proliferation in the presence of different ratios of tumor and T cells and concentrations of the drug. Using PK/PD modeling, the authors were able to estimate the concentration of the tri‐molecule (tumor‐T‐cell‐drug) formed in the in vitro assay system and its relationship to the in vitro tumor cell killing. From the model, the in vitro tri‐molecule concentration corresponding to 20% tumor cell killing (EC20 = 1.2 × 10−6 nM) was determined and used as the basis for the MABEL determination (Table 4.3). The authors then predicted the in vivo tri‐molecule concentration in patient tumors using a PK/PD model developed through incorporating several mechanistic processes: human plasma PK predicted from monkey PK using allometry; soluble P‐cadherin level in the circulation (turnover rate constant predicted from monkey data and allometrically scaled to humans); drug distribution kinetics into the tumor; and target‐related parameters including P‐cadherin and CD3 expressions, T‐cell populations in blood and tumors, tumor cell population, etc. Based on the established in vivo human PK/PD model, a MABEL dose following a one hour infusion was predicted to be 1.9 ng/kg/week. At this dose, the average steady‐state tri‐molecule concentration in tumors was equal to the in vitro concentration corresponding to 20% tumor cell killing.
Table 4.3 P‐cadherin LP‐DART FIH starting dose selection using MABEL approach.
Source: Adapted from Chen et al. 2016 [31].
| In vitro assay | Efficacy variable | MABEL | Starting dose a (ng kg−1wk−1) | |
| PK/PD‐driven approach | Kinetic cytotoxicity assay | EC20, syn = 1.2 × 10−6 nM |
Maximum tumor synapse Conc. < EC20, syn |
1.9 |
| PK‐driven approach | Cytokine release assay | EC20, CRA = 0.025 ng ml−1 | C max < EC20, CRA | 1.5 |
| Cytotoxicity assay | EC20, CTL = 0.01 ng ml−1 | C ave < EC20 CTL | ||
| Receptor occupancy (RO) | Binding assay | EC10, RO = 6 (P‐cad) and 134 (CD3) ng ml−1 | C max < EC10, RO |
360 (P‐cad) 8300 (CD3) |
CTL, clearance.
a One hour infusion.
Beyond the mechanistic PK/PD‐modeling‐based MABEL dose projection, the authors further explored other methodologies. One approach was the in vitro cytotoxicity EC20 and in vitro cytokine (IL‐6) release EC20 (Table 4.3). Because cytotoxicity was generally believed to be driven by the cumulative drug exposure (i.e. C ave, average drug concentration) and cytokine release is often determined by the C max, a starting dose was calculated to be 1.5 ng/kg/week, to ensure that the C ave less than the in vitro cytotoxicity EC20 and the C max below the cytokine release EC20. Lastly, the RO of PF‐06671008 to P‐cadherin and CD3 was calculated using the respective K d of 0.47 and 11.4 nM. The drug concentration corresponding to 10% RO for P‐cadherin and CD3 was calculated to be 6 and 134 ng ml−1, respectively. As a result, the corresponding MABEL dose based on the predicted human PK was projected to be 360 (P‐cadherin) and 8300 (CD3) ng/kg/week, respectively. In this case, a 10% RO for an immune agonist appears to be too high in comparison with a MABEL dose that was derived from the in vitro cytokine release data. Based on these integrated analyses, a dose of 1.5 ng/kg/week was proposed as the FIH starting dose.
4.6 Discussion and Conclusion
Among the four examples discussed above, the CD28, CD40L, and CD3 receptors, the respective target of BMS‐931699, BMS‐986004, and PF‐06671008, are well‐known high‐risk targets in the clinic, whereas MOXR0916 is a novel agonist of OX40, a costimulatory receptor on the T cells, that may have unforeseen agonistic toxicity. The nonclinical safety evaluation of BMS‐931699 and BMS‐986004 demonstrated safety in cynomolgus monkeys with the NOAEL established at their top toxicology doses. As a result, the MRSDs of BMS‐931699 and BMS‐986004 derived from the NOAEL exceeded the doses to achieve maximal PD responses, and, thus, are not suitable for the FIH starting doses. MOXR0916 was well tolerated in monkeys up to the highest tested dose of 30 mg kg−1 [49]. However, healthy monkeys were not considered predictive of MOXR0916 on‐target toxicity because OX40 was transiently expressed on activated T cells and healthy monkeys enrolled in the study have negligible activated T cells. In case of PF‐06671008, nonclinical safety data have not been reported. Nevertheless, the investigators suggested that because PF‐06671008 was a highly potent agonistic molecule, the HNSTD approach may lead to high receptor occupancy and could cause toxicity from exaggerated pharmacology [31]. Taken together, the four case studies clearly suggest that despite nonclinical toxicology studies were conducted in monkeys, the FIH starting doses of these IMPTs were selected based on the pharmacological assessment to mitigate on‐target toxicity resulting from exaggerated pharmacology.
A variety of MABEL approaches have been used to set the FIH starting dose by employing in vitro binding and activity data, in vivo rodent and monkey RO, efficacy and PD data. RO is often determined in nonclinical studies as an important indicator of the concentration required for in vivo target engagement. In the retrospective analysis of TGN1412 starting dose, a 10% RO is justified for the MABEL of high‐risk agonistic antibodies [7,27]. Since then, the 10% RO (or slightly lower and higher values) derived either from ex vivo whole blood assay or sometime the in vitro binding constant (K d) has been used extensively as the MABEL [6]. By no means, however, the MABEL should be simply calculated from an in vitro assay and based on an arbitrary cut‐off value of 10%. For example, in some cases of immune antagonists, FIH starting dose with a C max to target 10% RO result in a microgram dose range, which were 100–1000‐fold less than the doses given to patients with acceptable/manageable toxicities and may lead to years of dose escalation to the therapeutic dose [3]. While obtaining safety data is the main goal of Phase 1 trials, there is a clear need to minimize patient receiving subtherapeutic doses. In other cases, especially the agonistic CD3‐bispecific antibodies, the 10% RO may already lead to agonistic effect [3]. Therefore, prior to use RO for the MABEL, the connectivity of target engagement (RO) to the pharmacological effect needs to be established. As illustrated in Cases I and II, the RO of BMS‐931699 and BMS‐986004 to the respective CD28 and CD40 receptor was quantitatively correlated with the suppression of the TDAR response using PK/PD modeling, with a 10% RO for minimal TDAR response. In contrast, in the case of MOXR0916, the sponsors used the efficacy of PR0307205, the mouse surrogate anti‐OX40 antibody, in mouse tumor models rather than the whole blood RO to select the starting dose due to the lack of connectivity of systemic RO with the safety and pharmacological effects.
Although two of the four case examples were related to the immune‐oncology, the underlying principles could be readily adopted for the development of immune‐mediated inflammatory diseases.
In addition to the main focus of this chapter on the FIH starting dose selection, innovative FIH trial designs are critical to provide another venue that allows a rapid attainment of therapeutically active doses while maintaining patient safety. For example, employing a sentinel cohort approach in the FIH may be considered to help mitigate the risk of first dosing in humans. In BMS‐931699 FIH trail, there were two sentinel cohorts for each dose panel planned: on day 1, one subject receives BMS‐931699 and another received placebo in a blinded setting; an additional BMS‐931699 and placebo subject pair were randomized on day 2. The remaining subjects of that dose panel were dosed simultaneously on day 3. The sentinel cohorts allowed 24 hours monitoring of the first treated subject before treating the second subject; therefore, avoided the situation of TGN1412 where six subjects were simultaneously dosed and developed irreversible life‐threatening side effects [40]. Recently, in order to ensure cancer patient safety without a compromise of the FIH study duration, the FIH protocol of novel IMPTs with very low starting dose (such as 0.0001 mg kg−1) adopts a single patient cohort or intra‐patient dose escalation design, as opposed to the traditional 3 + 3 design, in the first few cohorts until a specific safety event/therapeutic goal is reached. The dose escalation in single patient or intra‐patient in the first several cohorts either follows a one‐log dose increment of Bayesian adaptive design to achieve fast dose escalation or skip cohorts. These innovative designs significantly shorten the FIH phase I study from traditional five years to two years [3,52].
In summary, we described the application of MABEL in FIH starting dose selection for IMPTs in this chapter. Our goal is not limited to whether or not using the MABEL in the FIH starting dose selection; rather, is to understand the dose/exposure‐response curve of pharmacology and toxicology data for these immune modulators in the nonclinical setting, with an aim to better inform the FIH dose selection and study design. In this regard, PK/PD modeling‐based approaches, advocated in the MABEL determination, provide a useful framework to integrate all the nonclinical pharmacology and toxicology data and understand dose/exposure–response relationships in a quantitative manner. With further improvements in the knowledge of disease mechanisms and target functions, the FIH starting dose selection will be tailored to ensure the safety and the speed to achieve therapeutic benefits in patients.
References
- 1 Hahn, A.W., Gill, D.M., Pal, S.K., and Agarwal, N. (2017). The future of immune checkpoint cancer therapy after PD‐1 and CTLA‐4. Immunotherapy 9 (8): 681–692.
- 2 Wolfe, R.M. and Ang, D.C. (2017). Biologic therapies for autoimmune and connective tissue diseases. Immunol. Allergy Clin. N. Am. 37 (2): 283–299.
- 3 Saber, H., Gudi, R., Manning, M. et al. (2016). An FDA oncology analysis of immune activating products and first‐in‐human dose selection. Regul. Toxicol. Pharmacol. 81: 448–456.
- 4 Brennan, F.R., Morton, L.D., Spindeldreher, S. et al. (2010). Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2: 233–255.
- 5 Brennan, F.R., Cauvin, A., Tibbitts, J., and Wolfreys, A. (2014). Optimized nonclinical safety assessment strategies supporting clinical development of therapeutic monoclonal antibodies targeting inflammatory diseases. Drug Dev. Res. 75 (3): 115–161.
- 6 Suh, H.Y., Peck, C.C., Yu, K.S., and Lee, H. (2016). Determination of the starting dose in the first‐in‐human clinical trials with monoclonal antibodies: a systematic review of papers published between 1990 and 2013. Drug Des. Devel. Ther. 8 (10): 4005–4016.
- 7 Duff, G. 2006. Expert scientific group on phase one clinical trials final report. http://webarchive.nationalarchives.gov.uk/20130105143109/http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_073165.pdf (accessed 21 November 2017).
- 8 Muller, P.Y. and Brennan, F.R. (2009). Safety assessment and dose selection for first‐in‐human clinical trials with immunomodulatory monoclonal antibodies. Clin. Pharmacol. Ther. 85 (3): 247–258.
- 9 Stebbings, R., Findlay, L., Edwards, C. et al. (2007). “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J. Immunol. 179 (5): 3325–3331.
- 10 Rudmann, D.G., Alston, J.T., Hanson, J.C., and Heidel, S. (2013). High molecular weight polyethylene glycol cellular distribution and PEG‐associated cytoplasmic vacuolation is molecular weight dependent and does not require conjugation to proteins. Toxicol. Pathol. 41 (7): 970–983.
- 11 Michot, J.M., Bigenwald, C., Champiat, S. et al. (2016). Immune‐related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54: 139–148.
- 12 Fraser, G., Smith, C.A., Imrie, K., and Meyer, R., and the Hematology Disease Site Group of Cancer Care Ontario's Program in Evidence‐Based Care (2007). Alemtuzumab in chronic lymphocytic leukemia. Curr. Oncol. 14: 96–109.
- 13 Kimby, E. (2005). Tolerability and safety of rituximab (Mabthera). Cancer Treat. Rev. 31: 456–473.
- 14 FDA (2010). Guidance for industry S9 nonclinical evaluation for anticancer pharmaceuticals.
- 15 Haley, P.J. (2017). The lymphoid system: a review of species differences. J. Toxicol. Pathol. 30 (2): 111–123.
- 16 FDA (2005). Guidance for industry: Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers (accessed 21 November 2017) https://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf#search=‘guidekines+for+industry+sfe+starting’.
- 17 Deng, R., Iyer, S., Theil, F. et al. (2011). Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs 3 (1): 61–66.
- 18 Ling, J., Zhou, H., Jiao, Q., and Davis, H. (2009). Interspecies scaling of therapeutic monoclonal antibodies: initial look. J. Clin. Pharmacol. 49 (12): 1382–1402.
- 19 Wang, W. and Prueksaritanout, T. (2010). Prediction of human clearance of therapeutic proteins: simple allometric scaling method revisited. Biopharm. Drug Dispos. 31 (4): 253–263.
- 20 Singh, A.P., Krzyzanski, W., Martin, S.W. et al. (2015). Quantitative prediction of human pharmacokinetics for mAbs exhibiting target‐mediated disposition. AAPS J. 17 (2): 389–399.
- 21 Glassman, P.M. and Balthasar, J.P. (2016). Physiologically‐based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 43 (4): 427–446.
- 22 Day, L. (2016). Ten years after the 'Elephant Man' drug trial. Lexology.
- 23 Suntharalingam, G., Perry, M.R., Ward, S. et al. (2006). Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. New Engl. J. Med. 355: 1018–1028.
- 24 Hünig, T. (2012). The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 12 (5): 317–318.
- 25 Eastwood, D., Findlay, L., Poole, S. et al. (2010). Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T‐cells. Br. J. Pharmacol. 161 (3): 512–526.
- 26 Römer, P.S., Berr, S., Avota, E. et al. (2011). Preculture of PBMCs at high cell density increases sensitivity of T‐cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood 118 (26): 6772–6782.
- 27 Tyrsin, D., Chuvpilo, S., Matskevich, A. et al. (2016). From TGN1412 to TAB08: the return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 34 (4): 45–48.
- 28 Yang, Z., Wang, H., Salcedo, T.W. et al. (2015). Integrated pharmacokinetic/pharmacodynamic analysis for determining the minimal anticipated biological effect level of a novel anti‐CD28 receptor antagonist BMS‐931699. J. Pharmacol. Exp. Ther. 355 (3): 506–515.
- 29 Dudal, S., Hinton, H., Giusti, A.M. et al. (2016). Application of a MABEL approach for a T‐cell‐bispecific monoclonal antibody: CEA TCB. J. Immunother. 39 (7): 279–289.
- 30 Vugmeyster, Y., Rohde, C., Perreault, M. et al. (2013). Agonistic TAM‐163 antibody targeting tyrosine kinase receptor‐B: applying mechanistic modeling to enable preclinical to clinical translation and guide clinical trial design. MAbs 5 (3): 373–383.
- 31 Chen, X., Haddish‐Berhane, N., Moore, P. et al. (2016). Mechanistic projection of first‐in‐human dose for bispecific immunomodulatory P‐cadherin LP‐DART: an integrated PK/PD modeling approach. Clin. Pharmacol. Ther. 100 (3): 232–241.
- 32 Agoram, B.M. (2009). Use pharmacokinetic/pharmacodynamic modelling for starting dose selection in first‐in‐human trials of high‐risk biologics. Br. J. Clin. Pharmacol. 67 (2): 153–160.
- 33 Muller, P., Milton, M., Lloyd, P. et al. (2009). The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr. Opin. Biotechnol. 20 (6): 722–729.
- 34 Bugelski, P.J., Achuthanandam, R., Capocasale, R.J. et al. (2009). Monoclonal antibody‐induced cytokine‐release syndrome. Expert. Rev. Clin. Immunol. 5 (5): 499–521.
- 35 Brennan, F.R. and Kiessling, A. (2017). In vitro assays supporting the safety assessment of immunomodulatory monoclonal antibodies. Toxicol. In Vitro 17: 30064.
- 36 Kirton, C.M., Gliddon, D.R., Bannish, G. et al. (2011). In vitro cytokine release assays: reducing the risk of adverse events in man. Bioanalysis 3 (23): 2657–2663.
- 37 Vessillier, S., Eastwood, D., Fox, B. et al. (2015). Cytokine release assays for the prediction of therapeutic mAb safety in first‐in man trials – whole blood cytokine release assays are poorly predictive for TGN1412 cytokine storm. J. Immunol. Methods 424: 43–52.
- 38 Stewart, R., Hammond, S.A., Oberst, M., and Wilkinson, R.W. (2014). The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. Immunother. Cancer 2: 29–39.
- 39 EMA (2007). Guideline on requirements for first‐in‐man clinical trials for potential high‐risk medicinal products.
- 40 Shi, R., Honczarenko, M., Zhang, S. et al. (2017). Pharmacokinetic, pharmacodynamic, and safety profile of a novel anti‐CD28 domain antibody antagonist in healthy subjects. J. Clin. Pharmacol. 57 (2): 161–172.
- 41 Suchard, S.J., Davis, P.M., Kansal, S. et al. (2013). A monovalent anti‐human CD28 domain antibody antagonist: preclinical efficacy and safety. J. Immunol. 191 (9): 4599–4610.
- 42 Boumpas, D.T., Furie, R., Manzi, S. et al., BG9588 Lupus Nephritis Trial Group(2003). A short course of BG9588 (anti‐CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 48 (3): 719–727.
- 43 Patel, V.L., Schwartz, J., and Bussel, J.B. (2008). The effect of anti‐CD40 ligand in immune thrombocytopenic purpura. Br. J. Haematol. 141 (4): 545–548.
- 44 Kuwana, M., Nomura, S., Fujimura, K. et al. (2004). Effect of a single injection of humanized anti‐CD154 monoclonal antibody on the platelet‐specific autoimmune response in patients with immune thrombocytopenic purpura. Blood 103 (4): 1229–1236.
- 45 Sidiropoulos, P. and Boumpas, D. (2004). Lessons learned from anti‐CD40L treatment in systemic lupus erythematosus patients. Lupus 13 (5): 391–397.
- 46 Xie, J.H., Yamniuk, A.P., Borowski, V. et al. (2014). Engineering of a novel anti‐CD40L domain antibody for treatment of autoimmune diseases. J. Immunol. 192 (9): 4083–4092.
- 47 Kim, S.C., Wakwe, W., Higginbotham, L.B. et al. (2017). Fc‐silent anti‐CD154 domain antibody effectively prevents nonhuman primate renal allograft rejection. Am. J. Transplant. 17 (5): 1182–1192.
- 48 Wakefield, I.D., Harari, O., Hutto, D. et al. (2010). An assessment of the thromboembolic potential of CDP7657, a monovalent Fab' PEG anti‐CD40L antibody, in rhesus macaques. Arthritis Rheum. (62 Suppl): 1243.
- 49 Prell, R., Halpern, W., Beyer, J. et al. (2014). Nonclinical safety assessment of a humanized anti‐OX40 agonist antibody, MOXR0916. Eur. J. Cancer 50 (suppl 6): 136.
- 50 Sukumaran S. (2014). Translational Pharmacology and First‐in‐Human Dose Projections for Cancer Immunotherapy Drugs: Case Study of MOXR0916 (Anti‐OX40 Antibody) https://zerista.s3.amazonaws.com/item_files/dd34/attachments/39146/original/144_nbc‐15‐85.pdf (accessed 21 November 2017).
- 51 Root, A.R., Cao, W., Li, B. et al. (2016). Development of PF‐06671008, a highly potent anti‐P‐cadherin/anti‐CD3 bispecific DART molecule with extended half‐life for the treatment of cancer. Antibodies 5: 6.
- 52 David Guédé, Reigner, B., Vandenhende, F. et al. (2014). Bayesian adaptive designs in single ascending dose trials in healthy volunteers. Br. J. Clin. Pharmacol. 78 (2): 393–400.