8
Tutorial: Numerical (NONMEM) Implementation of the Target‐Mediated Drug Disposition Model
8.1 Introduction
The target‐mediated drug disposition (TMDD) model is a nonlinear system of differential equations, with multiple fixed and random effect parameters that describe complex biological processes. Differential equations of the TMDD model describe processes with very different characteristic timescales, from few minutes for binding processes to several weeks for the elimination of the drug. Numerical methods for solving these equations are numerically unstable, unless the step size is taken to be extremely small. In mathematics such systems are called “stiff.” System parameters are often poorly identifiable either due to stiffness of the differential equations (for the full TMDD model) or due to limitations of the available data. This results in long computational run times and numerical difficulties, requiring careful attention to the selection of software, estimation methods, and its parameters. This chapter is intended to describe how to apply the NONMEM software application [1] and NONMEM estimation methods to the TMDD equations. NONMEM is the NONlinear Mixed Effect Modeling package originally developed by Stuart Beal, Lewis Sheiner, and Alison Boeckmann and is now being developed by Robert Bauer. While several other software packages for nonlinear mixed effect modeling are available, NONMEM remains the standard in the pharmaceutical industry. The chapter should be studied in conjunction with Chapter 7 as it does not reproduce the theoretical aspects of the TMDD model and focuses on details of implementation.
8.2 Notations and Data
As with any analysis, a TMDD analysis should start with a thorough understanding of available data. One important aspect is the units of dose and concentrations. TMDD equations include three different analyte species (unbound drug concentration C, unbound target concentration R, and drug–target complex concentration RC) with different molar weights. These equations are formulated in molar units to assure appropriate mass balance. To allow seamless work with the data, dose values, and concentration values used in the data, files should be converted to molar units, as mentioned in Section 7.3. The alternative approach is to correct for molar unit differences in the control stream. However, it is more cumbersome to keep track of units in the control stream.
Another important aspect is understanding of the assay properties and distinction between free (unbound) and total (bound and unbound) drug and target concentrations. Total drug and target concentrations will be denoted as Ctot = C + RC and Rtot = R + RC, respectively. To perform the analysis, it is important to understand whether the data represent unbound, bound, or total drug and target concentrations. As assays for therapeutic proteins are much more difficult than assays for small molecule drugs, assay properties are not always well understood. Thus, collaboration between bioanalytical scientists and modelers is often crucial in understanding the data. In addition to drug and target concentrations, data may include target occupancy (R/Rtot) or some activity measure that can be interpreted as a ratio of the unbound target concentration to the target concentration at baseline (R/R0).
TMDD equations as presented in Chapter 7 are written in units of concentrations, while in NONMEM implementation, it is easier to write in amounts in order to keep track of mass balance of the drug and the target in various model compartments. This distinction between drug and target concentrations and amounts should be understood and carefully followed.
The analysis of any specific drug should always start with the investigation of underlying biological processes. When the assumptions of the standard TMDD model do not hold, the equations should be modified to reflect mechanistic understanding of the system.
8.3 NONMEM Code for TMDD Model and Approximations
8.3.1 Full TMDD Model
The full TMDD model equations are described in Section 7.2. Selected details of the NONMEM implementation are presented below.
$PK ; PK block... ; code to assign model parametersKa=THETA(1)*EXP(ETA(1)) ; via THETA and ETA parametersBase = Ksyn/Kdeg ; derived parameters... ;A_0(4) = Base ; initial conditions that differ from zero$DES ; differential equations blockDADT(1) = -Ka*A(1) ; SC depot drug amountDADT(2) = Ka*A(1)-(Kel+Kpt)*A(2)+Ktp*A(3) ; continued on the next line-Kon*A(2)*A(4)+Koff*A(5)*Vc ; central compartment drug amountDADT(3) = Kpt*A(1)-Ktp*A(4) ; tissue compartment drug amountDADT(4) = Ksyn-Kdeg*A(4)-Kon*A(2)/Vc*A(4)+Koff*A(5) ; target concentrationDADT(5) = Kon*A(2)/Vc*A(4)‐(Koff+Kint)*A(5) ;drug‐target complex concentration$ERROR ; error model blockCfree = A(2)/Vc ; free (unbound) drug concentrationRC = A(5) ; drug‐target complex concentrationCtot = Cfree +RC ; total drug concentrationR = A(4) ; free (unbound) target concentrationRtot = R + RC ; total target concentrationY = LOG(Cfree) + EPS(1) ; error model for free drug concentrationIF(TYPE.EQ.2) Y = LOG(Rtot) + EPS(2) ; Error model for total targetconcentration
The $PK block is used to assign model parameters via NONMEM key words of THETA() for fixed effects and ETA() for random effect parameters.
The $DES block specifies differential equations (with DADT(i) denoting the expression for the time derivative of the compartment i variable). In this example, Compartments 1–5 represent, respectively, drug in the depot compartment (DADT(1), for subcutaneous administration), drug in the central compartment (DADT(2), with volume Vc), drug in the peripheral compartment (DADT(3)), unbound target in the central compartment (DADT(4)), and drug–target complex in the central compartment (DADT(5)). Note that the first three equations are written in amounts, while the last two represent concentrations. Compartments 2, 4, and 5 are assumed to have the same volume as they represent different substances (drug, target, and drug–target complex) in the same central compartment. The equations for the drug describe diffusion exchange with the peripheral compartment (described by the rate constants Kpt and Ktp), elimination (with the elimination rate Kel), binding of the drug to the target (with binding proportional to the drug and target concentrations with the second‐order association rate Kon), and dissociation of the drug–target complex (with the dissociation rate Koff). The target R is produced in the central compartment with the synthesis rate Ksyn (per unit volume) and eliminated with the degradation rate Kdeg. The drug–target complex is produced by association of the drug and the target with the production rate Kon·R·C, is eliminated with the internalization rate Kint, and dissociates back into the drug and the target with the dissociation rate Koff.
The drug is administered either subcutaneously (dose D1 into the depot compartment) or intravenously (IV bolus dose D2 and/or infusion with the rate Int(t) into the central compartment). Dose administration is set in the data file and in the $INPUT block of the control stream, and we refer the reader to the NONMEM user guides [1] for detailed instructions on how it should be coded.
The $ERROR block specifies what is measured and the corresponding error model. In the example code above, it is assumed that free drug and total target concentrations were observed (measured). Note that the observed variable is not necessarily one of the equation variables. In this control stream, the equations are written in terms of free (unbound) drug and target concentrations, while the observed total target concentration is the combination of the equation variables.
The system of equations should be supplemented by the initial conditions (assigned in $THETA, $OMEGA, and $SIGMA blocks). It is important to set initial conditions reasonably close to the true values (although even the range of the true values are often unknown at the start of the analysis) as most numerical methods are not able to find the global minimum, and setting initial conditions too far from the true minimum may result in the model being stopped in a local minimum. To avoid this scenario, typical values of the parameters for similar compounds and similar targets should be used. Alternatively, simpler models should be tested first to find the range of the main parameters, and more complex models should be built sequentially on the foundation of less complex models, each time starting from the final values of the simpler models. Yet another alternative is to test the wide range of initial conditions, but this random search can be complicated by instability of the model with incorrect initial conditions.
The code above specifies the exponential error model. As the exponential error model is not directly supported by NONMEM software, it is implemented as an additive error in log‐transformed variables. For this implementation, the data file should be populated by natural logs of observed concentrations. TYPE variable should be provided for each data record, and TYPE = 2 denotes data records with target concentration observations. An example of the data file is provided in the online supplemental material.
8.3.2 Quasi‐Steady‐State and Rapid Binding Approximations
The quasi‐steady‐state (QSS) approximation of the TMDD model is described in Section 7.5.1. Selected details of the NONMEM implementation are presented below.
$PK ; PK block... ; code to assign model parametersA_0(4) = Base ; initial conditions that differ from zero$DES ; differential equations blockCt =A(2)/Vc ; total drug concentrationD= Ct-A(4)-Kss ; abbreviated notation to shorten next lineC = 0.5*(D +SQRT(D**2+4*Kss*Ct)) ; unbound drug concentrationDADT(1) = -Ka*A(1) ; SC depot drug amountDADT(2) = Ka*A(1)-(Kel+Kpt)*C*Vc+Ktp*A(3) ; continued on the next line-Kint*A(4)*C*Vc /(Kss+C) ; total drug amountDADT(3) = Kpt*C*Vc -Ktp*A(3) ; unbound drug amountDADT(4) = Ksyn-Kdeg*A(4)-(Kint-Kdeg)*A(4)*C/(Kss+C) ; total targetconcentration$ERROR ; error model blockCtot = A(2)/Vc ; total drug concentrationRtot = A(4) ; total target concentrationDD = Ctot-A(4)-Kss ; abbreviated notation to shorten next lineCfree=0.5*(DD+SQRT(DD**2+4*Kss*Ctot)) ; free drug concentrationRC = Rtot*Cfree/(Kss+Cfree) ; total drug‐target complex concentrationR = Rtot*Kss/(Kss+Cfree) ; free (unbound) target concentrationY = LOG(Cfree) + EPS(1) ; error model for free drug concentrationIF(TYPE.EQ.2) Y = LOG(Rtot) + EPS(2) ; error model for total targetconcentration
Values A(1)–A(4) represent, respectively, drug amount in SC depot compartment, total drug amount in the central compartment, free drug amount in the peripheral compartment, and the total target concentration in the central compartment. As for the full TMDD model, the $ERROR block specifies what is measured and the corresponding error model. In the example code above, it is assumed that free drug and total target concentrations were observed (measured). As in the previous section, the observed variable (Cfree in this case) is not necessarily one of the equation variables (Ctot is the equation variable in this case).
The system of QSS equations should also be supplemented by initial conditions. In our experience with the clinical data, the QSS model is more stable and robust than the full TMDD model. While it is still important to set initial conditions close to the true values, the model usually can recover the true minimum even if started far from the solution.
When elimination rate of the target (Kdeg) and internalization rate of the drug–target complex (Kint) are equal, total target concentration is constant, and the control stream can be simplified as follows:
$DES ; differential equations blockCt =A(2)/Vc ; total drug concentrationD= Ct-Rtot-Kss ; abbreviated notationC = 0.5*(D+SQRT(D**2+4*Kss*Ct)) ; unbound drug concentrationDADT(1) = -Ka*A(1) ; SC depot drug amountDADT(2) = Ka*A(1)-(Kel+Kpt)*C*Vc+Ktp*A(3) ; continued on the next line-Kint*Rtot*C*Vc /(Kss+C) ; total drug amountDADT(3) = Kpt* C*Vc -Ktp*A(3) ; unbound drug amount$ERROR ; error model blockCtot = A(2)/Vc ; total drug concentrationDD = Ctot-Rtot-Kss ; abbreviated notationCfree=0.5*(DD+SQRT(DD**2+4*Kss*Ct)) ; free drug concentrationRC = Rtot*Cfree/(Kss+Cfree) ; total drug‐target complex concentrationR = Rtot*Kss/(Kss+Cfree) ; free (unbound) target concentrationY = LOG(Cfree) + EPS(1) ; error model for free drug concentration
In this code, Rtot is the estimated model parameter that should be defined (via THETA and ETA variables) in the $PK block.
8.3.3 Michaelis–Menten Approximation
The Michaelis–Menten approximation of the TMDD model is described in Section 7.5.2. Selected details of the NONMEM implementation are presented below.
$PK ; PK block... ; code to assign model parametersA_0(4) = Base ; initial conditions that differ from zero$DES ; differential equations blockC =A(2)/Vc ; drug concentrationDADT(1) = -Ka*A(1) ; SC depot drug amountDADT(2) = Ka*A(1)-(Kel+Kpt)*A(2)+Ktp*A(3) ; continued on the next line-Kint*A(4)*A(2) /(Kss+C) ; drug amountDADT(3) = Kpt*A(2) -Ktp*A(3) ; drug amountDADT(4) = Ksyn-Kdeg*A(4)-(Kint-Kdeg)*A(4)*C/(Kss+C) ; total targetconcentration$ERROR ; error model blockCfree = A(2)/Vc ; free (equal to total) drug concentrationRtot = A(4) ; total target concentrationRC = Rtot*Cfree/(Kss+Cfree) ; total drug‐target complex concentrationR = Rtot*Kss/(Kss+Cfree) ; free (unbound) target concentrationOCC = Cfree/(Kss+Cfree) ; receptor occupancyY = LOG(Cfree) + EPS(1) ; error model for free drug concentrationIF(TYPE.EQ.2) Y = OCC + EPS(2) ; error model for receptor occupancy
In this approximation, there is no distinction between free and total drug concentrations as the concentration of the drug–target complex is assumed to be negligible relative to the free drug concentration. As earlier, the Compartment A(4) represents the total target concentration. The $ERROR block specifies what is measured and the corresponding error model. In the example code above, it is assumed that the free drug and receptor occupancy were observed (measured). As in the previous section, the observed variable (receptor occupancy OCC in this case) is not necessarily one of the equation variables (Rtot is the equation variable in this case). For drugs that can be described by Michaelis–Menten equations, target measurements are rarely available. Target characteristics are computed for illustration of the rare situation where the receptor occupancy measurements are available. A simple additive error model is used for the receptor occupancy in this example.
When the elimination rate of the target Kdeg and internalization rate of the drug–target complex Kint are equal, the total target concentration is constant, and the MM approximation is reduced to MM equations. This simplifies the control stream as follows:
$DES ; differential equations blockDADT(1) = -Ka*A(1) ; SC depot drug amountDADT(2) = Ka*A(1)-(Kel+Kpt)*A(2)+Ktp*A(3)-Vmax*A(2)/(Kss+A(2)/Vc)DADT(3) = Kpt*A(2)-Ktp*A(3) ; drug amount$ERROR ; error model blockCfree = A(2)/Vc ; drug concentrationY = LOG(Cfree) + EPS(1) ; error model for drug concentration
8.4 How to Select Correct Approximation
As many different models may describe the same data, an important question is how to balance the flexibility of the model and its ability to describe the data precisely with identifiability of model parameters given the data. In this section, we discuss several approaches to this problem.
8.4.1 Bottom Up Approach
This is the application of the standard modeling approach where we start from the simplest available model. In the case of monoclonal antibodies (the most common case for the TMDD model), one can start with a linear model. The linear model is likely to converge and provide parameter estimates that can be used as initial values for more complex models. After the linear model is investigated, the next level of complexity is to use the model with parallel linear and Michaelis–Menten elimination followed by the QSS approximation of the TMDD model, first with fixed and then with variable total target concentrations. The next level of complexity is to use the full TMDD model. Note however, that we have not seen any data where the full TMDD model was needed (and/or well estimated). Moreover, simulations from the full TMDD model indicate that PK of monoclonal antibodies with TMDD and clinically relevant PK sampling can always be described by QSS equations [2].
Moving up in the level of model complexity, we need tools to evaluate and compare the models, so we know where to stop. The simplest way is to compare the minimum objective function value of hierarchical models and select the model based on some formal statistical criteria (e.g. the likelihood ratio test). While useful in some cases, this approach is likely to result in overparameterized models that are hard to work with. Instead (or in addition), the following diagnostic tools are recommended.
Individual concentration–time plots:
- Individual plots with overlaid observations, population, and individual predictions on arithmetic and semi-log scales may reveal increase of elimination (slope on the semilog plots) at low concentrations and/or at low doses indicating nonlinearity. Dose‐dependent bias in these plots relative to the population predictions may indicate need for a nonlinear model.
Plots of random effects vs. dose:
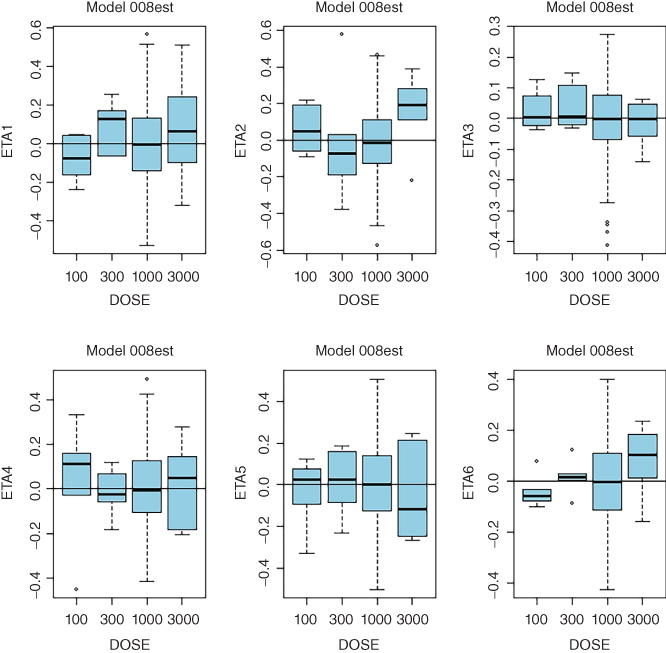
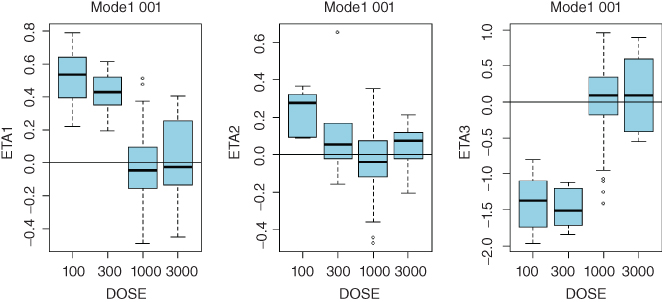
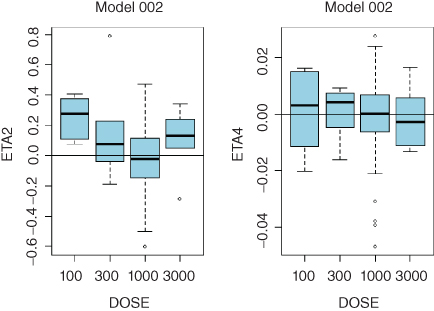
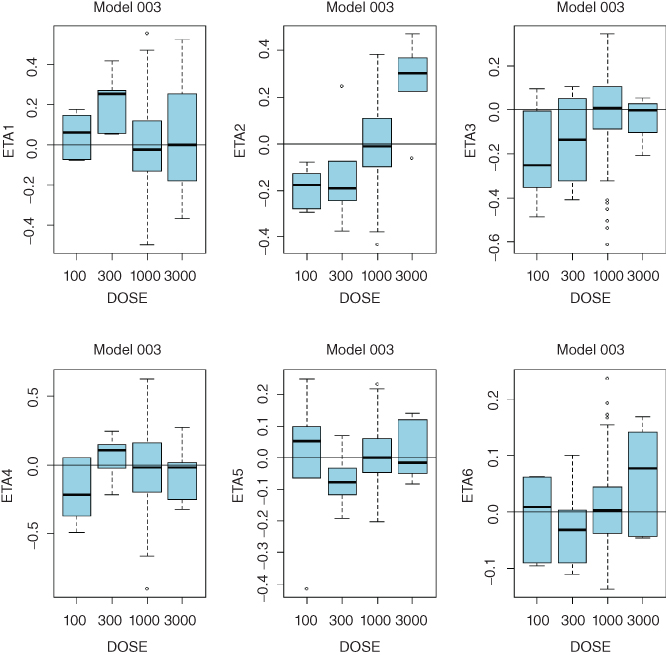
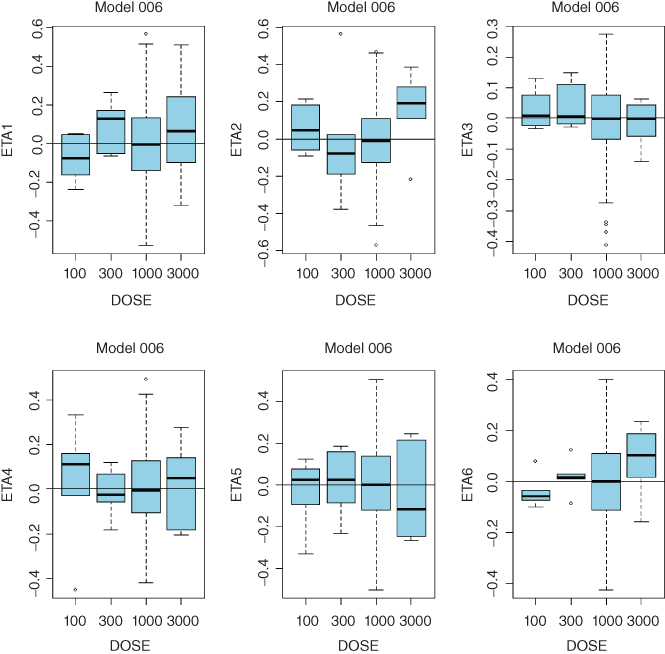
- Consistent trends in any of these plots may indicate that a more complex model is needed. While clearance is expected to decrease with dose in TMMD models, systematic trends for any of the model parameters may point to the need to increase complexity of the model. Sample diagnostic plots presented in the Appendix 8 illustrate incremental improvement of the model when the model complexity is increased from the linear model up to the full TMDD model. Plots for the QSS model are virtually identical to those of the full TMDD model.
Goodness of fit(GOF) plots stratified by dose:
- For nonlinear models, all GOF plots should be stratified by dose. Dose‐dependent bias in the plots of observed data versus population predictions or any other GOF plots may indicate that a more complex model is needed.
Precision of parameter estimates:
- Increase of model complexity may result in inability of the data to support estimation of all model parameters. This will be manifested in steep increase in relative standard errors (RSEs) of parameter estimates. Large RSE may indicate overparameterization and the need to go back to a simpler model structure.
Consistency of results with model assumptions and biology:
- After the model structure is selected, it is important to check whether model features are consistent with biology of the underlying system. If not, it could be hard to defend the model as the mechanistic description of the data is often the main goal of the analysis.
8.4.2 Approach Based on Biological Considerations
The alternative (or complementary) approach to model selection is based on biological considerations, i.e. on the properties of the drug and the target. The main questions to investigate are (i) whether the target is soluble or membrane‐bound; (ii) whether the internalization (elimination) rate of the drug–target complex is slow or fast; (iii) whether accumulation of the drug–target complex is small or significant relative to the total drug concentrations.
If the target is a low‐molecular‐weight, soluble molecule with a relatively fast turnover, we may expect (i) likely accumulation of the drug–target complex; (ii) availability of the free or total drug concentration measurements; (iii) availability of total target or drug–target complex concentration measurements. For these data, the QSS approximation of the TMDD model would be a model of choice.
If the target is membrane‐bound and with relatively fast turnover, we may expect (i) fast elimination of the drug–target complex; (ii) availability of only the free drug concentration measurements; (iii) low levels of total target and drug–target complex concentrations, and no measurements of these quantities. For these data, the MM approximation of the TMDD model or the model with parallel linear and MM elimination would be expected to provide an adequate description of the data.
8.5 Numerical Implementation
Details of numerical implementation of the TMDD model approximations in NONMEM Version 7.2 were studied in detail in [3]. We refer the reader to the original paper for details. Here we briefly summarize the main results of that work (and our unpublished experience) related to the choice of the numerical integration routine and parallel computing.
8.5.1 Choice of ADVAN Subroutines
NONMEM versions 7.2 and 7.3 have four PREDPP subroutines (ADVAN6, ADVAN8, ADVAN9, and ADVAN13) that solve differential equations. Comparison of the performance of these routines for identical models (the QSS approximation of the TMDD model) showed that different ADVAN subroutines provided similar if not identical parameter estimates. ADVAN13 provided the fastest and most robust solution. ADVAN6 required the same or slightly longer run time, but sometimes failed even when ADVAN13 was successful. ADVAN8 and ADVAN9 were significantly less efficient. There were cases when models with ADVAN6 converged successfully or provided successful covariance step while ADVAN13 counterparts converged with rounding errors, but the opposite effects were also observed. Performance of these subroutines may depend on details of the data and stiffness of the underlying differential equations. Therefore, results may be project‐specific. We would recommend the use of ADVA13 or ADVAN6 subroutines, with switching between them as needed for a specific dataset. In NONMEM Version 7.4, two new subroutines (ADVAN14 and ADVAN15) were introduced. In our tests, performance of ADVAN14 and ADVAN15 with TMDD models was similar to that of ADVAN13 and ADVAN9, respectively.
8.5.2 Parallel Computing
The run time of TMDD models can be extremely long, especially for large data sets. Therefore, it is beneficial if not necessary to use parallel computing option provided by NONMEM. The user chooses the number of processors that is used for each problem. As NONMEM parallelization works by distribution of whole‐subject computations across processors, it makes no sense to use more processors than number of subjects in the data set. However, this is a very crude estimate as efficiency of the overall run may depend on a particular implementation of the hardware configuration, network speed, and specific details of the data. For each data set and each hardware configuration, there is a point beyond which efficiency of parallelization is greatly reduced. Moreover, the overall run time may even increase with addition of extra processors. For long‐run time problems, it is helpful to test parallel computing at the start of each project, and then use this tool appropriately. It is also beneficial to check whether NONMEM default size parameters provide sufficient memory allocation to avoid data exchange with the hard drive. It can be verified by checking that files FILE07 to FILE39 in the NONMEM run directory have size of zero. If any of these files is not empty, $SIZE option should be changed as recommended in the manual. To guarantee reproducibility of the model runs for MCMC methods independently of the computer load and number of used processors, estimation step option RANMETHOD with the descriptor P is recommended (e.g. RANMETHOD=P). This option allows NONMEM to associate the seed of the random number generator with each subject, thus allowing for the same sequence of random numbers to be used for MCMC procedures independently of parallelization options.
8.6 Summary
No manuscript or course can replace a hands‐on experience in the field of population PK and PK–PD modeling. However, the control streams and simulated examples provided here in conjunction with the theoretical background provided in Chapter 7 should allow the reader enough details to start and successfully complete modeling projects for a drug with TMDD.
References
- 1 Beal, S., Sheiner, L.B., Boeckmann, A., and Bauer, R.J. (1989–2014). NONMEM User's Guides. Ellicott City, MD: Icon Development Solutions.
- 2 Gibiansky, L., Gibiansky, E., Kakkar, T., and Ma, P. (2008). Approximations of the target‐mediated drug disposition model and identifiability of model parameters. J. Pharmacokinet. Pharmacodyn. 35 (5): 573–591.
- 3 Gibiansky, L., Gibiansky, E., and Bauer, R. (2012). Comparison of NONMEM 7.2 estimation methods and parallel processing efficiency on a target‐mediated drug disposition model. J. Pharmacokinet. Pharmacodyn. 39 (1): 17–35. https://doi.org/10.1007/s10928‐011‐9228‐y.
Appendix Diagnostic Plots
- Linear Model 001:
- ETA1–ETA3: random effects on CL, Vc, and Q. DOSE: nominal dose
- Model 002 with Parallel Linear and Michaelis–Menten Elimination:
- ETA1–ETA5: random effects on CL, Vc, Q, Vmax, and KSS. DOSE: nominal dose

- QSS Model 003 with Constant Target Concentration:
- ETA1–ETA5: random effects on CL, Vc, Q, KSS, kint, and Rtot. DOSE: nominal dose
- QSS Model 006:
- ETA1–ETA6: random effects on CL, Vc, Q, kint, ksyn, and kdeg. DOSE: nominal dose
- Full TMDD Model 008est:
- ETA1–ETA6: random effects on CL, Vc, Q, kint, ksyn, and kdeg. DOSE: nominal dose