
1
Disease Interception in Autoimmune Diseases: From a Conceptual Framework to Practical Implementation
Anish Suri
Janssen R&D, Beerse, BE; Current Address: Cue Biopharma, 21 Erie Street, Cambridge, MA
1.1 Introduction to Disease Interception
The hallmark of autoimmunity, and perhaps many immune‐mediated inflammatory diseases, is the inappropriate recognition of self‐tissue resulting in subsequent effector reactions that ultimately damage the host. Numerous components may underlie these outcomes and likely include lack of central immune tolerance to self, breakdown of peripheral tolerance mechanisms that control inflammation, relevant stress signals that enhance and/or modify antigenicity of self‐tissues, and a complex interplay between the host and the environment; including the emergent immuno‐regulatory role of the microbiome. Much of the historical therapeutic success has focused on approaches that broadly dampen or modulate inflammatory mediators (e.g. success of anti‐cytokine antibodies directed against tumor necrosis factor (TNF), or interleukins such as IL‐6, IL‐17, IL‐1 in rheumatoid arthritis (RA), inflammatory bowel disease (IBD), psoriasis or small molecule kinase inhibitors) with little to no understanding of the earliest molecular triggers that underpin disease initiation [1,2]. While anti‐inflammatory approaches have yielded symptomatic benefits for the patients, a significant unmet medical need still exists particularly from the viewpoint of sustained disease remission and cessation of tissue‐destructive biological processes. To this end, the ultimate therapeutic goal for autoimmune diseases would be to restore immune homeostasis as evidenced by inhibition of self‐reactivity leading to reestablishment of a self‐tolerant state.
1.1.1 What is Disease Interception and How Does This Impact Our Prospective Thinking Toward Novel Solutions for Patients Suffering from Autoimmune Diseases?
Simply stated, disease interception refers to intervention from the time of the earliest stages of dysregulation in an individual bearing the appropriate genetic and/or environmental risks for a particular disease, until the time the disease is clinically evidenced (Figure 1.1). The temporal period for disease interception precedes any clinically symptomatic stage, hence defining a distinct timeframe for therapeutic intervention. The phenotypic alterations that constitute the early aberrant perturbations should be well‐defined so as to identify the at‐risk or “dysregulated” prospective patients with a high degree of confidence, allowing for implementation of appropriate disease interception strategies to alter the course of, or cease the progression of disease. This concept is particularly powerful since it provides strategic and scientific differentiation from current therapeutic approaches focused on the chronic stages of autoimmune disorders. Since most current therapies are broadly anti‐inflammatory or immune‐suppressive, they may not be an optimal choice for application in disease interception approaches – based from a viewpoint of distinct trigger mechanisms and safety considerations in a population not yet manifesting clinical symptoms. This presents new opportunities for translating scientific understanding of earliest mechanisms of immune dysregulation into novel therapeutics that are likely to offer superior, and perhaps lasting, clinical benefit. In addition, practical application of disease interception strategies to clinical practice will challenge new paradigms in clinical development approaches, regulatory endpoints, and health economics. A perspective on challenges and opportunities for disease interception has been discussed in a recent commentary by Hait and Lebowitz [3]. Some of the key points include challenges with validated targets and surrogate endpoints to demonstrate a beneficial outcome, as well as the positive precedence with current approved drugs that are already applied in an at‐risk patient population in a disease interception setting (e.g. statins and cardiovascular disease). Additional regulatory and commercial perspectives would also need to be factored in when thinking about future markets and opportunity in this space.

Figure 1.1 Graphical progression of an individual from a normal state toward a diseased state. Disease interception refers to the earliest phenotypic alterations that result as a consequence of genetic susceptibility of the individual coupled with an appropriate environmental trigger. Listed below are some key considerations for successful execution of disease interception strategies.
1.2 Disease Interception in Autoimmune Diseases
The stages of an autoimmune response can be broken down into phases that span the gamut starting from a predisposed healthy individual that experiences an environmental trigger or insult resulting in early signals of breakdown of tolerance (Figure 1.2); this stage subsequently progresses, via amplification of immune cell activation coupled with compromised regulatory mechanisms, to manifest as clinically evident symptomatic disease, which ultimately results in destruction of target tissue and additional comorbidities [4–7]. While the final effector reactions resulting in chronic inflammation have been the focus of research for many, the understanding of the earliest stages of immune dysregulation offers a novel opportunity for disease interception.
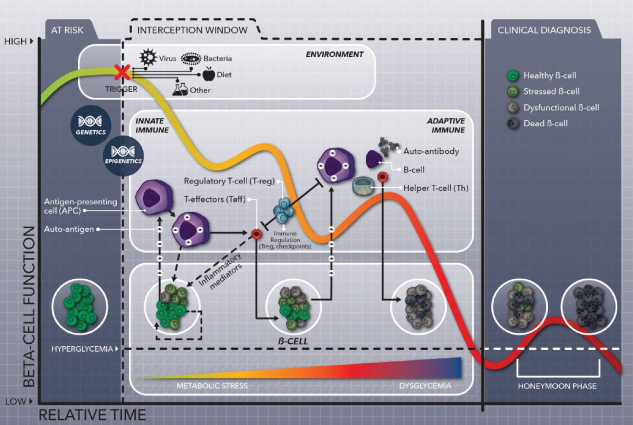
Figure 1.2 Window for disease interception in type 1 diabetes. Progression of at‐risk individuals toward diabetes is initiated by a combination of genetic risk coupled with environmental triggers, which ultimately result in the sensitization of the immune system against islet β‐cell antigens. Persistent and sustained activation of the autoimmune response ultimately results in destruction of islet β‐cells resulting in hyperglycemia. The interception window, as shown above, provides an opportunity wherein environmental triggers and/or immune components could be selectively targeted to arrest any further progression of the pathogenic state. (See insert for color representation of this figure.)
The single highest genetic susceptibility for many organ‐specific autoimmune diseases (including RA, type 1 diabetes [T1D], multiple sclerosis [MS], ankylosing spondylitis, etc.) maps to the major histocompatibility complex (MHC) molecules, also known as human leukocyte antigens (HLAs) [8,9]. The expression of disease‐associated MHC molecules has a marked impact on the repertoire of effector and regulatory T cells selected in the thymus, as well on the nature of potential antigenic self‐peptides that are selected and displayed in the periphery. In seropositive RA, for example, a notable correlation exists between expression of disease‐associated HLA‐DR molecules (0401, 0404, and 0101) and production of antibodies against modified citrullinated self‐proteins (also known as antibodies to citrullinated protein antigens, ACPAs) [10,11]. Individuals expressing this phenotype are at a high risk of developing RA, and exhibit a more severe form of destructive arthritis when compared to seronegative RA. The risk is further exacerbated by environmental factors particularly smoking and perhaps periodontal disease [12]. Taken together, the genotype of the individual along with the phenotypic alterations that result in the production of ACPAs identifies a subset that fits the criteria for disease interception. Importantly, ACPA seropositivity in predisposed adult individuals is detectable many years before the onset of arthritis, hence providing a time frame during which concepts of disease interception can be tested in a clinical setting [13]. However, there exists a need to implement screening approaches at a population level that incorporate various factors to increase the predictability of identifying individuals at‐risk for RA. Machine‐learning methodologies coupled with patient‐centered databases incorporating genetic, clinical, and environmental factors may allow for development of predictive algorithms that can identify cohorts at highest risk [14–16]. On a related note, recent trials in very early RA exploring agents, such as abatacept (cytotoxic T‐lymphocyte‐associated protein 4 [CTLA4]‐Ig fusion protein), that target‐specific components of the immune system have been initiated with the intent of scoping the impact of early intervention on outcome and remission [17,18]. Findings from such human experiments will be important in providing guidance as to whether such agents, perhaps with higher specificity, can be deployed in future disease interception approaches.
Another example is T1D, also known as autoimmune diabetes, which results from immune‐mediated destruction of pancreatic islet beta cells leading to persistent hyperglycemia (Figure 1.2). Analogous to RA, T1D patients exhibit MHC‐linked genetic susceptibility that triggers the production of antibodies directed against islet beta cells, which precedes onset of clinical disease [19–21]. Here again, a direct correlation exists between the specificity of antibodies and the likely risk of the predisposed individual progressing toward diabetes – the highest risk being for subjects that express antibodies directed against two or more key islet beta cell antigens [22]. The temporal window between sero‐conversion and development of disease can span several years. The large unmet medical need in T1D, exacerbated by a lack of significant innovation since the discovery of insulin in 1922, coupled with a defined window of dysregulation in the prediabetic stage provides attractive opportunities for testing the disease interception paradigm (Figure 1.2).
Similar paradigms can be formulated for many other autoimmune diseases wherein genetic susceptibility coupled with environmental factors and/or triggers can identify at‐risk subjects to test the concepts of disease interception. However, several points need to be considered before applications in clinical practice: (i) What are the strategies that need to be employed to find at‐risk subjects for various diseases at a population level? (ii) How robust (and predictive?) are the markers that are used to define the predisease stage in autoimmune diseases wherein the phenotypic alterations are evident in absence of the symptomatic disease? (iii) How deep is the understanding of the mechanistic basis that lead to the early perturbations of the immune component to initiate self‐reactivity? (iv) What are the therapeutic options for the disease interception approaches – conventional pharmacological targeting via small‐ or large‐molecules, lifestyle alterations, vaccinations (infectious triggers) or microbiota‐related influences? (v) What are the safety and regulatory considerations in gaining approval for such approaches? (vi) What are the clinical measures to demonstrate successful intervention (absence of disease may be too long of an outcome to measure for several diseases, hence novel endpoints that strongly correlate with disease and can be measured in a short‐period of time need to be considered).
1.3 Progress in Modulation of the Adaptive Immune Response in Autoimmune Inflammatory Diseases
A number of cell types and factors influence the outcome of inflammatory immune responses in context of autoimmune diseases. However, the fundamental property of self vs. nonself recognition of tissue mostly resides within the cells of the adaptive immune system – T cells in particular. This is a key consideration when thinking about disease interception since the primary decision concerning “self vs. nonself” is likely the first step in initiation and subsequent perpetuation of autoimmune reactions. Ideally, a deep understanding of key autoantigens and the corresponding repertoire of T and B cells would provide a rich substrate to explore antigen‐specific immuno‐modulatory approaches to intercept disease, especially soon after the first signals of aberrant recognition of self are evident. However, much of this information, both from the identity of key driver antigens and the specificity of the adaptive immune repertoire, remain poorly understood. On the other hand, clinical approaches broadly targeting adaptive immune cells have provided encouraging results, which is supportive of continued efforts for future antigen‐specific strategies for disease interception and prevention [23].
Engagement of T cells with antigen presenting cells (APCs) leads to activation of T cell‐dependent immune responses. Two key signals determine T cell activation: signal 1 is the T cell receptor (TCR) recognition of an antigenic peptide presented by the MHC molecules on the APC; in addition a costimulatory signal 2, prototypical of which is the CD28 on T cells interacting with CD80/86 on APCs is required for optimal T cell activation [24–26]. The need for two signals is especially important for naive T cell activation and proliferation, whereas memory T cell responses at times may not be as much dependent upon costimulation [27–29]. T cell activation is counteracted by a series of negative regulatory molecules, also known as checkpoint inhibitors. Blocking of checkpoint inhibitors has demonstrated notable success in cancer immunotherapies in recent clinical trials [30,31]. The balance between activation and inhibitory signals ultimately determines the fate and effector function of T cells. Modulation of T cell activation has demonstrated success in the clinic: the earliest approaches to dampening T cell activation involved the generation of antibodies against CD3, the signaling complex associated with the TCR (reviewed in detail in [32]); blocking of costimulation to inhibit self‐reactive T cells has being applied successfully in autoimmune diseases and prevention of solid organ transplant rejection [33–38]; in contrast, enhancing anti‐tumor T cell responses via blockade of checkpoint inhibitors, such as CTLA‐4 or PD‐1/PD‐L pathways, has ignited the field of immuno‐oncology [39–42].
Since the initial identification of CD28, numerous additional molecules, most belonging to the B7 or TNF/TNF‐R superfamily, have also been shown to provide costimulatory signals to sustain T cell activation [7]. As the earliest triggers in breakdown of tolerance are mapped, it will be critical to understand which receptors and cell‐types play a key role in this reaction. These may provide novel costimulatory therapeutic targets in trials testing disease interception. Selective blockade of key molecules involved in the initial activation of autoreactive T cells may lead to quenching of the autoimmune reaction or significantly delay/dampen the course of disease. The treatment would have to be specific and preferably for a defined period so as to not predispose the at‐risk individual for broad immune‐suppression. The contrast of this approach to antigen‐specific immune therapies is discussed in the later sections.
Progress in exploring costimulation blockade in very early stages of RA is already under way. An example is the Arthritis Prevention in the Preclinical Phase of Rheumatoid Arthritis with Abatacept (APIPPRA) study led by Professor Andrew Cope, which is testing the effects of CTLA4‐Ig, abatacept, on the outcome of disease (http://www.isrctn.com/ISRCTN46017566). CD28 and CTLA‐4 are both expressed by T cells and compete for the same ligands on APCs, namely CD80 and CD86. While CD28 provides a positive signal, CTLA‐4 counteracts this axis via cell‐intrinsic or cell‐extrinsic inhibitory mechanisms (reviewed in detail in [43]). CTLA4‐Ig prevents CD28 engagement of CD80 and CD86, hence inhibiting the positive costimulatory signaling cascade. Several lines of human evidence support a key role for the CD28–CD80/86 axis in autoimmunity: (i) CTLA4‐Ig, abatacept, is an approved fusion protein drug for established RA and juvenile idiopathic arthritis [33,36]; (ii) human genetic mutations in CTLA‐4 or components involved in cell‐surface expression of CTLA‐4 result in onset of diverse autoimmune phenotypes [44–46]; and (iii) application of anti‐CTLA‐4 monoclonal antibody (mAb) for cancer immunotherapy results in the induction of autoimmune manifestations, notably skin and gastrointestinal tract pathologies, in many patients [47]. The last two points highlight the key role for CTLA‐4 in maintaining peripheral tolerance. Importantly, recent data demonstrates that regulatory T cells (Treg) express high levels of CTLA‐4, which is critical for their ability to control self‐reactive T cells [48,49]. CTLA‐4‐Ig treatment, while dampening effector T cells also compromises Treg function – an outcome that is not desirable for autoimmune diseases. In support of this conclusion, Poirier et al. demonstrated that selective blockade of CD28 was superior to CTLA4‐Ig treatment, especially since the former preserved CTLA‐4‐dependent Treg activity [50]. If translatable to the clinic, this mechanism should provide differentiated and superior therapeutic benefit to the patient. Previous clinical experience with a bivalent anti‐CD28 agonist mAb resulted in severe adverse events due to systemic T cell activation [51]. However, more recently, monovalent anti‐CD28 antagonists, which are devoid of any agonist activity, are being tested in the clinic to scope utility in autoimmune inflammatory diseases [52,53]. From the disease interception perspective, such molecules could be employed in early stages of T cell dysfunction to cease activation of autoreactive T cells while propagating an active Treg‐dependent circuit that holds autoimmunity at bay.
Besides blocking costimulatory pathways, another option for modulation of autoreactive T cells involves agonizing inhibitory pathways that limit T cell activation. As noted before, the importance of inhibitory pathways, examples include checkpoint inhibitors like CTLA‐4 or PD‐1/PD‐L molecules, is best exemplified by the recent success in cancer immunotherapy wherein blockade of these molecules has resulted in robust antitumor immune responses in a subset of patients [30]. However, the important role of these negative regulators in maintaining peripheral tolerance is highlighted by the notable incidence of autoimmune diseases in cancer patients treated with checkpoint blockade inhibitors [47,54]. Reverse application of this approach, i.e. activating inhibitory pathways instead of blocking, may provide a fruitful avenue for early intervention or disease interception in autoimmune diseases [55–57]. This strategy harnesses the evolutionary selection of inhibitory pathways that control self‐reactivity and perhaps could be promising from a safety consideration if no signals for increased malignancies are evident. The choice of which negative pathways to agonize is dependent upon deep phenotypic analyses of the earliest T cell specificities that are triggered. Recent technologies like mass cytometry (CyTOF) coupled with single‐cell approaches may provide critical tools to dissect the temporal and spatial regulation of costimulatory and checkpoint regulators during the earliest stages of immune dysregulation [58,59]. Such datasets should provide a rational basis for choice of potential targets for disease interception approaches. To this end, a recent report by Rao et al. utilized CyTOF‐based phenotypic characterization of synovial T cells in RA to identify a pathogenic subset of autoreactive T cells that expressed high levels of PD‐1 [59].
Other approaches for dampening adaptive immunity have involved cell‐depletion via targeted antibodies or modulation of lymphocyte trafficking to sites of inflammation. For example, depletion of B cells has been achieved via molecules targeting B cell lineage‐specific markers; this approach has demonstrated efficacy in RA and has been evaluated in numerous additional autoimmune diseases [60,61]. Positive data in psoriasis, and more recently in T1D, was evident with molecules like alefacept (lymphocyte function‐associated antigen 3, LFA3‐Ig) that inhibits T cell activation (via blocking CD2–LFA3 interaction) and induced T cell depletion [62–64]. More recently, agents that target lymphocyte migration and trafficking have also demonstrated clinical success: vedolizumab (approved for Crohn's disease and ulcerative colitis) and natalizumab (approved for MS and Crohn's disease) are examples of two mAbs that block α‐4 integrin‐dependent adhesive interactions to prevent lymphocyte extravasation to inflamed tissues [65–68]. On the other hand, sphingosine‐1 phosphate receptor‐agonists (such as fingolimod approved for MS) inhibit egress of primed lymphocytes from secondary lymphoid tissues to sites of inflammation [69,70]. While such mechanisms have demonstrated benefit in established chronic disease, the application of these in disease interception may be challenging if the need for peripheral localization at effector tissue sites may not be as pronounced during the earliest stages of dysregulation. Furthermore, lineage‐specific depletion strategies or broad inhibition of immune surveillance or trafficking mechanisms may present challenges related to broad immune‐suppression and potential safety concerns (for example the risk of progressive multifocal leukoencephalopathy [PML] with natalizumab [71]).
1.4 The Complex Interplay between the Specificity of the Pathogenic Immune Repertoire and Its Sculpting by the Environment – Implications for Disease Interception
A challenge, and perhaps a significant opportunity, revolves around the diversity and specificity of the immune repertoire that reacts with self. Besides autoimmune diseases, the concept can be extended to any antigen‐specific immune response including infectious immunity, allergies, tumor immunity, and perhaps even in some cases of neurodegenerative and metabolic diseases. Developing tools to identify and monitor the pathogenic repertoire provide novel opportunities for the earliest detection and interception (Figure 1.3). Recent technological advances, notably high‐throughput next‐generation sequencing, single‐cell technological platforms and advances in computational biology, have ushered a rapidly progressing field that holds much promise [72–75]. These developments have sparked efforts to parse the diversity of the TCR and B cell receptor (BCR) to identify those that may be involved in various stages of pathogenesis. Understanding of clonal populations of lymphocytes that dominate a particular stage of health or disease will be crucial for any future approaches aimed at antigen‐specific immunotherapies. The complexity around analyzing the immune repertoire stems from the vast diversity that is present within an individual. The theoretical maximal combinatorial diversity of the adaptive immune repertoire based on the different variable gene segments that encode the TCR or BCR along with junctional diversity has been proposed to be >1015 [76]. However, functional and quantitative measurements in humans put this number closer to 10–25 × 106 [77,78]. The primary evolutionary pressure to maintain a diverse immune repertoire is to provide protective immunity from different microbial pathogens. While this may have been the selection pressure from a species survival viewpoint, continuing evolving environmental conditions in an industrialized world coupled with dietary and lifestyle modifications may exert new pressures on the adaptive immune system.

Figure 1.3 Utilizing the pathogenic immune repertoire for identifying earliest stages of dysregulation to enable disease interception approaches. The specificity of the immune repertoire could also be used to develop antigen‐specific therapeutic approaches as well as novel biomarkers indicating the initial stages of breakdown of tolerance.
Although both T and B cells play an important role in antigen‐specific responses, we will focus mostly on the T cell diversity in context of autoimmune diseases – largely based on dominant MHC associations and the fact that many aspects of B cell activation are dependent upon signals from T cells. T cell development initiates in the thymus where precursors from the bone‐marrow mature into single‐positive CD4+ or CD8+ T cell lineages. Thymic selection ensures that the selected T cell repertoire is self‐restricted and self‐tolerant. The initial sculpting of the repertoire is influenced by peptides derived from various peripheral self‐proteins that are expressed ectopically in the thymus under the influence of the AutoImmune REgulator (AIRE) gene [79]. Human mutations in the AIRE gene lead to autoimmune polyendocrinopathy syndrome type 1 (APS‐1), also known as autoimmune polyendocrinopathy‐candidiasis‐ectodermal dystrophy (APECED), which involves a range of autoimmune diseases [80,81]. The nature of peptides selected in thymus is dependent upon the biochemical features of the MHC molecules expressed within an individual. Hence, MHC specificity along with the available protein cargo in the thymus determines the final outcome of the epitopes displayed for T cell repertoire selection – both effector T cells and Treg. Once selected, the T cells migrate into circulation and reside mostly within primary lymphoid organs where productive encounters with specific antigen result in clonal expansion and acquisition of effector function [82].
Biochemical and structural studies of disease‐associated MHC molecules have identified unique features in the peptide‐binding groove that offers insights into selection of auto‐antigenic peptides for display. For example, HLA‐DR molecules associated with RA (such as 0401, 0101, 0404) express a unique constellation of amino acids, known as the “shared epitope,” involving positions 11, 71, and 74 of the beta chain [83,84]. The shared epitope residues are integral in defining the structural and biochemical features of the P4 pocket of the peptide‐binding groove of HLA‐DR molecules [85]. As mentioned before, a hallmark of seropositive RA is generation of ACPAs against citrullinated self‐proteins. The modification of arginine to citrulline in proteins is carried out by peptidyl arginine deiminases (PADs) enzymes – of which there are several isoforms; however, PAD‐4 which is expressed mostly in immune cells is thought to be the primary one involved in RA [85]. Moreover, CD4+ T cells specific for citrullinated peptides are detectable and notably expanded in RA subjects [12]. Recent studies have demonstrated that the modification of arginine to citrulline enables peptide binding to DR alleles expressing the shared epitope. Native peptides with arginine exhibit poor to no binding to RA‐associated DR molecules, while modification to citrulline allows for favorable interactions with the P4 pocket hence resulting in measurable binding with relatively high affinity [86,87]. This posttranslational modification may explain selection of unique peptides that are displayed to autoreactive T cells. Upon activation, such T cells may constitute the earliest dysregulated cells that ultimately lead to joint destruction and RA; in addition, this T cell repertoire may also likely provide help to trigger B cells to produce ACPAs and aid with their differentiation to plasma cells. From a disease interception perspective, selective modulation of PAD activity may be an opportunity to lessen the antigenic burden that triggers the initial autoreactive T cells and B cells. Unique structural interactions of citrulline with the shared epitope expressing DR molecules may provide additional opportunities for modulation of TCR recognition of self‐peptides [86]. Interestingly, environmental risk factors for RA such as smoking or periodontal disease have been shown to activate PAD enzymes, which may link the genetic risk to disease triggers [12]. A recent study in RA patients with periodontitis specifically identified a unique bacterial species in the gums that was able to induce citrullination [88]. The elegant mechanism in this report involved a bacterial pore‐forming protein that increased intracellular Ca2+ in host immune cells thereby activating PAD enzymes. Strikingly, many of the citrullinated proteins detected in joints of RA patients were also found in the gum tissues of RA patients harboring the microbe [88]. Such associations may support a vaccination approach as a future consideration in individuals at risk for developing RA.
T1D is another notable example wherein the unique polymorphism of the disease‐susceptible HLA‐DQ molecules translates into structural and biological outcomes [89]. T1D‐related HLA‐DQ molecules (such as DQ8 or DQ2) contain a conserved mutation of aspartic acid to alanine at position 57 of the beta chain [19,20]. This change alters the charge and shape of the P9 pocket of the peptide‐binding groove of the DQ molecules, which ultimately select for peptides with a unique motif dominated by acidic amino acids toward the c‐terminus [90,91]. The dominant T cell epitope from insulin has an acidic amino acid (glutamic acid) at the P9 position, which favors binding to the DQ molecule [89]. And similar to microbial associations as triggers in RA, recent data involving at‐risk cohorts of children identified a link between enterovirus infections and onset of T1D [92]. This finding adds to previous observations that have made similar connections [93–95]. The precise molecular mechanisms underlying infections as a putative trigger still remain unclear, although several possibilities ranging from enhanced inflammatory responses via localized infection of the islet beta cells to molecular mimicry between viral proteins and host proteins have been suggested. A clear case for molecular mimicry in initiation of an autoimmune response was evident from the rare induction of narcolepsy in a subset of individuals that received the flu vaccine [96]. In this instance, vaccination of individuals expressing HLA‐DQ6 triggered the production of antibodies against flu, which cross reacted with the hypocretin receptor, ultimately leading to narcolepsy. It was subsequently determined that the flu vaccine induced antibodies against the viral nucleoprotein segment that bore high homology to an epitope from the host hypocretin receptor [96]. Future assessments, via in silico and functional measurements, may identify similar situations wherein individuals expressing certain HLA alleles may be at risk for induction of immune responses against self‐tissues.
What are the strategies that could be applied for identification of the T cell repertoire involved in autoimmune diseases? Several points need to be considered: (i) the frequency of antigen‐specific T cells in circulation is rare, hence sampling the tissue‐resident repertoire may yield more useful information; (ii) the identity and hierarchy of antigens that initiate and sustain autoimmune responses is still not well understood; (iii) the importance of correlating antigen‐specificity to effector phenotype – to better understand whether a particular fraction of the repertoire tends to be more self‐reactive over other specificities that may be represented among regulatory T cell‐like phenotypes; and (iv) the application of tools and technologies that can be deployed for immune monitoring of at‐risk and patient populations to correlate dynamic changes in repertoire specificities and/or phenotype at different stages of progression from dysregulation to onset of disease.
Restricted clonality in inflamed tissue‐resident T cells has been demonstrated in several diseases. In RA, for example, a study by de Vries and coworkers pointed to the presence of a few clonotypes in the joint tissue that constituted a significant fraction of T cells in the inflamed joint [97]. Other studies have identified specific TCR clonotypes in the joint and have successfully tracked the same in blood [98,99]. A recent report by Rao et al. used CyTOF to identify a subset of CD4+ T cells that expressed high levels of PD‐1 and were unique to the synovium of seropositive RA [59]. The same study demonstrated that these cells could provide robust help to B cells to support their activation and differentiation into plasma cells. Taken together, these studies provide early evidence of restricted clonality and unique phenotypic markers that should be evaluated at a broader population level to strengthen the association with disease predisposition, induction, and progression. Another important dimensionality of the TCR repertoire is to understand shared patterns and structural features that may predict antigenicity, and whether such information can be deployed prospectively to track the evolution and expansion of a specific immune response. Recent publications from the Davis and Thomas labs have provided evidence that shared sequence patterns and structural motifs can be identified among antigen‐specific T cells across individuals, and in some instances TCR sequences correlated with expression of specific HLA molecules [100,101].
In addition, approaches to deciphering antigenic specificity of expanded clonotypes need to be seriously considered for tolerogenic applications to alter the course of disease. In order to determine epitope specificity, both the alpha and beta subunits of the TCR need to be identified – most reports analyzing clonality of the T cell repertoire focus on the TCR beta chain since that information is sufficient to establish clonal hierarchies [74]. Recent progress in single cell technologies has successfully identified TCR alpha and beta subunits, hence enabling downstream interrogation of antigenic targets [102,103]. Unbiased screening of T cells from diseased tissue may identify clones that react with known epitopes as well as additional specificities that need to be deciphered, as noted by Kent and coworkers in a recent report analyzing the islet‐resident T cell repertoire in T1D [104]. Deorphanizing TCR specificity has been explored via screening of combinatorial peptide libraries displayed by HLA molecules using various expression systems [105–107]. Autoimmune diseases with dominant HLA associations at least provide a bias insofar as the HLA substrate that one may employ to undertake a combinatorial peptide screen. In the absence of this, all HLA molecules expressed by an individual would need to be considered when screening, which makes the task significantly more challenging. To this end, a report by Prinz and coworkers successfully identified the antigen of a CD8+ TCR from a psoriasis patient restricted to HLA‐Cw06 molecule [108]; similarly, Birnbaum et al. established a combinatorial library yeast display system to evaluate the diversity of ligands that could be recognized by a human HLA‐DR2‐restricted TCR specific for an epitope from myelin basic protein (MBP) [105]. Surprisingly, Birnbaum et al. identified several ligands with homology to the MBP but were derived from different microbial sources. This raised the intriguing possibility whether any of these microbes could be the initial trigger for T cells that ultimately cross react with MBP to initiate MS [105]. Parallel developments in oncology have focused on parsing the diversity and specificity of the antitumor T cell response, particularly in the context of checkpoint blockade therapy [109–112]. Learnings from this field will be extremely useful for applications in autoimmune diseases.
The impact of the environment, particularly the host microbiome, in influencing the activation and effector function of the antigen‐specific T cell repertoire is crucial. As mentioned previously, microbial triggers or mechanisms of molecular mimicry may play a significant role in the initiation of the pathogenic autoimmune response [88,96,113]. In cancer immunotherapy, recent observations have made the correlation between microbiome composition and response to checkpoint blockade therapy [114,115]. How precisely does the microbiome influence the antitumor T cell response? Perhaps akin to autoimmunity, there may be molecular mimicry between certain microbial determinants and tumor antigens, which in this case would be beneficial to the host. Alternatively, specific microbial products or components may provide stimulatory signals to immune cells or alter the microenvironment that ultimately allows for optimal priming and activation of a potent antitumor T cell response. On the flip‐side, certain microbial signals may enhance regulatory T cells, which could be beneficial for autoimmune diseases – one such example is the findings with clostridia strains modulating gut mucosal immune responses via Treg activation [116,117]. From a disease interception viewpoint, understanding of the host microbiome from genesis and tracking changes as the individual develops will be critical to pinpointing perhaps the earliest inflection points of dysregulation. Related to this may be the opportunity to track the relevant immune repertoire of lymphocytes that are sensitized by specific microbial populations and translate these to opportunities in clinical intervention.
1.5 Clinical Application and Concluding Remarks
The ultimate value for disease interception is the successful application at a population level as a part of the standard health‐care practice. In the near future, integration of multitude datasets and measurements should enable personalized risk profiles for dominant autoimmune diseases. To this end, several relevant datasets could be considered: (i) genome sequencing coupled with computational approaches to understand transcriptional regulation; (ii) tracking immune repertoire diversity and specificity from genesis to pathogenesis; and (iii) host microbiome signatures and environmental perturbations – including infections, lifestyle modifications, etc., and their impact on above parameters.
Much progress has been made in the identification of tools and technologies that allow for deep interrogation of cellular immune components. For example, the advent of CyTOF has enabled simultaneous measurements of many markers to enable deep phenotyping and identification of novel populations of cells relevant to disease [118,119]. Similarly, bar‐coding strategies have exponentially expanded the application of peptide‐MHC multimers to detect antigen‐specific T cells in immune responses [120]. Development of sensitive array technologies can now be employed for measuring proteins, lipid, and other metabolites from body fluids. While most of these technologies have been developed and utilized as research tools, the future evolution will likely involve integration, commoditization, and miniaturization to enable application at a population level to drive the next generation of diagnostics and biomarkers (indicative of health, early dysregulation and disease). In addition, new information obtained from deconvolution of antigen specificities in autoimmune diseases will open up novel therapeutic avenues for disease interception.
In conclusion, the concept of disease interception can be realistically applied in practice based on the knowledge of the at‐risk individual coupled with reliable markers identifying the earliest stages of dysregulation preceding onset of disease. Several therapeutic options can be pursued; however, antigen‐specific approaches may provide an opportunity to reset or reestablish tolerance. And to successfully accomplish this, an understanding of the immune repertoire is imperative since that may identify transition states from genesis to pathogenesis for many autoimmune diseases.
Acknowledgments
I would like to thank Dr. Ben Wiegand, Dr. Murray McKinnon, and Dr. Dan Baker for input and critical reading of this manuscript, and for providing constructive feedback for clarity. Much gratitude to many talented colleagues, past and present, that have worked tirelessly on the themes outlined in this review.
References
- 1 Hirahara, K., Schwartz, D., Gadina, M. et al. (2016). Targeting cytokine signaling in autoimmunity: back to the future and beyond. Curr. Opin. Immunol. 43: 89–97.
- 2 Schwartz, D.M., Bonelli, M., Gadina, M., and O'Shea, J.J. (2016). Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat. Rev. Rheumatol. 12: 25–36.
- 3 Hait, W.N. and Lebowitz, P.F. (2016). Disease interception: myths, mountains, and mole hills. Cancer Prev. Res. 9: 635–637.
- 4 Abramson, J. and Husebye, E.S. (2016). Autoimmune regulator and self‐tolerance – molecular and clinical aspects. Immunol. Rev. 271: 127–140.
- 5 Paun, A. and Danska, J.S. (2015). Immuno‐ecology: how the microbiome regulates tolerance and autoimmunity. Curr. Opin. Immunol. 37: 34–39.
- 6 Perry, J.S. and Hsieh, C.S. (2016). Development of T‐cell tolerance utilizes both cell‐autonomous and cooperative presentation of self‐antigen. Immunol. Rev. 271: 141–155.
- 7 Zhang, Q. and Vignali, D.A. (2016). Co‐stimulatory and co‐inhibitory pathways in autoimmunity. Immunity 44: 1034–1051.
- 8 Cho, J.H. and Feldman, M. (2015). Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat. Med. 21: 730–738.
- 9 Trowsdale, J. and Knight, J.C. (2013). Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet. 14: 301–323.
- 10 Huizinga, T.W., Amos, C.I., van der Helm‐van Mil, A.H. et al. (2005). Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA‐DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 52: 3433–3438.
- 11 Klareskog, L., Stolt, P., Lundberg, K. et al. (2006). A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA‐DR (shared epitope)‐restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 54: 38–46.
- 12 Malmstrom, V., Catrina, A.I., and Klareskog, L. (2017). The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat. Rev. Immunol. 17: 60–75.
- 13 Sokolove, J., Bromberg, R., Deane, K.D. et al. (2012). Autoantibody epitope spreading in the pre‐clinical phase predicts progression to rheumatoid arthritis. PLoS ONE 7: e35296.
- 14 Kruppa, J., Ziegler, A., and Konig, I.R. (2012). Risk estimation and risk prediction using machine‐learning methods. Hum. Genet. 131: 1639–1654.
- 15 Lin, C., Karlson, E.W., Canhao, H. et al. (2013). Automatic prediction of rheumatoid arthritis disease activity from the electronic medical records. PLoS One 8: e69932.
- 16 van de Stadt, L.A., Witte, B.I., Bos, W.H., and van Schaardenburg, D. (2013). A prediction rule for the development of arthritis in seropositive arthralgia patients. Ann. Rheum. Dis. 72: 1920–1926.
- 17 Emery, P., Burmester, G.R., Bykerk, V.P. et al. (2015). Evaluating drug‐free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active‐controlled AVERT study of 24 months, with a 12‐month, double‐blind treatment period. Ann. Rheum. Dis. 74: 19–26.
- 18 Peterfy, C., Burmester, G.R., Bykerk, V.P. et al. (2016). Sustained improvements in MRI outcomes with abatacept following the withdrawal of all treatments in patients with early, progressive rheumatoid arthritis. Ann. Rheum. Dis. 75: 1501–1505.
- 19 Morel, P.A., Dorman, J.S., Todd, J.A. et al. (1988). Aspartic acid at position 57 of the HLA‐DQ beta chain protects against type I diabetes: a family study. Proc. Natl. Acad. Sci. U.S.A. 85: 8111–8115.
- 20 Todd, J.A., Bell, J.I., and McDevitt, H.O. (1987). HLA‐DQ beta gene contributes to susceptibility and resistance to insulin‐dependent diabetes mellitus. Nature 329: 599–604.
- 21 Todd, J.A., Bell, J.I., and McDevitt, H.O. (1988). HLA antigens and insulin‐dependent diabetes. Nature 333: 710.
- 22 Ziegler, A.G., Rewers, M., Simell, O. et al. (2013). Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309: 2473–2479.
- 23 Townsend, M.J., Monroe, J.G., and Chan, A.C. (2010). B‐cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol. Rev. 237: 264–283.
- 24 Esensten, J.H., Helou, Y.A., Chopra, G. et al. (2016). CD28 costimulation: from mechanism to therapy. Immunity 44: 973–988.
- 25 Jenkins, M.K., Ashwell, J.D., and Schwartz, R.H. (1988). Allogeneic non‐T spleen cells restore the responsiveness of normal T cell clones stimulated with antigen and chemically modified antigen‐presenting cells. J. Immunol. 140: 3324–3330.
- 26 Jenkins, M.K., Taylor, P.S., Norton, S.D., and Urdahl, K.B. (1991). CD28 delivers a costimulatory signal involved in antigen‐specific IL‐2 production by human T cells. J. Immunol. 147: 2461–2466.
- 27 Croft, M., Bradley, L.M., and Swain, S.L. (1994). Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen‐presenting cell types including resting B cells. J. Immunol. 152: 2675–2685.
- 28 London, C.A., Lodge, M.P., and Abbas, A.K. (2000). Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 164: 265–272.
- 29 Metz, D.P., Farber, D.L., Taylor, T., and Bottomly, K. (1998). Differential role of CTLA‐4 in regulation of resting memory versus naive CD4 T cell activation. J. Immunol. 161: 5855–5861.
- 30 Sharma, P. and Allison, J.P. (2015). Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161: 205–214.
- 31 Wolchok, J.D., Hodi, F.S., Weber, J.S. et al. (2013). Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N.Y. Acad. Sci. 1291: 1–13.
- 32 Kuhn, C. and Weiner, H.L. (2016). Therapeutic anti‐CD3 monoclonal antibodies: from bench to bedside. Immunotherapy 8: 889–906.
- 33 Genovese, M.C., Becker, J.C., Schiff, M. et al. (2005). Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med. 353: 1114–1123.
- 34 Lovell, D.J., Ruperto, N., Mouy, R. et al. (2015). Long‐term safety, efficacy, and quality of life in patients with juvenile idiopathic arthritis treated with intravenous abatacept for up to seven years. Arthritis Rheumatol. 67: 2759–2770.
- 35 Ruperto, N., Lovell, D.J., Quartier, P. et al. (2010). Long‐term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum. 62: 1792–1802.
- 36 Ruperto, N., Lovell, D.J., Quartier, P. et al. (2008). Abatacept in children with juvenile idiopathic arthritis: a randomised, double‐blind, placebo‐controlled withdrawal trial. Lancet 372: 383–391.
- 37 Vincenti, F., Blancho, G., Durrbach, A. et al. (2010). Five‐year safety and efficacy of belatacept in renal transplantation. J. Am. Soc. Nephrol. 21: 1587–1596.
- 38 Vincenti, F., Larsen, C., Durrbach, A. et al. (2005). Costimulation blockade with belatacept in renal transplantation. N. Engl. J. Med. 353: 770–781.
- 39 Brahmer, J.R., Tykodi, S.S., Chow, L.Q. et al. (2012). Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366: 2455–2465.
- 40 Garon, E.B., Rizvi, N.A., Hui, R. et al. (2015). Pembrolizumab for the treatment of non‐small‐cell lung cancer. N. Engl. J. Med. 372: 2018–2028.
- 41 Hodi, F.S., O'Day, S.J., McDermott, D.F. et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363: 711–723.
- 42 Topalian, S.L., Hodi, F.S., Brahmer, J.R. et al. (2012). Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N. Engl. J. Med. 366: 2443–2454.
- 43 Walker, L.S. and Sansom, D.M. (2011). The emerging role of CTLA4 as a cell‐extrinsic regulator of T cell responses. Nat. Rev. Immunol. 11: 852–863.
- 44 Kuehn, H.S., Ouyang, W., Lo, B. et al. (2014). Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345: 1623–1627.
- 45 Lo, B., Zhang, K., Lu, W. et al. (2015). AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349: 436–440.
- 46 Schubert, D., Bode, C., Kenefeck, R. et al. (2014). Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20: 1410–1416.
- 47 Bertrand, A., Kostine, M., Barnetche, T. et al. (2015). Immune related adverse events associated with anti‐CTLA‐4 antibodies: systematic review and meta‐analysis. BMC Med. 13: 211.
- 48 Friedline, R.H., Brown, D.S., Nguyen, H. et al. (2009). CD4+ regulatory T cells require CTLA‐4 for the maintenance of systemic tolerance. J. Exp. Med. 206: 421–434.
- 49 Ise, W., Kohyama, M., Nutsch, K.M. et al. (2010). CTLA‐4 suppresses the pathogenicity of self antigen‐specific T cells by cell‐intrinsic and cell‐extrinsic mechanisms. Nat. Immunol. 11: 129–135.
- 50 Poirier, N., Azimzadeh, A.M., Zhang, T. et al. (2010). Inducing CTLA‐4‐dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci. Transl. Med. 2: 17ra10.
- 51 Suntharalingam, G., Perry, M.R., Ward, S. et al. (2006). Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355: 1018–1028.
- 52 Poirier, N., Blancho, G., Hiance, M. et al. (2016). First‐in‐human study in healthy subjects with FR104, a pegylated monoclonal antibody fragment antagonist of CD28. J. Immunol. 197: 4593–4602.
- 53 Suchard, S.J., Davis, P.M., Kansal, S. et al. (2013). A monovalent anti‐human CD28 domain antibody antagonist: preclinical efficacy and safety. J. Immunol. 191: 4599–4610.
- 54 Cappelli, L.C., Shah, A.A., and Bingham, C.O. 3rd (2017). Immune‐related adverse effects of cancer immunotherapy‐implications for rheumatology. Rheum. Dis. Clin. North Am. 43: 65–78.
- 55 Kim, J.H., Choi, Y.J., Lee, B.H. et al. (2016). Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL‐17A production from programmed cell death 1‐high T cells. J. Allergy Clin. Immunol. 137: 1466–1476.e3.
- 56 Song, M.Y., Hong, C.P., Park, S.J. et al. (2015). Protective effects of Fc‐fused PD‐L1 on two different animal models of colitis. Gut 64: 260–271.
- 57 Wang, G., Hu, P., Yang, J. et al. (2011). The effects of PDL‐Ig on collagen‐induced arthritis. Rheumatol. Int. 31: 513–519.
- 58 Ermann, J., Rao, D.A., Teslovich, N.C. et al. (2015). Immune cell profiling to guide therapeutic decisions in rheumatic diseases. Nat. Rev. Rheumatol. 11: 541–551.
- 59 Rao, D.A., Gurish, M.F., Marshall, J.L. et al. (2017). Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542: 110–114.
- 60 Dorner, T., Isenberg, D., Jayne, D. et al. (2009). Current status on B‐cell depletion therapy in autoimmune diseases other than rheumatoid arthritis. Autoimmun. Rev. 9: 82–89.
- 61 Edwards, J.C., Szczepanski, L., Szechinski, J. et al. (2004). Efficacy of B‐cell‐targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 350: 2572–2581.
- 62 Ellis, C.N. and Krueger, G.G. (2001). Treatment of chronic plaque psoriasis by selective targeting of memory effector T lymphocytes. N. Engl. J. Med. 345: 248–255.
- 63 Rigby, M.R., DiMeglio, L.A., Rendell, M.S. et al. (2013). Targeting of memory T cells with alefacept in new‐onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Diabetes Endocrinol. 1: 284–294.
- 64 Rigby, M.R., Harris, K.M., Pinckney, A. et al. (2015). Alefacept provides sustained clinical and immunological effects in new‐onset type 1 diabetes patients. J. Clin. Invest. 125: 3285–3296.
- 65 Feagan, B.G., Rutgeerts, P., Sands, B.E. et al. (2013). Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369: 699–710.
- 66 Ghosh, S., Goldin, E., Gordon, F.H. et al. (2003). Natalizumab for active Crohn's disease. N. Engl. J. Med. 348: 24–32.
- 67 Miller, D.H., Soon, D., Fernando, K.T. et al. (2007). MRI outcomes in a placebo‐controlled trial of natalizumab in relapsing MS. Neurology 68: 1390–1401.
- 68 Sandborn, W.J., Feagan, B.G., Rutgeerts, P. et al. (2013). Vedolizumab as induction and maintenance therapy for Crohn's disease. N. Engl. J. Med. 369: 711–721.
- 69 Cohen, J.A., Barkhof, F., Comi, G. et al. (2010). Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 362: 402–415.
- 70 Kappos, L., Radue, E.W., O'Connor, P. et al. (2010). A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 362: 387–401.
- 71 McGuigan, C., Craner, M., Guadagno, J. et al. (2016). Stratification and monitoring of natalizumab‐associated progressive multifocal leukoencephalopathy risk: recommendations from an expert group. J. Neurol. Neurosurg. Psychiatry 87: 117–125.
- 72 Georgiou, G., Ippolito, G.C., Beausang, J. et al. (2014). The promise and challenge of high‐throughput sequencing of the antibody repertoire. Nat. Biotechnol. 32: 158–168.
- 73 Newell, E.W. and Davis, M.M. (2014). Beyond model antigens: high‐dimensional methods for the analysis of antigen‐specific T cells. Nat. Biotechnol. 32: 149–157.
- 74 Robins, H. (2013). Immunosequencing: applications of immune repertoire deep sequencing. Curr. Opin. Immunol. 25: 646–652.
- 75 Robinson, W.H. (2015). Sequencing the functional antibody repertoire – diagnostic and therapeutic discovery. Nat. Rev. Rheumatol. 11: 171–182.
- 76 Nikolich‐Zugich, J., Slifka, M.K., and Messaoudi, I. (2004). The many important facets of T‐cell repertoire diversity. Nat. Rev. Immunol. 4: 123–132.
- 77 Arstila, T.P., Casrouge, A., Baron, V. et al. (1999). A direct estimate of the human alphabeta T cell receptor diversity. Science 286: 958–961.
- 78 Arstila, T.P., Casrouge, A., Baron, V. et al. (2000). Diversity of human alpha beta T cell receptors. Science 288: 1135.
- 79 Anderson, M.S. and Su, M.A. (2016). AIRE expands: new roles in immune tolerance and beyond. Nat. Rev. Immunol. 16: 247–258.
- 80 Finnish‐German APECED Consortium (1997). An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD‐type zinc‐finger domains. Nat. Genet. 17: 399–403.
- 81 Nagamine, K., Peterson, P., Scott, H.S. et al. (1997). Positional cloning of the APECED gene. Nat. Genet. 17: 393–398.
- 82 Von Boehmer, H. (2014). Deciphering thymic development. Front. Immunol. 5: 424.
- 83 Gregersen, P.K., Silver, J., and Winchester, R.J. (1987). The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 30: 1205–1213.
- 84 Raychaudhuri, S., Sandor, C., Stahl, E.A. et al. (2012). Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44: 291–296.
- 85 Darrah, E., Giles, J.T., Ols, M.L. et al. (2013). Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci. Transl. Med. 5: 186ra65.
- 86 Scally, S.W., Petersen, J., Law, S.C. et al. (2013). A molecular basis for the association of the HLA‐DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 210: 2569–2582.
- 87 Hill, J.A., Southwood, S., Sette, A. et al. (2003). Cutting edge: the conversion of arginine to citrulline allows for a high‐affinity peptide interaction with the rheumatoid arthritis‐associated HLA‐DRB1*0401 MHC class II molecule. J. Immunol. 171: 538–541.
- 88 Konig, M.F., Abusleme, L., Reinholdt, J. et al. (2016). Aggregatibacter actinomycetemcomitans‐induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 8: 369ra176.
- 89 Lee, K.H., Wucherpfennig, K.W., and Wiley, D.C. (2001). Structure of a human insulin peptide‐HLA‐DQ8 complex and susceptibility to type 1 diabetes. Nat. Immunol. 2: 501–507.
- 90 Suri, A., Walters, J.J., Gross, M.L., and Unanue, E.R. (2005). Natural peptides selected by diabetogenic DQ8 and murine I‐A(g7) molecules show common sequence specificity. J. Clin. Invest. 115: 2268–2276.
- 91 Suri, A., Walters, J.J., Rohrs, H.W. et al. (2008). First signature of islet beta‐cell‐derived naturally processed peptides selected by diabetogenic class II MHC molecules. J. Immunol. 180: 3849–3856.
- 92 Honkanen, H., Oikarinen, S., Nurminen, N. et al. (2017). Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus‐induced autoimmunity. Diabetologia 60: 424–431.
- 93 Christen, U., Bender, C., and von Herrath, M.G. (2012). Infection as a cause of type 1 diabetes? Curr. Opin. Rheumatol. 24: 417–423.
- 94 Knip, M. and Simell, O. (2012). Environmental triggers of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2: a007690.
- 95 Kondrashova, A. and Hyoty, H. (2014). Role of viruses and other microbes in the pathogenesis of type 1 diabetes. Int. Rev. Immunol. 33: 284–295.
- 96 Ahmed, S.S., Volkmuth, W., Duca, J. et al. (2015). Antibodies to influenza nucleoprotein cross‐react with human hypocretin receptor 2. Sci. Transl. Med. 7: 294ra105.
- 97 Klarenbeek, P.L., de Hair, M.J., Doorenspleet, M.E. et al. (2012). Inflamed target tissue provides a specific niche for highly expanded T‐cell clones in early human autoimmune disease. Ann. Rheum. Dis. 71: 1088–1093.
- 98 Rossetti, M., Spreafico, R., Consolaro, A. et al. (2017). TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann. Rheum. Dis. 76: 435–441.
- 99 Spreafico, R., Rossetti, M., van Loosdregt, J. et al. (2016). A circulating reservoir of pathogenic‐like CD4+ T cells shares a genetic and phenotypic signature with the inflamed synovial micro‐environment. Ann. Rheum. Dis. 75: 459–465.
- 100 Dash, P., Fiore‐Gartland, A.J., Hertz, T. et al. (2017). Quantifiable predictive features define epitope‐specific T cell receptor repertoires. Nature 547: 89–93.
- 101 Glanville, J., Huang, H., Nau, A. et al. (2017). Identifying specificity groups in the T cell receptor repertoire. Nature 547: 94–98.
- 102 Han, A., Glanville, J., Hansmann, L., and Davis, M.M. (2014). Linking T‐cell receptor sequence to functional phenotype at the single‐cell level. Nat. Biotechnol. 32: 684–692.
- 103 Han, A., Newell, E.W., Glanville, J. et al. (2013). Dietary gluten triggers concomitant activation of CD4+ and CD8+ alphabeta T cells and gammadelta T cells in celiac disease. Proc. Natl. Acad. Sci. U.S.A. 110: 13073–13078.
- 104 Babon, J.A., DeNicola, M.E., Blodgett, D.M. et al. (2016). Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 22: 1482–1487.
- 105 Birnbaum, M.E., Mendoza, J.L., Sethi, D.K. et al. (2014). Deconstructing the peptide‐MHC specificity of T cell recognition. Cell 157: 1073–1087.
- 106 Judkowski, V., Pinilla, C., Schroder, K. et al. (2001). Identification of MHC class II‐restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166: 908–917.
- 107 Pinilla, C., Martin, R., Gran, B. et al. (1999). Exploring immunological specificity using synthetic peptide combinatorial libraries. Curr. Opin. Immunol. 11: 193–202.
- 108 Arakawa, A., Siewert, K., Stohr, J. et al. (2015). Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 212: 2203–2212.
- 109 Gubin, M.M., Zhang, X., Schuster, H. et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature 515: 577–581.
- 110 Kvistborg, P., Philips, D., Kelderman, S. et al. (2014). Anti‐CTLA‐4 therapy broadens the melanoma‐reactive CD8+ T cell response. Sci. Transl. Med. 6: 254ra128.
- 111 Linnemann, C., van Buuren, M.M., Bies, L. et al. (2015). High‐throughput epitope discovery reveals frequent recognition of neo‐antigens by CD4+ T cells in human melanoma. Nat. Med. 21: 81–85.
- 112 Stronen, E., Toebes, M., Kelderman, S. et al. (2016). Targeting of cancer neoantigens with donor‐derived T cell receptor repertoires. Science 352: 1337–1341.
- 113 Wucherpfennig, K.W. and Strominger, J.L. (1995). Molecular mimicry in T cell‐mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80: 695–705.
- 114 Sivan, A., Corrales, L., Hubert, N. et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science 350: 1084–1089.
- 115 Vetizou, M., Pitt, J.M., Daillere, R. et al. (2015). Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science 350: 1079–1084.
- 116 Atarashi, K., Tanoue, T., Oshima, K. et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500: 232–236.
- 117 Narushima, S., Sugiura, Y., Oshima, K. et al. (2014). Characterization of the 17 strains of regulatory T cell‐inducing human‐derived Clostridia. Gut Microbes 5: 333–339.
- 118 Bjornson, Z.B., Nolan, G.P., and Fantl, W.J. (2013). Single‐cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 25: 484–494.
- 119 Maecker, H.T., Nolan, G.P., and Fathman, C.G. (2010). New technologies for autoimmune disease monitoring. Curr. Opin. Endocrinol. Diabetes Obes. 17: 322–328.
- 120 Davis, M.M., Altman, J.D., and Newell, E.W. (2011). Interrogating the repertoire: broadening the scope of peptide‐MHC multimer analysis. Nat. Rev. Immunol. 11: 551–558.