
10
Application of Pharmacometrics and Systems Pharmacology to Current and Emerging Biologics in Inflammatory Bowel Diseases
Sihem Ait‐Oudhia Yi Ting (Kayla) Lien Sumit Basu Lawrence Lesko and Stephan Schmidt
University of Florida, Center for Pharmacometrics and Systems Pharmacology, Department of Pharmaceutics, College of Pharmacy, Orlando, FL, USA
10.1 Introduction
Inflammatory bowel diseases (IBDs) represent a group of intestinal disorders. Together, they are the third highest risk factor for colorectal cancer [1–4]. In the United States, IBDs affect approximately 1.3 million people. The majority of patients is between 15 and 30 years old [5,6]. IBDs are complex disorders, which evolve through several convoluted immunological pathways. They are characterized by processes of remission and relapse, which ultimately result in functional impairment of the gut wall. IBDs are typically classified into two major types: ulcerative colitis (UC) and Crohn's disease (CD). This classification is essentially based on the location of the inflammation in the gastrointestinal tract (GIT) [7,8]. The Center for Disease Control and Prevention defines IBD as “a broad term that describes conditions characterized by chronic inflammation of the GIT with the two most common IBD being UC and CD.” Currently, there is no gold standard criteria for the diagnosis of IBD neither for distinguishing between UC and CD [9–13]. For example, one way to decipher UC from CD is through examination of their distinct and overlapping clinical and pathological features. For instance, CD may localize anywhere on the GIT from the mouth to the rectum in a disrupted pattern, whereas UC localizes specifically in the rectum and the colon in an uninterrupted pattern [14]. Moreover, the pattern for inflammation varies between UC and CD. While in UC the inflammation is confined in the mucosa, in CD, it mainly localizes in the transmural space in the intestinal tissue and can affect all layers of the intestinal tissues.
10.1.1 Pathophysiology of IBD
The pathophysiology of IBD involves many factors in the GIT, which can be anatomical, histological, and immunological resulting in a loss of its homeostasis. Therefore, IBDs compromise the integrity of the intestinal mucosa, which in normal conditions, is usually protected by host mucosal immunity and commensal bacteria. Table 10.1 summarizes the intrinsic and extrinsic risk factors that have been associated with IBD [15,16]. A general depiction of the multilevel intrinsic and extrinsic factors (molecular, cellular, and tissue) involved in the pathophysiology of IBD along with the mechanisms of action of selected U.S. Food and Drug Administration (FDA)‐approved drugs in IBD (Table 10.2) are represented in Figure 10.1. In brief, the host immune system fails to recognize pattern recognition receptors (PRRs), such as Toll‐like receptors, which are found on epithelial and immune cells in the intestine [21]. This inability to differentiate between pathogenic and commensal bacteria causes overproduction of inflammatory cytokines (e.g. tumor necrosis factor (TNF)‐α and interleukins, IL‐12 and IL‐23) activated by inflammatory transcription factors, such as nuclear factor kappa B (NF‐κB) [ 16 ,29]. Yet, the impaired barrier of the GIT system in these genetically predisposed IBD patients allows microbial products and pathogenic bacteria to invade the intestinal mucosa and be presented as antigens to naive CD4 T‐cells. This interaction triggers a cascade of immunological activations, whereby T‐cells bind to colonic endothelial cells through the mucosal vascular cell adhesion molecule‐1 (VCAM‐1) or mucosal addressin cell adhesion molecule‐1 (MAdCAM‐1) [17,30]. VCAM‐1 and MAdCAM‐1 function as docking molecules for the α4β1 (only for VCAM‐1) and the α4β7 integrin dimer [ 17 , 30 ]. These integrins are secondary adhesion molecules whose expression is increased in IBD [20]. They prevent lymphocytes from leaving the intestinal tissue, which subsequently triggers an inflammatory immune response [30] .
Table 10.1 Summary of risk factors for the development of IBD.
| Intrinsic factors | Extrinsic factors |
Genetic factors
|
Diet |
| Antimicrobial peptides | Infections |
| Pathogenic bacteria handling | Stress |
| Autophagy | Medications (e.g. NSAIDs and antibiotics) |
| Cytokines and chemokines | Surgical interventions (e.g. appendectomy) |
NOD2, nucleotide‐binding oligomerization domain‐containing protein 2; IBD5, inflammatory bowel disease 5; IL23R, Interleukin 23 receptor; NSAIDs, nonsteroidal anti‐inflammatory drugs.
Source: Adapted from Ananthakrishnan 2015 [15] and Neurath 2014 [16] .
Table 10.2 Survey of FDA‐approved biologics in IBD and their targets (biomarkers).
| Drug | Year approved by FDA | Biomarker/target | Application | References |
| Entyvio (vedolizumab) | 2014 | α4β7 integrin | UC and CD | [17] |
| Simponi (golimumab) | 2013 | Soluble and transmembrane forms of human TNF‐α | UC | [18] |
| Stelara (ustekinumab) | 2009 | IL‐12 and IL‐23 cytokines | CD | [19] |
| Cimzia (certolizumab pegol) | 2008 | Human TNF‐α | CD | [20] |
UC, ulcerative colitis; CD, Crohn's disease; IL, interleukin; TNF‐α, tumor necrosis factor alpha.
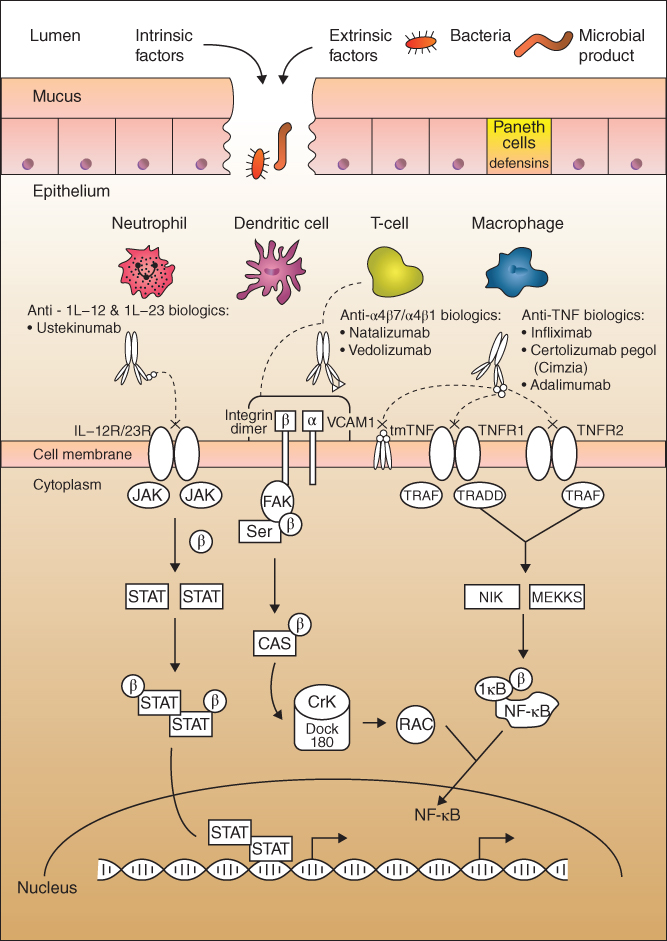
Figure 10.1 Schematic representation of the multiple factors involved in the pathophysiology of inflammatory bowel diseases. Briefly, a bridge in the gastro‐intestinal mucosa barriers may be caused by multiple intrinsic and or extrinsic factors such as invasion of pathogenic bacteria and microbial products and other immune and environmental factors. The impaired barrier further triggers a heightened response from the innate immune cells such as macrophages, neutrophils, T‐cells, and dendritic cells to cause more inflammation. Several signaling proteins in the immune‐inflammatory pathways have been associated with the onset and progression of IBD. They include: IL, interleukin; JAK, Janus kinase; STAT, signaling transducer and activator of transcription; P, phosphate group, VCAM‐1, vascular cell adhesion molecule‐1; FAK, focal adhesion kinase; Src, Src‐family kinases; CAS, cellular apoptosis susceptibility; Crk, chicken tumor virus number 10 regulator of kinase; Rac, Ras‐related C3 botulinum toxin substrate; tmTNF, transmembrane tumor necrosis factor; TNFR1, tumor necrosis factor receptor type 1; TNFR2, tumor necrosis factor receptor type 2; TRAF, tumor necrosis factor receptor‐associated factors; TRADD, tumor necrosis factor receptor type 1‐associated death domain protein; NIK, NF‐kappa‐B‐inducing kinase; IκB, inhibitor of kappa B. (See insert for color representation of this figure.)
Source: Adapted from Neurath 2014 [16] , Shih and Targan 2008 [21] , Singh et al. 2016 [22], Barbalho et al. 2016 [23], de Mattos 2015 [24], Guagnozzi and Caprilli 2008 [25], Koutruba et al. 2010 [26], Pedersen et al. 2014 [27], and Khor et al. 2011 [28].
10.1.2 Current Advances in Biomarkers for IBD
To date, the quest for diagnostic biomarkers for IBD remains challenging. Currently used diagnostics rely on the analysis of blood and fecal samples. They are also referred to as “surrogate biomarkers” as they do not reveal the origin of the inflammation causing IBD. Therefore, prognosis of the IBD trajectory using these surrogate biomarkers is imprecise and necessitates the use of additional diagnostic measures, such as specific clinical symptoms of IBD, radiological, endoscopy, and pathological tests, and many others [ 18 , 19 ]. In this section, we will focus on the most commonly used surrogate biomarkers for IBD: (i) plasma C‐reactive protein (CRP), (ii) fecal calprotectin, (iii) atypical perinuclear antineutrophil cytoplasmic antibodies (pANCA), and (iv) outer membrane porin C (OmpC) are also being covered. A comprehensive overview of all FDA‐approved drugs and its target/biomarkers for IBD is provided in Table 10.2 [ 14 , 18 ,31,32].
10.1.2.1 C‐reactive Protein (CRP)
CRP is a circulating serum protein whose plasma concentration increases up to 350‐fold in IBD [ 18 , 19 ,33]. It is produced by the liver, where its synthesis and release are upregulated by the proinflammatory cytokines IL‐6, TNF‐α, and IL‐1β [ 14 , 18 , 19 , 33 ]. Yet, the role of CRP during the acute phase of inflammation in IBD is not yet fully understood. It is thought that it acts through the activation of the complement cascade by triggering the C1q complex and by binding to the phosphocholine‐containing antigens. As a surrogate biomarker to IBD, it is proven a more accurate predictor for the diagnosis and monitoring of CD progression than UC [ 13 ,34].
10.1.2.2 Fecal Calprotectin
Calprotectin is a calcium and zinc‐binding protein first discovered by Fagerhol [35]. It is found in the cytoplasm of neutrophils, granulocytes, monocytes, and macrophages, but it is released only by activated, stressed, or damaged neutrophils, where it constitutes more than 60% of the cytosolic proteins content [36–39]. Thus, the presence of calprotein in the stools reflects the occurrence of an active inflammation in the GIT, which makes this protein an ideal diagnostic and prognostic biomarker for IBD [38, 39 ]. Several studies have shown that calprotectin is a more sensitive and specific biomarker than CRP, or the atypical pANCA and the OmpC [ 31 , 32 ]. Despite its improved sensitivity and selectivity for IBD, elevated concentrations of fecal calprotectin only indicates that there is activation of the innate immune response in the GIT [40,41]. Hence, it is not specific to IBD only, but rather a very helpful indicator of any mucosal disease activity. It is routinely used in the clinical settings for the monitoring of drug therapy response to biologics in IBD, such as with anti‐TNF‐α treatment [42,43].
CRP and calprotectin are examples of biomarkers used in the general diagnosis of IBD, but they do not specifically decipher between UC and CD. In order to distinguish these two diseases in cases where their clinical and pathological features are similar, other serological biomarkers can be used. They are generally classified in two groups: (i) the group of antibodies targeting microbial antigens and (ii) the group of autoantibodies. In this section, we will cover one example from each group.
10.1.2.3 Atypical Perinuclear Antineutrophil Cytoplasmic Antibodies (pANCA)
Antineutrophil cytoplasmic antibodies (ANCAs) are a group of antibodies targeted against cytoplasmic antigens localized in the azurophil granules of neutrophils. pANCA is a subset of ANCA classified as atypical due to their unknown target antigens and are found in IBD patients [44]. These patients demonstrate atypical perinuclear staining using enzyme immunoassays of immunoglobulin G (IgG) class antineutrophil cytoplasmic antibodies unlike ANCA [45]. On its own, pANCA can be used for indication of UC when diagnosing between CD and UC. However, differential diagnosis of CD and UC is more favorable when combined testing of serological biomarkers is utilized as diagnostic accuracy increases. For example, pANCA becomes a more robust marker when combined with other biomarkers and/or other clinical diagnosis means, such as enteroscopy, and not as a standalone biomarker for a better reliability and prediction of IBD diagnosis [46]. A commonly used combination includes anti‐Saccharomyces cerevisiae antibodies (ASCAs) since ASCAs indicate the presence of CD. Moreover, ASCA sensitivity increases for CD when used in combination with pANCA.
It was found that a combination of pANCA+ and ASCA− is associated with UC and have high specificity (between 80% and 95%), whereas pANCA−/ASCA+ is associated with CD specificity of about 88–93% [47,48]. However, the sensitivity of these assays is low. Thus, one should not assume the lack of UC or CD in negative results. Currently, there are interests in using pANCA as predictors for drug response to anti‐TNFs. The studies have not shown conclusive results, but these studies suggest that pANCA+/ASCA− combination could predict nonresponse effect to anti‐TNF drugs [49–52].
10.1.2.4 Anti‐outer Membrane Porin C (OmpC)
OmpC is an outer membrane protein originally isolated from Escherichia coli. Anti‐OmpC are anti‐microbial antibodies specific to microorganisms, such as yeast, bacteria, and fungi [53]. They are of increasing clinical utility as serological markers for IBD diagnosis with a success rate of 50% of positive results [ 52 –56] either alone as diagnostic tools or in combination with ASCA [57]. Table 10.2 summarizes all FDA‐approved drugs and their targets, which may serve as biomarkers for IBD [ 14 , 18 , 31 , 32 ].
10.1.2.5 Other Mediators of Inflammation
In response to the intrusion of signals, such as microbial products or pathogenic bacteria, cells from the innate immune system including macrophage and monocytes are rapidly activated and release various proinflammatory cytokines including TNF‐α, IL‐1β, and IL‐6 [ 18 ,58,59](Figure 10.1 ). TNF‐α plays a pivotal role in the pathogenesis of IBD in addition to many other inflammatory pathways [60]. It is secreted by various immune cells, such as macrophages, monocytes, and T‐cells. This proinflammatory mediator also impacts on the upregulation of other inflammatory cytokines, such as IL‐1β [61]. Increased plasma concentrations of IL‐1β are found to correlate with the increased risk of development of IBD [62,63]. High levels of IL‐6 plasma concentrations are found in UC and CD patients. Moreover, IL‐6 is proven to be a good predictive biomarker for IBD activity and progression, essentially during relapse and patient therapeutic response to steroids [64,65]. Furthermore, recent studies have also shown that these intrinsic proinflammatory cytokines levels are elevated in IBD patients due to other comorbidity factors, such as obesity [66]. The pathophysiologic link and its clinical impact interplay between obesity and IBD have shown to influence the changes in efficacy for specific IBD drugs in patients of different body mass index [67–72]. Hence, it might be salient to consider the impact of obesity as a covariate in drug development optimization modeling strategies.
10.2 Pharmacological Approaches for the Treatment of IBD
The overall approach for the treatment of IBD is to achieve remission and to prevent disease flares [ 8 , 22 ,73]. This can be accomplished by surgical and/or pharmacological approaches, depending on the disease. Since the last couple of decades, several therapeutic agents, either alone or in combination, have been used to prevent and treat IBD. In general, available therapeutic options for IBD can be classified into various categories including anti‐inflammatory drugs, immunosuppressants, antibiotics, and monoclonal antibodies (mAbs). In this book chapter, we will focus on the quantitative pharmacological approaches by means of targeted therapies, specifically mAbs. Indeed, there is a growing interest by the pharmaceutical industry to develop newer and safer targeted therapies in various types of diseases including IBD. Furthermore, drug companies are expanding from conventional mAbs to complex engineered recombinant antibodies acting on specific targets in the inflammatory pathways involved in IBD pathophysiology [ 73 ,74].
Combination therapy of mAbs with other traditional therapies (see above paragraph) is also common in IBD. This is essentially due to the multitude of immunological factors involved in its pathogenesis, such as the immune cells and their secreted cytokines. The rationale for combining selected anti‐inflammatory agents in IBD depends on various factors including the stage and severity of the disease and the pre‐ or postoperative status of the patient. Although, there is debate about whether or not the combination therapy of an immunosuppressant with a biologic should be pursued due to associated adverse events [75], the ultimate goal from these therapeutic approaches is to maximize drugs' efficacies (i.e. synergistic combinatorial effects) and minimize their toxicities (i.e. reduce patients' side effects) [ 73 , 74 ].
The increasing interest in biologics therapies in IBD is attributed to their high success rate in remission and mucosal healing [76]. This can be ascribed to the desirable characteristics of mAbs, such as target specificity and long elimination half‐life. Since IBD is thought to originate from a dysregulation in the mucosal immune system of the GIT, the purpose of using biologics in IBD is to be able to specifically inhibit immunological molecules, such as cytokines and their cognate receptors, along various immunological pathways. In this section of book chapter, we will focus on several mAbs that have received regulatory approval by the FDA for the indications of UC and CD.
10.2.1 Biologics for the Treatment of IBD
In comparison of the treatment options suggested above, the development of biologics has been accelerated owing to the tremendous advances in the biotechnology field coupled with the growing knowledge of the molecular pathophysiology of IBD. Apart from TNF‐α inhibitors, which have evolved over the years to become the standard of care biologics in IBD, quite few types of biologics, such as anti‐adhesion molecules, anti‐IL‐6R/IL‐2 antibodies, recombinant immunostimulators, and growth factors, have emerged as alternative treatment options to improve IBD patients' outcomes [77]. In this section, current and emerging biologics (Table 10.3) in the treatment of IBD are discussed.
Table 10.3 Summary of the clinical trials for biologics in inflammatory bowel disease.
| Drug | Clinical trial | Dose | Route of administration | Development status | References |
| Infliximab | Not applicable | 5 and 10 mg kg−1 | IV | Approved in EU and USA | [78] |
| ACT I, ACT II | 5 and 10 mg kg−1 | IV | Approved in EU and USA | [78] | |
| Adalimumab | GAIN, CLASSIC I, CHARM | 40/20, 80/40, 160/80 and 40 mg eow | SC | Approved in EU and USA | [79–81] |
| ULTRA‐1, ULTRA‐2 | 80/40, 160/80 and 40 mg eow | SC | Approved in EU and USA | [82,83] | |
| Certolizumab pegol | PRECiSE I, PRECiSE II | 100, 200, and 400 mg | SC | Approved in EU and USA | [84,85] |
| Golimumab |
PURSUIT‐SC, PURSUIT‐M |
200/100, 400/100, 50 and 100 mg | SC | Approved in EU and USA | [86–89] |
| Vedolizumab | GEMINI 1, GEMINI 2 | 300 mg | IV | Phase III | [ 83 ,90] |
10.2.1.1 Tumor Necrosis Factor Alpha (TNF‐α) Inhibition
Among all the inflammatory mediators, TNF‐α plays a pivotal role in IBD pathophysiology (Figure 10.1 ). Its inhibition provides a powerful treatment strategy for both UC and CD patients. Presently, two anti‐TNF‐α mAbs are approved in the United States and Europe, infliximab and adalimumab. Both are prescribed to patients suffering from moderate‐to‐severe IBD or to those who have relapsed after treatment with conventional therapies.
Infliximab
Among all biologics, infliximab is the most extensively studied and utilized mAb for the treatment of IBD. It is a chimeric IgG1 that binds and neutralizes the activity of both plasma soluble and membrane‐bound TNF‐α [91]. The downstream pharmacological effects resulting from this interaction include an enhanced cell‐mediated cytotoxic reaction leading to the programmed cell death of activated T‐cells. Clinical studies of infliximab in IBD patients have clearly demonstrated a superior overall response while administrated as a maintenance therapy rather than episodic treatment. The reason is that the use of episodic TNF‐α antagonists compared with regular dosing may lead to a higher incidence of anti‐drug antibody formation, and subsequently cause loss of therapeutic response [ 75 ,92].
In most pharmacodynamics (PD) studies, the healing of the mucosa due to reduction in the number of draining fistulas serves as a primary clinical endpoint since it was found a strong predictor of improvement of CD [93]. As an example, in a phase II clinical trial testing infliximab in patients with CD, this primary endpoint was achieved in 68% and 56% of patients after administration of 5 and 10 mg kg−1 infliximab at Week 0, 2, and 6 compared to placebo treatment (13%) [94]. In addition to that, the secondary endpoint (closure of all fistulas) was also achieved in 55% and 38% after administration of 5 and 10 mg kg−1 of infliximab in the same clinical study [94] . In another study, it was shown and concluded that the median remission time was prolonged for the patients treated over 40 weeks in comparison to placebo patients (14 weeks) [95]. The efficacy of infliximab monotherapy has also been compared with other conventional treatments, such as azathioprine monotherapy, where patients (n = 508) with moderate‐to‐severe CD were treated with infliximab either as a single agent or combined with azathioprine demonstrated more likely to have prolonged clinical remission compared to others receiving azathioprine alone [96].
In order to evaluate the clinical efficacy of infliximab for induction and maintenance therapy in patients suffering from UC, two randomized, double‐blind and placebo‐controlled studies (ACT 1 and ACT 2) were conducted [78] . Results from both studies indicate that patients in the treatment groups (5 or 10 mg kg−1 of infliximab) demonstrated a better clinical response (61–69%) than the ones in the placebo group (29–37%) [78] . Another study also demonstrated that patients suffering from moderately severe to severe UC as well as refractory to the corticosteroid treatment responded favorably to infliximab treatment [97].
Adalimumab
Adalimumab is a fully human, IgG1 type, anti‐TNF‐α mAb (first‐of‐its‐kind) that it is administered subcutaneously [91] . It binds to the TNF‐α and reduces the inflammatory responses to injury after modulating the downstream signaling pathway. Similar to infliximab, adalimumab is primarily used to manage the acutely ill or steroid‐dependent individuals suffering from CD and UC [77] . In general, infliximab is more advantageous over adalimumab in terms of remission in UC, although there is little difference is demonstrated in CD.
In two clinical studies, CLASSIC‐I and CLASSIC‐II trials, the short and long‐term efficacy and safety of adalimumab were investigated in patients (n = 276 in CLASSIC I and 299 in CLASSIC II) suffering from moderate‐to‐severe CD at the doses of 20 and 160 mg. Therapeutic efficacy was examined in terms of induction and maintenance of remission in CD patients [ 79 ,98]. The CLASSIC‐I trial concluded that the rates of remission at Week 4 for adalimumab treatment group are better than placebo (18–36% vs. 12%) [79] . The same clinical study further confirmed the optimal induction dosing regimen for adalimumab to be 160 mg as a starting dose followed by 80 mg as the second induction dose at the second week of treatment. Results showed that approximately 80% of patients receiving adalimumab weekly or every other week achieved remission at Week 56 compared to 44% of patients in the placebo group [ 79 , 98 ]. A larger clinical study, CHARM, investigated the clinical efficacy of adalimumab in the maintenance of a stable clinical response in CD patients (n = 854) at the loading dose of 80 mg and a maintenance dose of 40 mg given either weekly or bi‐weekly [80]. Results showed that after 26 weeks of treatment, patients' remission was significantly improved as compared to the placebo group (45% vs. 17%) [80] . Additionally, other clinical studies supported the fact that adalimumab has the ability to induce remissions more frequently than placebo in adult patients with CD who are refractory to infliximab monotherapy [ 81 ,99]. Based on these studies, adalimumab is considered a suitable alternative for patients suffering from CD who acquired resistance to infliximab and hence became nonresponder to this therapy. Contrary to CD, there are relatively few published clinical studies examining the efficacy and safety of adalimumab in moderately to severely active UC [ 82 ,100,101].
Certolizumab Pegol
In comparison to infliximab and adalimumab, certolizumab pegol (CZP) is regarded as a novel antibody scaffold biologic used in the treatment of IBD. It is an antigen‐binding fragment (Fab') portion of an IgG antibody attached to a 40 kDa polyethylene glycol moiety [91] . Although, this drug has been approved in the United States for use in patients with CD refractory to conventional treatments, in Europe, it is approved only in Switzerland for the treatment of CD [91] .
In two different clinical trials, PRECiSE 1 and 2, the therapeutic efficacy of CZP was critically evaluated, where patients with moderate‐to‐severe CD (n = 662) were randomly assigned to 400 mg CZP or placebo at Weeks 0, 2, and 4, then maintenance therapy was carried out for every 4 weeks [ 84 , 85 ]. It was shown that the response rate in the treatment group was significantly higher compared to the placebo groups after Week 6 (35% vs. 27%) and Week 26 (23% vs. 16%) in the PRECiSE 1 study. Similarly, the PRECiSE 2 study indicated that the maintenance of response was significantly higher in the treatment group (63% vs. 36%) [84] . In addition, another clinical study also investigated the long‐term effects (3.5 years) of CZP treatment, which also demonstrated prolonged remission rates for patients receiving CZP who weren't previously exposed to other TNF inhibitors [85] . In a different clinical study (WELCOME), the maintenance therapy with CZP helped a significant proportion of patients return to a normal life in comparison to others who received placebo [102]. Continuous treatment with CZP also improved the sustained perianal fistula closure compared to placebo. Although, usage of CZP in the treatment of CD share similar limitations as other TNF‐α antagonists, it is still considered to be an effective agent for adult patients suffering from moderate‐to‐severe CD.
Golimumab
Golimumab is a fully human IgG1 targeted against TNF‐α [ 86 ,103,104]. Preclinical findings showed that golimumab is superior to both infliximab and adalimumab because it targets both soluble and transmembrane forms of TNF‐α [104] , resulting in an increased inhibition of cytotoxicity induced by TNF‐α and human endothelial cell activation.
Golimumab was approved by the FDA and the European Medicines Agency (EMEA) in 2013 for the treatment of adult patients with moderate‐to‐severe active UC. The safety and efficacy of golimumab were determined in phase II and III clinical trials on patients with moderate‐to‐severe active UC and confirm golimumab utility for induction therapy [87,105]. The proportion of patients who achieved clinical remission at Week 6 in the lower‐ and higher‐dose treatment arms were comparable and much larger than in the placebo group [105] . Similarly, a placebo‐controlled maintenance trial evaluated the safety and efficacy of subcutaneous golimumab maintenance treatment in patients with moderate‐to‐severe active UC [87] . The results indicated that golimumab was able to maintain clinical responses through Week 54 compared to the placebo group [87] . The trial also provided additional evidence that clinical response and remission rates were better achieved in patients with high golimumab trough levels [87] . Due to its greater potential at inhibiting TNF‐α compared to other anti‐TNF‐α, golimumab was also tested in clinical trial to assess its efficacy in patients with severe‐to‐moderate active CD patients [88]. In addition to adult patients, golimumab was also tested in pediatric patients (age 12–19 years) who were resistant to various CD therapies [89] . Although all the patients responded to the first injection of golimumab, the therapeutic effect was not sustained. The observed suboptimal therapeutic responses due to golimumab treatment were attributed to the increased clearance of the drug due to its immunogenicity [106,107].
10.2.1.2 Side‐Effects of Anti‐TNF‐α Agents
Anti‐TNF‐α agents are associated with the emergence of opportunistic infections, malignancies, injection/infusion reactions, and autoimmunity. According to the FDA labels, most of these drugs are contraindicated in the presence of heart failure and acute infectious diseases. A meta‐analysis study of clinical trials using anti‐TNF‐α indicated that the frequency of occurrence of malignancy and infections didn't differ between treatment and placebo groups [108]. In addition, both regulatory agencies, the FDA and the European Crohn's and Colitis Organization, categorize anti‐TNF agents as safe during pregnancy.
10.2.2 Emerging Therapeutic Options for Inflammatory Bowel Disease
Reports indicated that although anti‐TNF‐α agents are effective and influential against IBDs, approximately 30% of patients with CD become nonresponders to induction therapy, and among initial responders, 50% will become nonresponder within a year of treatment [91] . During recent years, a large number of substances such as unfractionated or low molecular weight heparin, omega‐3 polyunsaturated fatty acids, microbes, and microbial products have been explored as possible alternatives to biologics in the management of IBD [109]. Discovery of these substances paved the way to examining novel biological targets, such as IL‐12 and IL‐23, recently proven involved in inflammation. This led to the development of ustekinumab, a fully human mAb targeted against the p40 subunit of IL‐23 and IL‐12 [110]. Table 10.3 summarizes these emerging therapeutic options for IBD. Their details are provided in the section below.
10.2.2.1 Anti‐adhesion (Anti‐integrin) Molecules
Several endothelial adhesion molecules, such as E‐selectin, ICAM‐1, ICAM‐2, VCAM‐1, and MAdCAM‐1, play important roles to potentiate inflammation by trafficking leucocytes as well as recruiting immune cells into the gut. Consequently, they interact with integrins on leukocytes inducing their migration from blood vessels to the site of inflammation. Due to their aforementioned properties, they are attractive targets for the development of new drugs [30] .
Natalizumab
It is a humanized IgG4 mAb against cell adhesion molecule α4‐integrin, which inhibits the VCAM and MAdCAM‐1 pathways of leucocyte adhesion and transmigration resulting in reduction of moderate‐to‐severe CD. This drug is used as an alternative therapy in patients who are refractory to anti‐TNF‐α agents. In a clinical trial using 300 mg natalizumab, a superior response and longer remission was observed after Week 8 posttherapy without demonstrating any side effects [111]. However, due to risk of natalizumab‐associated progressive multifocal leukoencephalopathy (PML), it was voluntarily withdrawn from the market in 2005 [112].
Vedolizumab
Vedolizumab is a humanized IgG1 mAb that binds to the integrin α4β7 or lymphocyte Peyer's patch adhesion molecule 1 (LPAM‐1) to selectively impede the adhesion of leukocytes to the gastro‐intestinal (GI) mucosa resulting in the elimination of release of cytokines as well as complement fixation. In a phase III trial (GEMINI III), intravenous administration of vedolizumab demonstrated clinical remission of the patients suffering from active CD compared to the patients receiving placebo [83] . However, vedolizumab, as compared to placebo, was associated with a slightly higher rate of serious adverse events (24.4% vs. 15.3%), infections (44.1% vs. 40.2%), and serious infections (5.5% vs. 3.0%) [83] . Similarly, in case of UC, vedolizumab when given intravenously was more effective than placebo when administered as an induction and or maintenance therapy [90] . Nevertheless, the frequency of adverse effects was similar in the treatment and placebo groups [90] . Similar to adults, vedolizumab is also found to be safe and effective in pediatrics population who are refractory to IBD. However, to date it is not a FDA‐approved for the treatment of IBD [113].
10.2.2.2 Anti‐ICAM‐1 Therapy
ICAM‐1 (CD54) is also another important inflammatory mediator of cell‐mediated inflammation, specifically cell trafficking. Alicaforsen (ISIS 2302) is a known antisense to ICAM‐1, which have shown promising results against UC patients. However, it did not demonstrate any signs of improvement in CD patients [114].
10.2.2.3 Anti‐IL‐6R Antibodies
Tocilizumab
It is a humanized IL‐6 receptor (IL‐6R) mAb that inhibits the activity of IL‐6 by blocking the expression of IL‐6R in both forms, plasma‐soluble and membrane‐bound. This interaction ultimately prevents the inflammatory processes resulting from IL‐6 overproduction. Due to its mechanism of action (MoA) through neutralizing the receptor of one of the most potent proinflammatory cytokines, it has been studied for potential use in the treatment of CD [115]. It demonstrated a higher reduction of CD activity index compared to placebo [116,117]. However, the efficacy of tocilizumab in CD warrants future investigation because clinical responses were not completely suppressed after four weeks of continued therapy [117] .
10.2.2.4 Immunostimulators
Since defects in the innate immune system might also cause CD, a number of agents known to stimulate the innate immune system have been tested in CD patients. Here we describe two protein therapeutics used as immunostimulators in IBD, the recombinant human granulocyte‐macrophage colony‐stimulating factor (GM‐CSF, sargramostim) and the recombinant human granulocyte‐colony stimulating factor (G‐CSF, filgrastim).
GM‐CSF
Clinical trials with IBD patients showed that while treated with sargramostim, patients experienced a better and longer clinical remission as well as improved health‐related quality of life [118]. However, sargramostim treatment causes different adverse effects ranging from musculoskeletal pain, injection site reactions, and dyspnea that which limits its systematic use in IBD and requires further investigations for this drug.
G‐CSF
Similar to GM‐CSF, G‐CSF is a recombinant protein therapeutic that has been tested in CD [119]. Contrary to the treatment with GM‐CSF, G‐CSF showed a higher clinical remission without exhibiting any significant side effect except for some mild bone pain [119] .
Growth Factors
Human growth factors are signaling molecules leading to ligand‐specific signal transduction that which are ultimately associated with several cellular functions including epithelial healing in response to injury. In general, deficiency of specific growth factors as well as impaired epithelial repair is known to associate with the emergence of IBD. In this regard, several growth factors, such as growth hormone, epidermal growth factor, keratinocyte growth factor, teduglutide, have shown promising results for the modulation of intestinal inflammation [120]. However, further studies are needed to determine the efficacy of the growth factors in IBDs.
10.2.2.5 T‐cell–Directed Therapies
T‐cell‐mediated immune responses directed against normal components of the gut flora are also another critical factor that can amplify as well as sustain the mucosal inflammation by generating excessive amount of cytokines. Different strategies have been taken to interfere with the accumulation of T‐cells along with the restoration of gut function to treat IBD patients. In this regard, different therapeutic targets have been selected, such as CD80 or CD86–CD28 co‐stimulatory signal (abatacept), CD2 (alefacept), CD11a (efalizumab), and CD20 (rituximab) [121].
10.2.2.6 Fontolizumab
Fontolizumab is a humanized IgG1 mAb‐directed against IFN‐γ. The aim from this therapy is to reduce the gut mucosal level of IFN‐γ. A clinical trial examining fontolizumab in CD patients showed that at 1 mg kg−1 fontolizumab either given intravenously or subcutaneously results in a better clinical response in comparison to placebo [122]. In addition, significant reduction of plasma CRP levels has also been shown after fontolizumab treatment.
10.2.2.7 Ustekinumab
Ustekinumab is a human IgG1‐based mAb‐directed against the p40 subunit of IL‐12 and IL‐23 shown to be involved in the pathophysiology of CD [123]. In a double‐blind, crossover clinical trial with moderate‐to‐severe CD patients, the clinical response rate to ustekinumab was shown to be more significant than placebo [124]. This finding was also supported by a separate open label clinical trial where ustekinumab exhibited an improved clinical response and reduced patients' serum CRP levels [125].
10.2.2.8 Inhibitors of T‐cell Proliferation
Selective blockade of the interaction between T cell lymphocytes and the vascular endothelium in the GIT is thought to be a promising therapeutic strategy to prevent progression of IBD [91] . A number of mAbs are being developed and currently tested in UC patients.
Visilizumab
Visilizumab is a humanized, IgG2, anti‐CD3 mAb for the treatment of UC. Visilizumab acts by decreasing the levels of circulating CD4+ T cells [126]. While administered at the dose of 10 µg kg−1, it exhibited an acceptable safety profile and revealed efficacious in patients with severe steroid‐refractory UC [126] .
Basiliximab
Basiliximab is a chimeric IgG1 mAb that targets CD25 (IL‐2A receptor) on the surface of the regulatory T cell lymphocytes. Successful results have been reported after single treatment in moderate steroid‐resistant UC [91] . In a clinical trial, it has been shown that a single dose of 40 mg basiliximab along with standard steroid therapy can achieve clinical remission in 50–65% of patients after 8–24 weeks of treatment [127].
The development of biologic agents and their use in IBD has improved patient quality of life considerably by modifying disease course and preventing complications and surgery. Control of inflammation can be achieved with all three available anti‐TNF‐α biologic agents, namely infliximab (IFX), adalimumab, and CZP. These agents are effective in both induction and maintenance of remission. Anti‐TNF‐α agents appear to be more effective in patients with a shorter disease history and who are naive to any of the anti‐TNF‐α agents. Patients with CD previously treated with IFX plus azathioprine or IFX monotherapy are more likely to have a steroid‐free clinical remission than those receiving azathioprine monotherapy [77] . Several strategies can minimize the risks associated with biologic therapies, such as the development of lymphoma, nonmelanoma skin cancer, and some complex infections caused by the mycobacteria and fungi. Proper treatment strategies include (i) careful examination of past history and physical examination and screening for latent tuberculosis, (ii) close clinical and therapeutic monitoring of patients during treatment, (iii) education of physicians and patients to allow the early detection of any adverse effect.
In addition, during the past decade, a better understanding of the underlying pathogenesis of IBD along with great advances in biotechnology has resulted in the introduction of many biologic therapies, other than anti‐TNF‐α. However, PML is the leading safety concern with natalizumab, which may limit its use in IBD [128]. Results from clinical trials on the use of mAbs against IL‐12 and IL‐6R in patients with CD produced relatively satisfactory results, although studies including large numbers of patients are needed [129]. In patients with CD‐refractory UC, basiliximab could be an effective agent. We must stress the fact that so far no studies have compared anti‐TNF‐α therapies head‐to‐head to other biological agents. Despite promising results on the use of growth factors in IBD treatment, future endeavors regarding their impact on this disease need further investigation. Ustekinumab, a mAb against IL‐12/23, seems to be promising in patients with active CD, especially in those with high baseline CRP values. Because the data on the efficacy and short‐ and long‐term safety of these new biologic agents are limited, their use in IBD patients remains to be justified.
10.3 Mathematical Models in IBD
Quantitative systems pharmacology (QSP) is a bourgeoning interdisciplinary field that aims to integrate and apply the theoretical and experimental tools of systems biology with basic pharmacology in drug development and regulatory decision‐making. The goal of systems pharmacology (SP) approaches is to explore the pharmacological and toxicological effects of a drug through its specific and high affinity binding to the target on a multiscale system level of the disease (i.e. molecular, cellular, organ, and whole body). Over the years, vast amounts of information about the regulatory relationships among genes, proteins, and drug intervention (i.e. small and large molecules) have been acquired in IBD (Figure 10.1 ). However, major knowledge gaps are yet to be filled in order to fight this chronic and devastating disease. There is an abundance of qualitative information related to the activation or blockade of signaling pathways from various biologics in IBD, but a dearth of quantitative and temporal information. One means to overcome this challenge and to reconcile between the limited data currently available is to build multilevel link models by bridging SP models to more parsimonious pharmacokinetics (PK)/PD models. Such approach may provide a framework for understanding and predicting clinical outcomes in patients exhibiting complex phenotypes, such as in IBD, and allow to address multiple challenges in IBD drug development, such as (i) predicting accurately the drug efficacy during the early stages of drug development and providing rationale for treatment designs, (ii) understanding the mechanisms behind the toxicities of IBD drugs, or their off‐target effects, and (iii) optimizing the clinical development with dose selection, identification of optimal combination dosing regimens, prediction of clinical outcomes, and selection of responsive patients as well as personalized treatments [130]. Figure 10.2 depicts a general workflow for the integration of multiscale systems biology data to build comprehensive SP and PK/PD models as useful tools to streamline IBD drug development and predict clinical outcome.
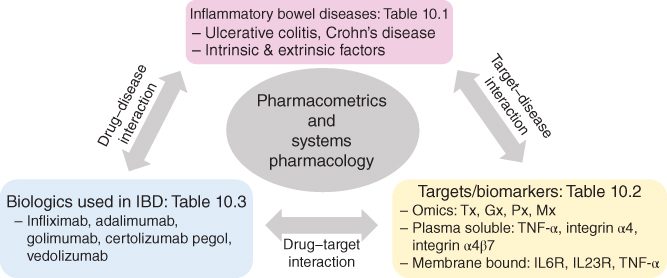
Figure 10.2 General workflow for the integration of multi‐scale systems biology data to build comprehensive systems pharmacology and PK/PD models as useful tools to streamline IBD drug development and predict clinical outcome.
In this section of this book chapter, we discuss examples of systems models for the three selected mAbs that are approved by the FDA: infliximab, adalimumab, and CZP, along with the specific challenges and unique opportunities for the knowledge gained from QSP and systems PK/PD modeling and simulation in IBD.
10.3.1 Infliximab
A mechanism‐based PK/PD model of anti‐inflammatory effect of infliximab on CD was developed by Furuya et al. [131]. In their work, the authors analyzed a sequential change of the Crohn's disease activity index (CDAI) with means of a PK/PD model integrating the time course of infliximab plasma concentrations and the turnover rate of circulating TNF‐α. A target‐mediated drug disposition (TMDD) model [132] well captured infliximab's plasma concentration time profiles. The unbound TNF‐α plasma concentration was used as a driver for the passage from noninflammatory to inflammatory state. The model assumed that the mAb–ligand complex was eliminated with the same fractional catabolic rate as the unbound ligand. This model assumption may have impacted on the estimated half‐life of TNF‐α at ∼30–40 days, which is considerably different from the known value (18.2 min–4.8 h) [133–135]. Nevertheless, the PD model predictions agreed well with the observed CDAI ratio data.
10.3.2 Adalimumab
A PK/PD model for adalimumab was developed by Ternant et al. [136], and employed for the therapeutic drug monitoring (TDM) of this mAb in CD patients. While the absorption of adalimumab from the subcutaneous depot compartment was captured by a zero‐order process, a one‐compartment model with linear disposition from the central compartment sufficed to characterize its plasma concentration time profiles reasonably well. The relationship between the drug and the plasma levels of CRP, a surrogate biomarker to the patients' inflammatory status, was captured with an indirect response model where adalimumab inhibits its production. This mechanistic model satisfactorily described the PK of adalimumab and its relationship to CRP in all 65 eligible CD patients [136] .
10.3.3 Certolizumab Pegol
A PK model for CZP in patients with CD was developed [137]. It consisted in a one‐compartment model with linear elimination and a first‐order absorption process from the subcutaneous compartment. This population PK modeling analysis was performed by pooling data from nine separate studies with the primary objective of screening for clinically relevant covariates. The results determined that body surface area (BSA) increased both apparent clearance (CL/F) (53%) and apparent volume of distribution (49%) in a linear manner across the range of BSA measurements. On the contrary, increment of albumin decreased CL/F in antibody‐negative patients (from 1.05 to 0.613 l d−1) in a nonlinear manner. Other covariates such as CRP and race had a minor influence on V/F. However, a further comprehensive PK/PD study is warranted in order to understand the clinical impact of these covariates on the disposition of CZP.
10.3.4 Vedolizumab
The PK and PD of vedolizumab were investigated in several clinical trials including phase I, II, and III in UC and CD patients [138]. The purpose of the PK analysis was to identify the clinically relevant determinants of the vedolizumab clearance and support the rationale of fixed dosing. The PK of the drug was described with a two‐compartment model with parallel linear and nonlinear eliminations pathways from the central compartment [124] . The main finding from this model is that covariate analysis indicated that only extreme albumin and body weight values were potentially clinically important predictors of clearance.
10.3.5 Challenges in Systems PK/PD Modeling of mAbs in IBD
The pharmacology of therapeutic mAbs is complex and depends not only on the structure of the antibody and the target antigen properties but also on intrinsic factors related to the patient and the disease. Although, neutralizing ADAs have been considered as a primary cause of therapeutic failure, other factors can also profoundly alter the PK and subsequently the PD of large molecule therapeutics such as (i) concomitant medications, (ii) serum albumin concentrations, (iii) presence of disease severity, (iv) type of disease and patient factors, and (v) patients compliance to therapy [139,140]. Collectively, these factors probably account for the large inter‐individual differences in PK and clinical efficacy observed after standard dosing of therapeutic mAbs [139] .
Whereas PK and PD are relatively well established as the predominant methods for identifying and optimizing dosing regimens during drug development and clinical trials, SP is an emerging area of research with great promise in drug target identification and in individualizing therapies. IBD is a complex disease that results from the complex interplay between several multiscale biological factors making it an ideal platform for SP modeling. However, the most challenging task of this mechanistic framework is to decipher biological implications using extremely large amount of experimental platforms. Other challenges for SP models in IBD would include the anticipated model complexity due to the multitude of factors involved in its pathophysiology, experimental and biological noises, redundant and interconnected pathways, multiple spatial and temporal scales, and inter‐patients' differences in biological responses. Yet, the relationships between PK and PD of the aforementioned therapeutic mAbs in IBD have been compromised by several problems, such as retrospective study designs that are not optimally designed to identify relevant PK/PD relationships, sample collection error, use of inappropriate modeling approaches, inclusion/exclusion of patients without the evidence of inflammation, failure to use objective PD endpoints, and failure to account for the effects of confounders [ 139 ,141]. However, the combination of clinical and imaging data with more comprehensive PK and PD studies is being employed to further understand the underlying PK/PD relationships of biologics in IBD providing guidance to drug dosage, monitoring of therapeutic responses, as well as clinical outcomes and challenges related to treated patients.
10.4 Role of FDA in the Drug Development of Biologics in the Treatment of IBD
Approximately 75% of all patients in the United States suffering from IBD are treated for the disease [142]. To date, the FDA has approved 12 different therapeutic agents for the treatment of CD and UC, although a number of other drugs are also used as “off‐label” medications. According to the reports, at least 40 drugs are in the drug development pipeline of IBD. The predicted value of the overall market for CD drugs in 2017 is as high as $4.7 billions (https://www.prlog.org/11371699‐crohns‐disease‐pipeline‐assessment‐and‐market‐forecasts‐to‐2017.html).
Generally, for biologics the most expensive and time‐consuming component in the FDA approval process is the planning of clinical trials, recruitment of healthy volunteers/patients and generation as well as analysis of the generated outcome data. In phase I studies, approximately 100 healthy volunteers are enrolled over 6–18 months to examine the safety of the new drug candidate at different doses. Depending on the result of this trial, the efficacy and safety of the drug is assessed in a well‐defined cohort of patients in open trials (phase IIA) followed by randomized controlled trials (phase IIB) or randomized trials alone. Once the efficacy and safety data from the phase II trial are regarded as positive and robust, sponsors will move to the next large and expensive phase III study such as the original infliximab trial in CD (ACCENT I), and the CLASSIC trial for adalimumab [ 79 ,143]. Finally, phase III studies are performed to further investigate the safety and dosing schedules of the drug candidate in defined populations, which are the basis of the prescribing and package‐insert information if the drug is approved for marketing. In general, only 30% of investigational drugs submitted to the FDA will complete phase III studies. This is exemplified in IBD with the phase III trials: ACCENT, CHARM, and PRECISE [ 80 , 84 , 143 ].
After the approval of the agent for the intended use, the candidate drug is first tested in individuals who were ineligible for controlled trials such as pediatric, geriatric, pregnant women, and patients with comorbidities to identify any kind of rare side effects. Usually, FDA requires drug sponsors to report all the adverse events associated with the new drug. For example, during postmarketing phase of natalizumab in patients with CD and multiple sclerosis, the risk of PML was detected [144]. Similarly, an increased number of cases of tuberculosis were identified with the early use of infliximab [145]. Although, a phase IV study can be requested by the FDA as part of a drug's postmarketing commitment; however, there is currently no mechanism to enforce this requirement once a drug is approved for marketing. To include risk evaluation mitigation strategies (REMS), an increase in the percentage of user fees have been devoted to the drug safety was included in the renewal of the prescription drug user fee act (PDUFA 2007). This area remains of particular relevance to IBD as novel targeted biologic agents come to market; case reports of hepatosplenic T‐cell lymphoma in young patients with IBD only appeared after 10 years of anti‐TNF use [146].
Among the biologics that have been approved for IBD, infliximab was initially approved as an orphan drug in 1998 for the treatment of moderate‐to‐severely active and fistulizing CD, on the basis of data from the ACCENT I and II trials [ 95 , 143 ]. Clinical endpoints were a reduction in CDAI of 70 points at 4 weeks, and maintenance of CDAI below 150 points at 30 and 54 weeks. The fistula endpoint (ACCENT II) was a closure of at least 50% of entero‐cutaneous fistulae present at baseline. This was observed at 14 and 54 weeks [95] . Subsequently, in 2005, infliximab received approval for UC based on the ACT1 and ACT2 studies [78] . In these studies, clinical endpoints were based on physician's assessment of clinical remission and endoscopic evaluation of mucosal healing. However, the timing, and CDAI scores, used to measure induction and maintenance of remission in CD by subsequent biologics have varied over the years. In assessing the induction of response/remission to therapy, the CDAI has been the primary endpoint in these clinical trials, with a drop of 70 or 100 points considered a “response.” The limitations of such an index are widely acknowledged in the IBD community, and it is common to see placebo “response” rates of 40–50% in clinical trials with this endpoint [147]. Comparative studies have reported that the CDAI actually correlates poorly with objective markers of inflammation such as endoscopy, raising the question of whether it actually measures manifestations of macroscopic intestinal inflammation or other processes. Some clinical trials used for FDA approval of biologics in CD included a plasma CRP concentration cutoff for inclusion, but this did not appear to significantly alter response rates [84] . There has been much emphasis in the clinical literature in recent years on mucosal healing as an endpoint for biologic therapies in CD, but these have not yet become part of the primary efficacy endpoint in clinical trials [148].
Recently, there has been a growing interest in FDA's criteria for approving generic drugs due to the patent expirations on a number of commonly used branded medications for IBD. To circumvent this issue, sponsors can apply for a 351 (K) abbreviated new drug application (ANDA) for generic medications, where independent evidence of safety and efficacy for the proposed generic drug is not needed. However, the ANDA applicant must demonstrate that the proposed generic contains the same active ingredient and is “highly biosimilar” to a reference listed drug (RLD). However, addressing these two requirements has proven to be difficult for the biologics owing to the fact that they are more complex molecules that may not allow exact replication. Posttranslational modifications and protein aggregations may prevent analysis of the physicochemical attributes of biosimilar products using traditional methods. The experience in Europe has been that potency of some biosimilars may not be as high as the original drugs, as was seen with the case of biosimilar epoetins [149]. Nonetheless, the push to reduce health‐care costs has necessitated legislative mechanisms for regulation of generic biologics. The Biologics Price Competition Act, a component of the 2010 Patient Protection and Affordable Care Act, legislates for an abbreviated approval pathway for biosimilars or follow‐on biologics (FOBs) in the United States. This law provides for 12 years of market exclusivity for pioneer agents, standards for biosimilarity and interchangeability, and exclusivity for the first FOB to prove interchangeability with a pioneer agent. How the FDA will determine such “biosimilarity” is currently undecided, and a matter of intense interest from the producers of currently approved biologics.
It is clear that developing safe and effective IBD agents for FDA approval can be a time‐consuming, costly, and somewhat risky process for all parties involved. For IBD drug developers, patent expirations, reimbursement pressures, rising research and development (R&D) costs, and increasing regulatory demands have led to a flattening of the rate of new drug development. The mean duration of the approval phase of drug development declined by more than 1 year over the last 30 years, whereas the duration of the clinical trial phase has increased. For regulators, an increased demand for better safety oversight has been coupled with industry demands for more efficient review processes and funding restraints.
Only few novel therapeutic options for secondary nonresponders to anti‐TNF‐α agents and alternative oral medications for mesalamine failures, such as tofacitinib, exist [150]. From a societal standpoint, robust methods for assessing the bioequivalence of generic agents and FOBs may open the door to less costly, but safe and effective medications for patients. The FDA, the scientific and medical communities, and industry must look increasingly toward more collaboration and innovation to achieve the goal of timely approval of safe and efficacious medications. Patients with IBD have benefited from the FDA's comprehensive review and approval of a number of medications to treat these conditions. This lengthy and expensive process continues to juggle the demands of industry for timely drug approval with the demands of patients and providers for “safe” therapies. Harmonization of clinical endpoints for clinical trials, and the universal inclusion of objective outcomes, would allow indirect comparisons of future therapies for IBD. It will require further evolution of the drug approval process, and the regulations that govern it, to address the issues of generic medications and FOBs in the near future.
10.5 Summary
Although this chapter focuses on systems models for biologics in IBD, it is useful to put these modeling efforts in a wider context. Constructing systems models for IBD is rather challenging as it requires a deep understanding of (i)the underlying complex biological system as well as associated dynamics, (ii) the question that is articulated, (iii) the answer that is sought, (iv) the availability of the physical and biological data, and (v) the development of the relevant mathematical and computational depictions that represents a comprehensible picture of the physical reality of interest. Therefore, moving toward the development of comprehensive systems models in IBD, the process starts by identifying the specific question or problem of interest. This very first step is critical to yield meaningful research results with a well‐founded and experimentally testable hypothesis, since a model is a specific representation of a biological system whose complexity depends on the scope and goals of the analysis.
References
- 1 Kulaylat, M.N. and Dayton, M.T. (2010). Ulcerative colitis and cancer. J. Surg. Oncol. 101: 706–712, https://doi.org/10.1002/jso.21505.
- 2 Canavan, C., Abrams, K.R., and Mayberry, J. (2006). Meta‐analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment. Pharmacol. Ther. 23: 1097–1104, https://doi.org/10.1111/j.1365‐2036.2006.02854.x.
- 3 Gillen, C.D., Walmsley, R.S., Prior, P. et al. (1994). Ulcerative colitis and Crohn's disease: a comparison of the colorectal cancer risk in extensive colitis. Gut 35: 1590–1592.
- 4 Rhodes, J.M. and Campbell, B.J. (2002). Inflammation and colorectal cancer: IBD‐associated and sporadic cancer compared. Trends Mol. Med. 8: 10–16, https://doi.org/10.1016/S1471‐4914(01)02194‐3.
- 5 Kappelman, M.D., Rifas‐Shiman, S.L., Kleinman, K. et al. (2007). The prevalence and geographic distribution of Crohn's disease and ulcerative colitis in the United States. Clin. Gastroenterol. Hepatol. 5: 1424–1429, https://doi.org/10.1016/j.cgh.2007.07.012.
- 6 Loftus, E.V. Jr. (2004). Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 126: 1504–1517.
- 7 Louis, E., Van Kemseke, C., and Reenaers, C. (2011). Necessity of phenotypic classification of inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 25 (Suppl. 1): S2–S7, https://doi.org/10.1016/s1521‐6918(11)70003‐8.
- 8 Papay, P. et al. (2013). Optimising monitoring in the management of Crohn's disease: a physician's perspective. J. Crohns Colitis 7: 653–669, https://doi.org/10.1016/j.crohns.2013.02.005.
- 9 Ochsenkuhn, T., Sackmann, M., and Goke, B. (2003). Inflammatory bowel diseases (IBD) – critical discussion of etiology, pathogenesis, diagnostics, and therapy. Radiologe 43: 1–8, https://doi.org/10.1007/s00117‐002‐0844‐9.
- 10 van Bodegraven, A.A. et al. (2010). Guideline ‘Diagnosis and treatment of inflammatory bowel disease in adults’. I. Diagnosis and treatment. Ned. Tijdschr. Geneeskd. 154: A1899.
- 11 Mowat, C. et al. (2011). Guidelines for the management of inflammatory bowel disease in adults. Gut 60: 571–607, https://doi.org/10.1136/gut.2010.224154.
- 12 Feakins, R.M. (2013). Inflammatory bowel disease biopsies: updated British Society of Gastroenterology reporting guidelines. J. Clin. Pathol. 66: 1005–1026, https://doi.org/10.1136/jclinpath‐2013‐201885.
- 13 Vermeire, S., Van Assche, G., and Rutgeerts, P. (2004). C‐reactive protein as a marker for inflammatory bowel disease. Inflamm. Bowel Dis. 10: 661–665.
- 14 Cho, J.H. and Weaver, C.T. (2007). The genetics of inflammatory bowel disease. Gastroenterology 133: 1327–1339, https://doi.org/10.1053/j.gastro.2007.08.032.
- 15 Ananthakrishnan, A.N. (2015). Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 12: 205–217, https://doi.org/10.1038/nrgastro.2015.34.
- 16 Neurath, M.F. (2014). Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14: 329–342, https://doi.org/10.1038/nri3661.
- 17 Shishido, S., Bönig, H., and Kim, Y.‐M. (2014). Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 4, https://doi.org/10.3389/fonc.2014.00099.
- 18 Fengming, Y. and Jianbing, W. (2014). Biomarkers of inflammatory bowel disease. Dis. Markers 2014: 11, https://doi.org/10.1155/2014/710915.
- 19 Andersen, V., Halfvarson, J., and Vogel, U. (2012). Colorectal cancer in patients with inflammatory bowel disease: can we predict risk? World J. Gastroenterol. 18: 4091–4094, https://doi.org/10.3748/wjg.v18.i31.4091.
- 20 Van Assche, G. and Rutgeerts, P. (2005). Physiological basis for novel drug therapies used to treat the inflammatory bowel diseases. I. Immunology and therapeutic potential of antiadhesion molecule therapy in inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 288: G169–G174, https://doi.org/10.1152/ajpgi.00423.2004.
- 21 Shih, D.Q. and Targan, S.R. (2008). Immunopathogenesis of inflammatory bowel disease. World J. Gastroenterol. 14: 390–400, https://doi.org/10.3748/wjg.14.390.
- 22 Singh, H., Grewal, N., Arora, E. et al. (2016). Vedolizumab: a novel anti‐integrin drug for treatment of inflammatory bowel disease. J. Nat. Sci. Biol. Med. 7: 4–9. https://doi.org/10.4103/0976‐9668.175016.
- 23 Barbalho, S.M., Goulart, R.D.A., Quesada, K. et al. (2016). Inflammatory bowel disease: can omega‐3 fatty acids really help? Ann. Gastroenterol. 29: 37–43.
- 24 de Mattos, B.R.R. et al. (2015). Inflammatory bowel disease: an overview of immune mechanisms and biological treatments. Mediators Inflamm. 493012, https://doi.org/10.1155/2015/493012: 2015.
- 25 Guagnozzi, D. and Caprilli, R. (2008). Natalizumab in the treatment of Crohn's disease. Biologics 2: 275–284.
- 26 Koutruba, N., Emer, J., and Lebwohl, M. (2010). Review of ustekinumab, an interleukin‐12 and interleukin‐23 inhibitor used for the treatment of plaque psoriasis. Ther. Clin. Risk Manag. 6: 123–141.
- 27 Pedersen, J., Coskun, M., Soendergaard, C. et al. (2014). Inflammatory pathways of importance for management of inflammatory bowel disease. World J. Gastroenterol. 20: 64–77, https://doi.org/10.3748/wjg.v20.i1.64.
- 28 Khor, B., Gardet, A., and Xavier, R.J. (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474: 307–317, https://doi.org/10.1038/nature10209.
- 29 Abraham, C. and Cho, J.H. (2009). IL‐23 and autoimmunity: new insights into the pathogenesis of inflammatory bowel disease. Annu. Rev. Med. 60: 97–110, https://doi.org/10.1146/annurev.med.60.051407.123757.
- 30 Ghosh, S. and Panaccione, R. (2010). Anti‐adhesion molecule therapy for inflammatory bowel disease. Therap. Adv. Gastroenterol 3: 239–258, https://doi.org/10.1177/1756283X10373176.
- 31 Sands, B.E. (2015). Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology 149: 1275–1285.e2, https://doi.org/10.1053/j.gastro.2015.07.003.
- 32 Lopez, R.N. et al. (2016). Faecal biomarkers in inflammatory bowel disease. J. Gastroenterol. Hepatol., https://doi.org/10.1111/jgh.13611 .
- 33 DeVoss, J. and Diehl, L. (2014). Murine models of inflammatory bowel disease (IBD): challenges of modeling human disease. Toxicol. Pathol. 42: 99–110, https://doi.org/10.1177/0192623313509729.
- 34 Henriksen, M. et al. (2008). C‐reactive protein: a predictive factor and marker of inflammation in inflammatory bowel disease. Results from a prospective population‐based study. Gut 57: 1518–1523, https://doi.org/10.1136/gut.2007.146357.
- 35 Fagerhol, M.K., Dale, I., and Andersson, T. (1980). A radioimmunoassay for a granulocyte protein as a marker in studies on the turnover of such cells. Bull. Eur. Physiopathol. Respir. 16 (Suppl.): 273–282.
- 36 Bjerke, K., Halstensen, T.S., Jahnsen, F. et al. (1993). Distribution of macrophages and granulocytes expressing L1 protein (calprotectin) in human Peyer's patches compared with normal ileal lamina propria and mesenteric lymph nodes. Gut 34: 1357–1363.
- 37 Lehmann, F.S., Burri, E., and Beglinger, C. (2015). The role and utility of faecal markers in inflammatory bowel disease. Therap. Adv. Gastroenterol. 8: 23–36, https://doi.org/10.1177/1756283x14553384.
- 38 Walsham, N.E. and Sherwood, R.A. (2016). Fecal calprotectin in inflammatory bowel disease. Clin. Exp. Gastroenterol. 9: 21–29, https://doi.org/10.2147/ceg.s51902.
- 39 Benitez, J.M. and Garcia‐Sanchez, V. (2015). Faecal calprotectin: management in inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 6: 203–209, https://doi.org/10.4291/wjgp.v6.i4.203.
- 40 Smith, L.A. and Gaya, D.R. (2012). Utility of faecal calprotectin analysis in adult inflammatory bowel disease. World J. Gastroenterol. 18: 6782–6789, https://doi.org/10.3748/wjg.v18.i46.6782.
- 41 Summerton, C.B., Longlands, M.G., Wiener, K., and Shreeve, D.R. (2002). Faecal calprotectin: a marker of inflammation throughout the intestinal tract. Eur. J. Gastroenterol. Hepatol. 14: 841–845.
- 42 (2006). Digestive disease week and the 107th Annual Meeting of the American Gastroenterological Association Institute, May 20–25, 2006, Los Angeles, California, USA. Abstracts. Gastroenterology 130: A1–A911.
- 43 Sipponen, T. et al. (2008). Fecal calprotectin, lactoferrin, and endoscopic disease activity in monitoring anti‐TNF‐alpha therapy for Crohn's disease. Inflamm. Bowel Dis. 14: 1392–1398, https://doi.org/10.1002/ibd.20490.
- 44 Gross, W.L., Schmitt, W.H., and Csernok, E. (1993). ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clin. Exp. Immunol. 91: 1–12.
- 45 Terjung, B. et al. (1998). Atypical antineutrophil cytoplasmic antibodies with perinuclear fluorescence in chronic inflammatory bowel diseases and hepatobiliary disorders colocalize with nuclear lamina proteins. Hepatology 28: 332–340, https://doi.org/10.1002/hep.510280207.
- 46 Kuna, A.T. (2013). Serological markers of inflammatory bowel disease. Biochem. Med. (Zagreb) 23: 28–42.
- 47 Linskens, R.K. et al. (2002). Evaluation of serological markers to differentiate between ulcerative colitis and Crohn's disease: pANCA, ASCA and agglutinating antibodies to anaerobic coccoid rods. Eur. J. Gastroenterol. Hepatol. 14: 1013–1018.
- 48 Mokrowiecka, A., Gasiorowska, A., and Malecka‐Panas, E. (2007). pANCA and ASCA in the diagnosis of different subtypes of inflammatory bowel disease. Hepatogastroenterology 54: 1443–1448.
- 49 Esters, N. et al. (2002). Serological markers for prediction of response to anti‐tumor necrosis factor treatment in Crohn's disease. Am. J. Gastroenterol. 97: 1458–1462, https://doi.org/10.1111/j.1572‐0241.2002.05689.x.
- 50 Siegel, C.A. and Melmed, G.Y. (2009). Predicting response to anti‐TNF agents for the treatment of Crohn's disease. Therap. Adv. Gastroenterol. 2: 245–251, https://doi.org/10.1177/1756283X09336364.
- 51 Bossuyt, X. (2006). Serologic markers in inflammatory bowel disease. Clin. Chem. 52: 171–181, https://doi.org/10.1373/clinchem.2005.058560.
- 52 Ferrante, M. et al. (2007). New serological markers in inflammatory bowel disease are associated with complicated disease behaviour. Gut 56: 1394–1403, https://doi.org/10.1136/gut.2006.108043.
- 53 Mitsuyama, K. et al. (2016). Antibody markers in the diagnosis of inflammatory bowel disease. World J. Gastroenterol. 22: 1304–1310, https://doi.org/10.3748/wjg.v22.i3.1304.
- 54 Landers, C.J. et al. (2002). Selected loss of tolerance evidenced by Crohn's disease‐associated immune responses to auto‐ and microbial antigens. Gastroenterology 123: 689–699.
- 55 Zholudev, A., Zurakowski, D., Young, W. et al. (2004). Serologic testing with ANCA, ASCA, and anti‐OmpC in children and young adults with Crohn's disease and ulcerative colitis: diagnostic value and correlation with disease phenotype. Am. J. Gastroenterol. 99: 2235–2241, https://doi.org/10.1111/j.1572‐0241.2004.40369.x.
- 56 Kohoutova, D., Drahosova, M., Moravkova, P. et al. (2014). Anti‐outer membrane protein C and anti‐glycoprotein 2 antibodies in inflammatory bowel disease and their association with complicated forms of Crohn's disease. BMC Gastroenterol. 14: 190, https://doi.org/10.1186/s12876‐014‐0190‐1.
- 57 Tamboli, C.P., Doman, D.B., and Patel, A. (2011). Current and future role of biomarkers in Crohn's disease risk assessment and treatment. Clin. Exp. Gastroenterol. 4: 127–140, https://doi.org/10.2147/ceg.s18187.
- 58 Nesbitt, A. et al. (2007). Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti‐tumor necrosis factor alpha agents. Inflamm. Bowel Dis. 13: 1323–1332, https://doi.org/10.1002/ibd.20225.
- 59 Michelon, M.A. and Gottlieb, A.B. (2010). Role of golimumab, a TNF‐alpha inhibitor, in the treatment of the psoriatic arthritis. Clin. Cosmet. Investig. Dermatol. 3: 79–84.
- 60 Sands, B.E. and Kaplan, G.G. (2007). The role of TNFalpha in ulcerative colitis. J. Clin. Pharmacol. 47: 930–941, https://doi.org/10.1177/0091270007301623.
- 61 Cao, W., Fiocchi, C., and Pricolo, V.E. (2005). Production of IL‐1beta, hydrogen peroxide, and nitric oxide by colonic mucosa decreases sigmoid smooth muscle contractility in ulcerative colitis. Am. J. Physiol. Cell Physiol. 289: C1408–C1416, https://doi.org/10.1152/ajpcell.00073.2005.
- 62 Balding, J. et al. (2004). Inflammatory bowel disease: the role of inflammatory cytokine gene polymorphisms. Mediators Inflamm. 13: 181–187, https://doi.org/10.1080/09511920410001713529.
- 63 Lacruz‐Guzman, D. et al. (2013). Influence of polymorphisms and TNF and IL1beta serum concentration on the infliximab response in Crohn's disease and ulcerative colitis. Eur. J. Clin. Pharmacol. 69: 431–438, https://doi.org/10.1007/s00228‐012‐1389‐0.
- 64 Louis, E., Belaiche, J., and Reenaers, C. (2010). Do clinical factors help to predict disease course in inflammatory bowel disease? World J. Gastroenterol. 16: 2600–2603.
- 65 Mudter, J. and Neurath, M.F. (2007). IL‐6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 13: 1016–1023, https://doi.org/10.1002/ibd.20148.
- 66 Harper, J.W. and Zisman, T.L. (2016). Interaction of obesity and inflammatory bowel disease. World J. Gastroenterol. 22: 7868–7881, https://doi.org/10.3748/wjg.v22.i35.7868.
- 67 Holtmann, M.H. et al. (2010). Significant differences between Crohn's disease and ulcerative colitis regarding the impact of body mass index and initial disease activity on responsiveness to azathioprine: results from a European multicenter study in 1,176 patients. Dig. Dis. Sci. 55: 1066–1078, https://doi.org/10.1007/s10620‐009‐0846‐9.
- 68 Schmajuk, G. et al. (2014). Identification of risk factors for elevated transaminases in methotrexate users through an electronic health record. Arthritis Care Res. (Hoboken) 66: 1159–1166, https://doi.org/10.1002/acr.22294.
- 69 Bhalme, M., Sharma, A., Keld, R. et al. (2013). Does weight‐adjusted anti‐tumour necrosis factor treatment favour obese patients with Crohn's disease? Eur. J. Gastroenterol. Hepatol. 25: 543–549, https://doi.org/10.1097/MEG.0b013e32835d1f15.
- 70 Lie, M.R., Peppelenbosch, M.P., West, R.L. et al. (2014). Adalimumab in Crohn's disease patients: pharmacokinetics in the first 6 months of treatment. Aliment. Pharmacol. Ther. 40: 1202–1208, https://doi.org/10.1111/apt.12969.
- 71 Cassano, N. et al. (2008). Influence of body mass index, comorbidities and prior systemic therapies on the response of psoriasis to adalimumab: an exploratory analysis from the APHRODITE data. J. Biol. Regul. Homeost. Agents 22: 233–237.
- 72 Briot, K., Gossec, L., Kolta, S. et al. (2008). Prospective assessment of body weight, body composition, and bone density changes in patients with spondyloarthropathy receiving anti‐tumor necrosis factor‐alpha treatment. J. Rheumatol. 35: 855–861.
- 73 Leitner, G.C. and Vogelsang, H. (2016). Pharmacological‐ and non‐pharmacological therapeutic approaches in inflammatory bowel disease in adults. World J. Gastrointest. Pharmacol. Ther. 7: 5–20, https://doi.org/10.4292/wjgpt.v7.i1.5.
- 74 Stein, R.B. and Hanauer, S.B. (1999). Medical therapy fo inflammatory bowel disease. Gastroenterol. Clin. North. Am. 28: 297–321, https://doi.org/10.1016/S0889‐8553(05)70058‐3.
- 75 McLean, L.P. and Cross, R.K. (2014). Adverse events in IBD: to stop or continue immune suppressant and biologic treatment. Expert Rev. Gastroenterol. Hepatol. 8: 223–240, https://doi.org/10.1586/17474124.2014.881715.
- 76 Cote‐Daigneault, J., Bouin, M., Lahaie, R. et al. (2015). Biologics in inflammatory bowel disease: what are the data? United European Gastroenterol. J. 3: 419–428, https://doi.org/10.1177/2050640615590302.
- 77 Bernstein, C.N. (2015). Treatment of IBD: where we are and where we are going. Am. J. Gastroenterol. 110: 114–126, https://doi.org/10.1038/ajg.2014.357.
- 78 Rutgeerts, P. et al. (2005). Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 353: 2462–2476, https://doi.org/10.1056/NEJMoa050516.
- 79 Hanauer, S.B. et al. (2006). Human anti‐tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC‐I trial. Gastroenterology 130: 323–333; quiz 591. https://doi.org/10.1053/j.gastro.2005.11.030.
- 80 Colombel, J.F. et al. (2007). Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology 132: 52–65, https://doi.org/10.1053/j.gastro.2006.11.041.
- 81 Sandborn, W.J. et al. (2007). Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann. Intern. Med. 146: 829–838.
- 82 Reinisch, W. et al. (2011). Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut 60: 780–787, https://doi.org/10.1136/gut.2010.221127.
- 83 Sandborn, W.J. et al. (2013). Vedolizumab as induction and maintenance therapy for Crohn's disease. N. Engl. J. Med. 369: 711–721, https://doi.org/10.1056/NEJMoa1215739.
- 84 Sandborn, W.J. et al. (2007). Certolizumab pegol for the treatment of Crohn's disease. N. Engl. J. Med. 357: 228–238, https://doi.org/10.1056/NEJMoa067594.
- 85 Schreiber, S. et al. (2007). Maintenance therapy with certolizumab pegol for Crohn's disease. N. Engl. J. Med. 357: 239–250, https://doi.org/10.1056/NEJMoa062897.
- 86 Hutas, G. (2008). Golimumab, a fully human monoclonal antibody against TNFalpha. Curr. Opin. Mol. Ther. 10: 393–406.
- 87 Sandborn, W.J. et al. (2014). Subcutaneous golimumab maintains clinical response in patients with moderate‐to‐severe ulcerative colitis. Gastroenterology 146: 96–109.e101, https://doi.org/10.1053/j.gastro.2013.06.010.
- 88 Ben‐Bassat, O., Iacono, A., Irwin, S.P. et al. (2012). Tu1327a Golimumab for treatment of moderate to severe anti‐TNF refaractory Crohn's disease: open label experience. Gastroenterology 142: S‐804.
- 89 Merras‐Salmio, L. and Kolho, K.L. (2016). Golimumab therapy in six patients with severe pediatric onset Crohn disease. J. Pediatr. Gastroenterol. Nutr. 63: 344–347, https://doi.org/10.1097/MPG.0000000000001165.
- 90 Feagan, B.G. et al. (2013). Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369: 699–710, https://doi.org/10.1056/NEJMoa1215734.
- 91 Triantafillidis, J.K., Merikas, E., and Georgopoulos, F. (2011). Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des. Devel. Ther. 5: 185–210, https://doi.org/10.2147/DDDT.S11290.
- 92 Roda, G., Jharap, B., Neeraj, N., and Colombel, J.F. (2016). Loss of response to anti‐TNFs: definition, epidemiology, and management. Clin. Transl. Gastroenterol. 7: e135, https://doi.org/10.1038/ctg.2015.63.
- 93 Fidder, H.H. and Hommes, D.W. (2010). Anti‐TNF and Crohn's disease: when should we start? Curr. Drug Targets 11: 143–147.
- 94 Present, D.H. et al. (1999). Infliximab for the treatment of fistulas in patients with Crohn's disease. N. Engl. J. Med. 340: 1398–1405, https://doi.org/10.1056/NEJM199905063401804.
- 95 Sands, B.E., Blank, M.A., Patel, K. et al. (2004). Long‐term treatment of rectovaginal fistulas in Crohn's disease: response to infliximab in the ACCENT II Study. Clin. Gastroenterol. Hepatol. 2: 912–920.
- 96 Colombel, J.F. et al. (2010). Infliximab, azathioprine, or combination therapy for Crohn's disease. N. Engl. J. Med. 362: 1383–1395, https://doi.org/10.1056/NEJMoa0904492.
- 97 Jarnerot, G. et al. (2005). Infliximab as rescue therapy in severe to moderately severe ulcerative colitis: a randomized, placebo‐controlled study. Gastroenterology 128: 1805–1811.
- 98 Sandborn, W.J. et al. (2007). Adalimumab for maintenance treatment of Crohn's disease: results of the CLASSIC II trial. Gut 56: 1232–1239, https://doi.org/10.1136/gut.2006.106781.
- 99 Triantafillidis, J.K. et al. (2010). Similar response to adalimumab in patients with active Crohn's disease either naive to biologic agents or with prior loss of response or intolerance to infliximab. Rev. Med. Chir. Soc. Med. Nat. Iasi 114: 85–90.
- 100 Peyrin‐Biroulet, L., Laclotte, C., Roblin, X., and Bigard, M.A. (2007). Adalimumab induction therapy for ulcerative colitis with intolerance or lost response to infliximab: an open‐label study. World J. Gastroenterol. 13: 2328–2332.
- 101 Taxonera, C. et al. (2011). Adalimumab induction and maintenance therapy for patients with ulcerative colitis previously treated with infliximab. Aliment. Pharmacol. Ther. 33: 340–348, https://doi.org/10.1111/j.1365‐2036.2010.04531.x.
- 102 Feagan, B.G., Coteur, G., Tan, S. et al. (2009). Clinically meaningful improvement in health‐related quality of life in a randomized controlled trial of certolizumab pegol maintenance therapy for Crohn's disease. Am. J. Gastroenterol. 104: 1976–1983, https://doi.org/10.1038/ajg.2009.199.
- 103 Lonberg, N. (2005). Human antibodies from transgenic animals. Nat. Biotechnol. 23: 1117–1125, https://doi.org/10.1038/nbt1135.
- 104 Shealy, D.J. et al. (2010). Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. MAbs 2: 428–439.
- 105 Sandborn, W.J. et al. (2014). Subcutaneous golimumab induces clinical response and remission in patients with moderate‐to‐severe ulcerative colitis. Gastroenterology 146: 85–95; quiz e14‐85. https://doi.org/10.1053/j.gastro.2013.05.048.
- 106 Vande Casteele, N. and Khanna, R. (2017). Therapeutic drug monitoring of Golimumab in the treatment of ulcerative colitis. Pharm. Res. 34: 1556–1563, https://doi.org/10.1007/s11095‐017‐2150‐2.
- 107 Jani, M., Barton, A., Warren, R.B. et al. (2014). The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology (Oxford) 53: 213–222, https://doi.org/10.1093/rheumatology/ket260.
- 108 Peyrin‐Biroulet, L. et al. (2008). Efficacy and safety of tumor necrosis factor antagonists in Crohn's disease: meta‐analysis of placebo‐controlled trials. Clin. Gastroenterol. Hepatol. 6: 644–653, https://doi.org/10.1016/j.cgh.2008.03.014.
- 109 Fiorino, G., Rovida, S., Correale, C. et al. (2010). Emerging biologics in the treatment of inflammatory bowel disease: what is around the corner? Curr. Drug Targets 11: 249–260.
- 110 Amiot, A. and Peyrin‐Biroulet, L. (2015). Current, new and future biological agents on the horizon for the treatment of inflammatory bowel diseases. Therap. Adv. Gastroenterol. 8: 66–82, https://doi.org/10.1177/1756283X14558193.
- 111 Targan, S.R. et al. (2007). Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology 132: 1672–1683, https://doi.org/10.1053/j.gastro.2007.03.024.
- 112 Bloomgren, G. et al. (2012). Risk of natalizumab‐associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 366: 1870–1880, https://doi.org/10.1056/NEJMoa1107829.
- 113 Ledder, O. et al. (2017). Vedolizumab in paediatric inflammatory bowel disease: a retrospective multi‐centre experience from the paediatric IBD porto group of ESPGHAN. J. Crohns Colitis 11: 1230–1237, https://doi.org/10.1093/ecco‐jcc/jjx082.
- 114 Philpott, J.R. and Miner, P.B. Jr. (2008). Antisense inhibition of ICAM‐1 expression as therapy provides insight into basic inflammatory pathways through early experiences in IBD. Expert Opin. Biol. Ther. 8: 1627–1632, https://doi.org/10.1517/14712598.8.10.1627.
- 115 Venkiteshwaran, A. (2009). Tocilizumab. MAbs 1: 432–438.
- 116 Ito, H. et al. (2004). A pilot randomized trial of a human anti‐interleukin‐6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 126: 989–996; discussion 947.
- 117 Korzenik, J.R. and Podolsky, D.K. (2006). Evolving knowledge and therapy of inflammatory bowel disease. Nat. Rev. Drug Discovery 5: 197–209, https://doi.org/10.1038/nrd1986.
- 118 Korzenik, J.R. et al. (2005). Sargramostim for active Crohn's disease. N. Engl. J. Med. 352: 2193–2201, https://doi.org/10.1056/NEJMoa041109.
- 119 Korzenik, J.R. and Dieckgraefe, B.K. (2005). An open‐labelled study of granulocyte colony‐stimulating factor in the treatment of active Crohn's disease. Aliment Pharmacol. Ther. 21: 391–400, https://doi.org/10.1111/j.1365‐2036.2005.02287.x.
- 120 Krishnan, K., Arnone, B., and Buchman, A. (2011). Intestinal growth factors: potential use in the treatment of inflammatory bowel disease and their role in mucosal healing. Inflamm. Bowel Dis. 17: 410–422, https://doi.org/10.1002/ibd.21316.
- 121 Sharma, R., Sharma, C.L., and Mahajan, A. (2008). Biological agents targeting beyond TNF‐alpha. Indian J. Crit. Care Med. 12: 181–189, https://doi.org/10.4103/0972‐5229.45079.
- 122 Reinisch, W. et al. (2010). Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double‐blind, placebo‐controlled, multiple‐dose study. Inflamm. Bowel Dis. 16: 233–242, https://doi.org/10.1002/ibd.21038.
- 123 Feagan, B.G. et al. (2016). Ustekinumab as induction and maintenance therapy for Crohn's disease. N. Engl. J. Med. 375: 1946–1960, https://doi.org/10.1056/NEJMoa1602773.
- 124 Sandborn, W.J. et al. (2008). A randomized trial of Ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with moderate‐to‐severe Crohn's disease. Gastroenterology 135: 1130–1141, https://doi.org/10.1053/j.gastro.2008.07.014.
- 125 Toedter, G.P. et al. (2009). Relationship of C‐reactive protein with clinical response after therapy with ustekinumab in Crohn's disease. Am. J. Gastroenterol. 104: 2768–2773, https://doi.org/10.1038/ajg.2009.454.
- 126 Plevy, S. et al. (2007). A phase I study of visilizumab, a humanized anti‐CD3 monoclonal antibody, in severe steroid‐refractory ulcerative colitis. Gastroenterology 133: 1414–1422, https://doi.org/10.1053/j.gastro.2007.08.035.
- 127 Creed, T.J. et al. (2006). Basiliximab for the treatment of steroid‐resistant ulcerative colitis: further experience in moderate and severe disease. Aliment. Pharmacol. Ther. 23: 1435–1442, https://doi.org/10.1111/j.1365‐2036.2006.02904.x.
- 128 Brandstadter, R. and Katz Sand, I. (2017). The use of natalizumab for multiple sclerosis. Neuropsychiatr. Dis. Treat. 13: 1691–1702, https://doi.org/10.2147/NDT.S114636.
- 129 Bradley, C.A. (2017). IBD: tofacitinib effective in ulcerative colitis. Nat. Rev. Gastroenterol. Hepatol. 14: 388, https://doi.org/10.1038/nrgastro.2017.66.
- 130 Sorger, P.K. et al. (2011). Quantitative and Systems Pharmacology in the Post‐genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms, 1–47. http://www.nigms.nih.gov/training/documents/systemspharmawpsorger2011.pdf (accessed 01 October 2018).
- 131 Furuya, Y. et al. (2007). Theory based analysis of anti‐inflammatory effect of infliximab on Crohn's disease. Drug Metab. Pharmacokinet. 22: 20–25, https://doi.org/10.2133/dmpk.22.20.
- 132 Mager, D.E. and Jusko, W.J. (2001). General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J. Pharmacokinet. Pharmacodyn. 28: 507–532.
- 133 Dekkers, P.E. et al. (1999). The effect of a metalloproteinase inhibitor (GI5402) on tumor necrosis factor‐alpha (TNF‐alpha) and TNF‐alpha receptors during human endotoxemia. Blood 94: 2252–2258.
- 134 Oliver, J.C. et al. (1993). Cytokine kinetics in an in vitro whole blood model following an endotoxin challenge. Lymphokine Cytokine Res. 12: 115–120.
- 135 Waage, A., Brandtzaeg, P., Halstensen, A. et al. (1989). The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 169: 333–338.
- 136 Ternant, D., Karmiris, K., Van Assche, G. et al. (2012). Adalimumab pharmacokinetics and concentration‐effect relationship in Crohn's disease. Fundam. Clin. Pharmacol. 26.
- 137 Vande Casteele, N. et al. (2017). Accounting for pharmacokinetic variability of certolizumab pegol in patients with Crohn's disease. Clin. Pharmacokinet. 56: 1513–1523, https://doi.org/10.1007/s40262‐017‐0535‐3.
- 138 Rosario, M. et al. (2015). Population pharmacokinetics‐pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn's disease. Aliment. Pharmacol. Ther. 42: 188–202, https://doi.org/10.1111/apt.13243.
- 139 Galluppi, G.R. et al. (2001). Integration of pharmacokinetic and pharmacodynamic studies in the discovery, development, and review of protein therapeutic agents: a conference report. Clin. Pharmacol. Ther. 69: 387–399.
- 140 Mould, D.R. (2012). Models for disease progression: new approaches and uses. Clin. Pharmacol. Ther. 92: 125–131, https://doi.org/10.1038/clpt.2012.53.
- 141 Ordas, I., Mould, D.R., Feagan, B.G., and Sandborn, W.J. (2012). Anti‐TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics‐based dosing paradigms. Clin. Pharmacol. Therapeut. 91: 635–646, https://doi.org/10.1038/clpt.2011.328.
- 142 Longobardi, T., Jacobs, P., and Bernstein, C.N. (2004). Utilization of health care resources by individuals with inflammatory bowel disease in the United States: a profile of time since diagnosis. Am. J. Gastroenterol. 99: 650–655, https://doi.org/10.1111/j.1572‐0241.2004.04132.x.
- 143 Hanauer, S.B. et al. (2002). Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet 359: 1541–1549, https://doi.org/10.1016/S0140‐6736(02)08512‐4.
- 144 Van Assche, G. et al. (2005). Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353: 362–368, https://doi.org/10.1056/NEJMoa051586.
- 145 Keane, J. et al. (2001). Tuberculosis associated with infliximab, a tumor necrosis factor alpha‐neutralizing agent. N. Engl. J. Med. 345: 1098–1104, https://doi.org/10.1056/NEJMoa011110.
- 146 Rosh, J.R., Gross, T., Mamula, P. et al. (2007). Hepatosplenic T‐cell lymphoma in adolescents and young adults with Crohn's disease: a cautionary tale? Inflamm. Bowel Dis. 13: 1024–1030, https://doi.org/10.1002/ibd.20169.
- 147 Renna, S., Orlando, A., Mocciaro, F., and Cottone, M. (2009). Placebo therapy in Crohn's disease. Eur. J. Intern. Med. 20: 572–578, https://doi.org/10.1016/j.ejim.2009.04.006.
- 148 Kane, S. (2008). Endoscopic healing should be a goal for everyone with ulcerative colitis. Inflamm. Bowel Dis., https://doi.org/10.1002/ibd.20779 .
- 149 Jelkmann, W. (2010). Biosimilar epoetins and other "follow‐on" biologics: update on the European experiences. Am. J. Hematol. 85: 771–780, https://doi.org/10.1002/ajh.21805.
- 150 Panes, J. et al. (2017). Tofacitinib for induction and maintenance therapy of Crohn's disease: results of two phase IIb randomised placebo‐controlled trials. Gut 66: 1049–1059, https://doi.org/10.1136/gutjnl‐2016‐312735.