
Chapter 9
Self-Assembly and Directed Assembly
Hejin Jiang, Yutao Sang, Li Zhang and Minghua Liu
The Chinese Academy of Sciences, Institute of Chemistry, CAS Key Laboratory of Colloid Interface and Chemical Thermodynamics, Zhongguancun North First Street 2, Beijing, 100190, China
9.1 Introduction
The properties of materials are determined not only by their individual components but also by their assembly manner, and, thus, self-assembly becomes an important approach to the bottom-up fabrication of materials. Self-assembly is a process in which a disordered system of preexisting components forms an organized structure or pattern as a consequence of specific, local interactions among the components themselves, without external direction [1]. One can find the self-assembly phenomena everywhere and at all scales [2]. Since the Nobel Prize was awarded to the concepts of supramolecular chemistry in 1987, this field has grown rapidly and enormously [3]. In 2005, one of the 25 big questions by Science “How far can we push chemical self-assembly?” brought further attention to the self-assembly systems [4]. Chemical self-assembly is one of the most important processes where chemistry, physics, biology, and materials are merged since it originated from chemistry and follows the physical and chemical principles, but is inspired from the bio-system and can be applied to the functional materials.
Chemical self-assembly started from the design of molecules. Different from the organic synthesis, which generally uses the covalent bond, self-assembly is based on the non-covalent bond or supramolecular “rule.” Thus, in the design of the molecules or building blocks suitable for self-assembly, two elements should be considered most. One is the molecular skeleton that contains the molecular information or function, the other is the intermolecular interaction or joint that connects various component molecules. Various interactions such as van der Waals interaction, hydrophobic interaction, π–π stacking, hydrogen bonding, and ion pairing are widely used. Besides those, other interactions including cation–π, anion–π, dipole–dipole, and halogen bonding are usually used for the self-assembly system. In addition, the metal coordination interaction can add further interaction and strengthen the assemblies, which are also widely used [5]. Not all molecules can be assembled and only those with appropriate molecular skeletons and joints can show the self-assembly process. Figure 9.1 illustrates some typical skeletons and “joints” for self-assembly. These joint pairs assure the self-assembly, while the skeleton part can derivate into more complicated structures [6]. Furthermore, the combination of various joint parts can assist the building blocks with diverse assembly capacity. Such a combination is not only useful for the connections of the molecules but also for larger entities such as nanoparticles. When the surface of the nanoparticles is modified with these “joint” parts, the assembly can be performed.

Figure 9.1 Typical intermolecular interactions and their possible assembly manner.
There are several important features for self-assembly. One is the dynamic properties. Self-assembly is a process where small molecules get together to form large, ordered entities. Such entities are connected by the non-covalent bond, thus forming equilibrium with the component molecules. The process can be reversible in many cases and the assembly and disassembly occurs simultaneously. Of course, the self-assembled entities can be separated from the system, which will become irreversible.
The second feature is the hierarchical issue with diverse nanostructures [7]. For example, in the case of a protein, amino acids form the peptide chain through covalent bonds. One peptide chain can fold into secondary structures like α-helix and β-sheet. These structures can further fold into tertiary or higher order structures to perform the functions. In the self-assembly system, the molecules get together to form a certain basic unit, which further forms higher order nanostructures. For example, in the case of a self-assembled nanotube, the building blocks usually form the flat or planar structure first and then roll into tubular structures. Under certain conditions, these nanotubes can be further bundled. Remarkably, in the self-assembly system, one kind of molecule can form possibly various kinds of nanostructures and show structural diversity.
The third feature is the supramolecular chirality in the self-assembled system [8]. Although not all the self-assembly systems show chirality, one can frequently encounter the chiral phenomena. Chirality is ubiquitous in nature and organisms as one of the most important structural characteristics. In a biological system, important chiral molecules, either amino acids in proteins or sugars in DNA, are actively involved in the self-assembly process and play important roles in the protein folding and DNA duplication. In the artificial self-assembly system, those chiral gelators are found to easier form the nanostructures. Even the achiral components can also self-assemble into nanostructures with handedness. Thus, the chiral issue is also important in understanding the running of the biological system and the fabrication of the self-assembled system.
On the other hand, although most of the self-assembly is performed spontaneously, there are many cases where it should be driven by external factors. Template, light, magnet, and ultrasound can usually trigger or direct self-assembly [9]. These external factors assist the building blocks to form a more large-scale ordered and hierarchical pattern. In addition, they provide the control on the assembly process and structures over a large-scale range. Thus, a new concept of nanoarchitectonics is proposed in the construction of functional materials with nano units, which contributes to materials innovation not only by the creation of individual nanostructures but also by including the creation of macroscopic materials based on a profound understanding of mutual interactions between the individual nanostructures and their arbitrary arrangements [10, 11].
In this section, we show how some typical molecules like amphiphiles, functional π-conjugates, peptides, DNA, and block polymers are self-assembled into ordered nanostructures. Further, we show how external factors drive the directed assembly to form large patterns. Here, we only focus on the nanoarchitectonics structural issues and the discussion on the function is neglected in most cases.
9.2 Amphiphile Self-Assembly
Amphiphiles refer to molecules that have both hydrophilic and hydrophobic groups, typically like soap molecules. As one of the most widely investigated building blocks in the self-assembly system, amphiphiles are widely investigated [12–16]. A simple amphiphile is a molecule that contains a head hydrophilic group and a long alkyl chain. However, depending on the saturation, length, number of hydrocarbons, and linearity of the alkyl chains, various amphiphiles can be derived. Headgroup is also an important variable factor; those with hydrophilic properties, charged species and even peptides, dendrons, and aromatics with hydrophilic groups can be derived into amphiphiles, thus providing the versatile molecule design and assembly in the amphiphilic assemblies. Besides the single component, complex type of amphiphiles such as the supra-amphiphile can be designed and largely expand the scope of the amphiphiles. Figure 9.2 illustrates some typical amphiphiles, and shows how amphiphiles can be self-assembled.
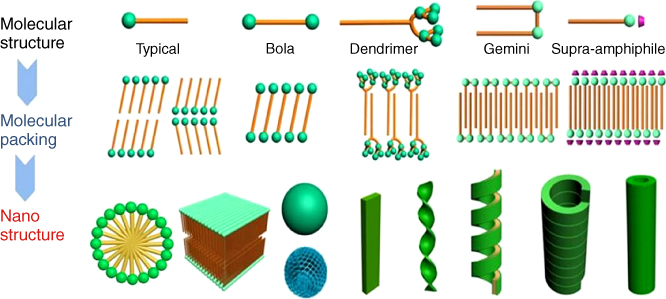
Figure 9.2 Various kinds of amphiphiles and the illustrations of the self-assembly of amphiphiles to diverse nanostructures through a bilayer or monolayer unit.
As in the typical assembly manner, amphiphiles essentially form monolayer or bilayer structures as the basic unit. There are various packing modes for the basic mono- and bilayers. The amphiphile can form in a head-to-head, tail-to-tail, or head-to-tail manner. The alkyl chains can be tilted or interdigitated in bilayers. The stability and geometry, including the shape, are quite important for further self-assembly into higher ordered nanostructures. Such a basic monolayer or bilayer can be planar and further stacked to form a multi-bilayer structure or curved to form micelles or vesicles depending on the packing parameter. When there are chiral elements in the headgroup, the amphiphiles are easy to self-assemble into helices or nanotubes.
9.3 π-Conjugated Molecule Self-Assembly
π-Conjugated molecules are one of the most important molecules that can form ordered nanostructures and show functions. In a biological system, self-assembly of porphyrin is significant for the light harvesting by the photosynthetic systems [17]. In developing new devices like solar cells, organic light-emitting diodes (OLEDs), and many others, the π-conjugated molecules play an important role [18, 19]. One of the most important features of the self-assembly of the π-conjugated molecules is the π–π stacking. Although the π–π stacking is a relatively weak non-covalent bond, its collective effect will be great since the π-conjugated molecules can be easily stacked. In addition, in assembling these systems, other non-covalent bonds such as the electrostatic and hydrogen bonds are simultaneously utilized, thus providing “strong” interactions.
There are two typical stacking modes of the π-conjugated molecules. Figure 9.3 illustrates the J-type and H-type aggregation of typical π-conjugated molecules. The J-aggregate and H-aggregate originally refer to the dye association modes based on the spectral shifts. In comparison with the absorption band of the molecular solution, the bathochromical or redshifted band is called J-band, named after Jelly, one of the first workers who investigated these shifts [22, 23]. The hypsochromical or blueshifted absorption band refers to H-bands, denoting the initial letter of hypsochromic. These absorption shifts due to aggregation have been explained in terms of molecular exciton coupling theory. The J- and H-aggregates are relative to the π-conjugated molecules packed in a head-to-tail and shoulder-to-shoulder way, respectively.
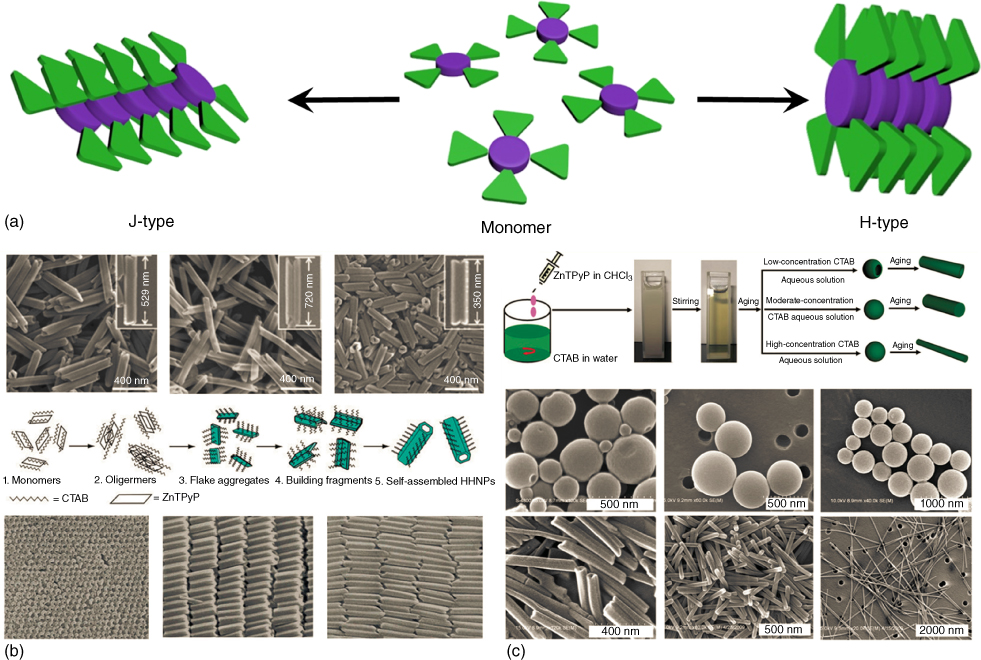
Figure 9.3 (a) Typical packing modes of the π-conjugated molecules. (b) Various nanostructures from the self-assembly of ZnTPyP in aqueous CTAB solution. (c) Surfactant-assisted self-assembly of ZnTPyP via an oil-in-water way from chloroform solution to aqueous CTAB solutions to provide diverse nanostructures.
((b, c) Hu et al. 2005 [20] and Qiu et al. 2010 [21]. Reproduced with permission of American Chemical Society.)
Porphyrin, as one of the most important π-conjugated molecules, shows diverse assembly capacity. Wan and coworkers demonstrated that zinc 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine (ZnTPyP) could be assembled to form hollow hexagonal nanoprisms, simply by dropping a DMF solution of ZnTPyP into an aqueous solution containing a well-known cetyltrimethylammoniumbromide (CTAB) surfactant [20]. The hollow hexagonal nanoprisms can be further aligned. It was suggested that the Zn–N axial coordination of the pyridyl N-atoms to zinc atoms of ZnTPyP, the crystal-packing force, and the intermolecular π–π stacking interactions between the porphyrins contributed greatly to the growth of the aggregates, while the CTAB adsorbed on the surface can help the alignment. Here, hierarchical self-assembly features were shown. Our group has further found that tuned nanostructures can be obtained via the used surfactant-assisted self-assembly method. When a chloroform solution of ZnTPyP was added into an aqueous micellar solution of CTAB, ZnTPyP-based nanostructures of diverse architectures, including hollow nanospheres, solid nanospheres, nanotubes, nanorods, and nanofibers could be assembled [21]. The morphologies of the formed ZnTPyP nanostructures displayed distinct dependence on aging time and surfactant concentration, which illustrated the diverse nanostructures through the self-assembly.
While π–π stacking is important in forming various nanostructures and shows new functions, the aggregation by avoiding the π–π stacking to increase the emission, or aggregation-induced emission (AIE) is a hot topic recently, which opened a new field in the self-assembly of π-conjugated molecules [24, 25].
9.4 Peptide Self-Assembly
Peptides are composed of amino acids linked through the covalent bond with diverse sequences and have gained widespread popularity. The self-assembly of peptides is a hot topic, which extends to the materials and biological sciences [26, 27]. Peptide chemistry is well established, and various building blocks based on peptides can be easily designed and synthesized. Peptides have many non-covalent bonding sites such as the H-bond between amides, π–π stacking in some cases, and H-bond and chargeable end groups depending on the pH value. These unique structures make peptides have a large capacity for self-assembly. While simple amino acids are generally difficult to assemble, starting from the dipeptide they showed strong ability to self-assemble [28]. The self-assembly of dipeptides has been widely investigated and can serve to understand the assembly mechanism. Various kinds of peptides can be designed with different sequences, lengths, and linearity; and their self-assembly is really diverse. The self-assembly of peptide amphiphiles showed similarity to the self-assembly of amphiphiles; and the ones composed of different sequences of the peptide showed unique properties as well as bioactivities, although the general schemes might be similar.
Figure 9.4 illustrates the self-assembly of peptides. Those peptides with 2–50 amino units have been widely investigated. They can assemble into nanofibers, nanotubes, particles, matrices, and membranes depending on the precise structures. In addition, the assembly can be regulated through the pH. While many studies are focused on the important peptide fragments from the biologically important proteins, an interesting example is the self-assembly of the Phe-Phe or FF dipeptide, which is regarded as the model compound for the understanding of Alzheimer disease. This dipeptide shows interesting self-assembly ability and forms diverse nanostructures, as shown in Figure 9.4.
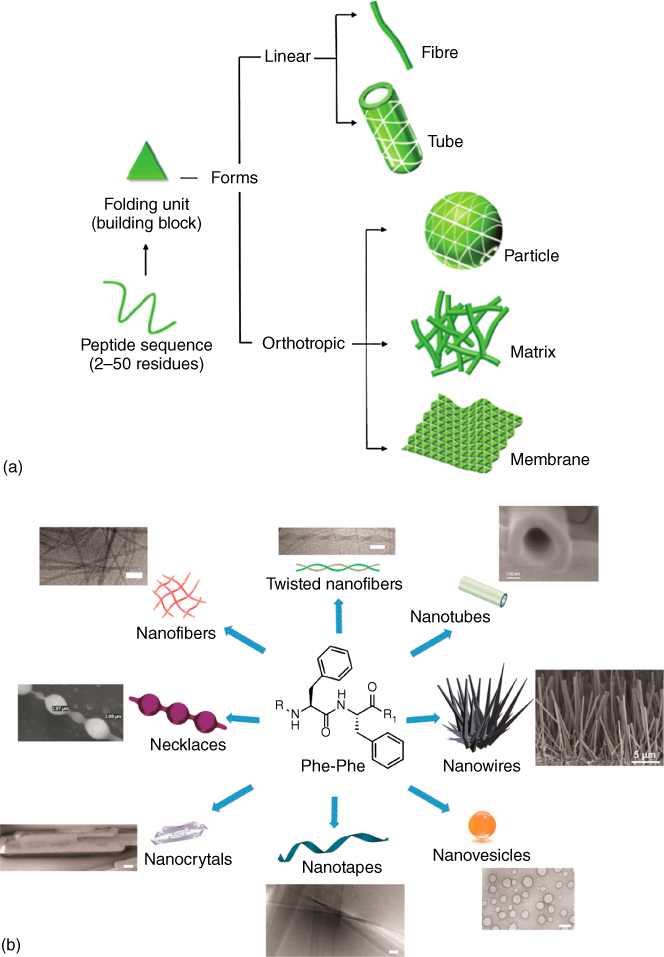
Figure 9.4 (a) Illustration of the self-assembly of peptides to form various nanostructures.
(Santis and Ryadnov 2015 [27]. Reproduced with permission of Royal Society of Chemistry.)
(b) Phe-Phe self-assembled into various kinds of nanostructures.(Marchesan et al. 2015 [29], http://www.mdpi.com/1420-3049/20/11/19658/htm. Licensed under CC BY 4.0.)
Peptides uniquely self-assemble into hierarchical and chiral nanostructures. As is well known, peptides are connected by amino acids through amide bonds. They can easily form various kinds of secondary structures, and these structures can further fold into higher order nanostructures. The other issue is that of supramolecular chirality. Since the component amino acids have a chiral center, they can easily form homochiral nanostructures. By introducing enantiomers with opposite chirality, the self-assembly manner of the peptides will change even with the same sequence. Thus, the chiral effect of the peptide self-assembly is also an important topic [30].
9.5 Self-Assembly of Block Polymers
Block copolymers (BCPs) have two or more homopolymer subunits linked by covalent bonds. Polymers with two or three blocks of two distinct chemical species (e.g., A and B) are called diblock copolymers and triblock copolymers, respectively. The chemistry difference between covalently bonded blocks leads to microphase separation of the incompatible blocks in a selective solvent, in which the solvent-selective block is compatible with the solvent, whereas the other block is incompatible. This microphase separation causes the self-assembly process of BCPs and the consequent self-assemblies display diversiform ordered morphologies, ranging from typical hollow spheres, cylinders, and gyroids to lamellae. These self-assembled structures can be affected by numerous parameters, such as variations in the copolymer composition, polydispersity of the copolymer chains, molecular weight of the copolymer, the initial copolymer concentration, and the nature of the solvent, temperature, and presence of additives.
9.5.1 Directed Self-Assembly (DSA) of BCPs
With the demand for microelectronic devices, defect-free patterns on a large scale or irregular device-oriented structures are being explored. Directed self-assembly (DSA) of BCPs is currently regarded as one of the most attractive next-generation lithography techniques, because of spontaneously forming ordered nanostructures with conventional photolithography, such as E-beam lithography, ArF lithography, and I-line lithography [9, 31, 32]. These surface patterns direct the orientation and ordering of BCP self-assembled nanodomains to obtain some ordered periodic nanopatterns. In general, the diblock copolymer, polystyrene-block-polymethylmethacrylate (PS-b-PMMA) is used as a standard model polymer. Periodic arrays of self-assembled lamellae, spheres, and cylinders with a typical feature size in the 3–50 nm can be obtained by this technique. For the past two decades, surface pattern DSA of BCPs has been attracting tremendous attention, because it provides distinct advantages such as molecular-scale pattern precision, dense areal packing of nanodomains, low-cost spontaneous pattern generation, and smooth/narrow interfacial width.
Recently, an advanced process for surface pattern DSA of BCPs has also been developed. For example, a selective DSA chemical patterning, a method that could simultaneously fabricate multiple, aligned BCPs within a single patterning layer based on traditional DSA, has been proposed [33]. A blend of BCPs consists of cylinder-forming material and material lamellar-forming assembles on specially designed surface chemical line gratings, leading to selectively forming either dot patterns or line patterns by locally adjusting the relative energy of these competing morphologies through spatial variations of the underlying chemical pattern. By this method, multicomponent blends of self-assembling materials are ordered in the presence of a spatially varying directing field (Figure 9.5).

Figure 9.5 Conventional and selective directed self-assembly. (a) Directed self-assembly utilizes a substrate prepattern to impart long-range order to both lamellar and cylindrical self-assembled block copolymer films. (b) In selective directed self-assembly, a blend of block copolymers (either cylindrical or lamellar) assembles on specially designed surface chemical line gratings, leading to the simultaneous formation of coexisting ordered morphologies in separate areas of the substrate.
(Stein et al. 2016 [33], https://www.nature.com/articles/ncomms12366?WT.feed_name=subjects_physical-sciences. Licensed under CC BY 4.0.)
9.5.2 Magnetic Fields Directing the Alignment of BCPs
Magnetic fields are also a very important factor to direct the self-assembly or alignment of BCPs. The diamagnetic anisotropy of the smectic mesophase formed by some units in the BCP cause the self-assembled BCP structures to be aligned by the magnetic field. For example, in a liquid crystalline brush-like diblock copolymer, NBCB-b-NBPLA, cyanobiphenyl species worked as magnetic-field-responsive units based on their positive diamagnetic anisotropy and parallel orientation at the interface with more polar BCP domains such as PLA [34, 35]. Then, aligned hexagonally packed cylindrical domains can be produced under magnetic fields, as illustrated in Figure 9.6. The system can be successively UV cross-linked, producing mechanically robust films. Further, vertically aligned nanopores were fabricated by selectively removing the packed cylindrical domains under mild etching conditions. More interestingly, this nanoporous material is stimuli-responsive, exhibiting reversible closure and opening in response to heating and cooling above a critical temperature. The approach using magnetic fields to provide a scalable route for membrane fabrication is well suited to large area processing.
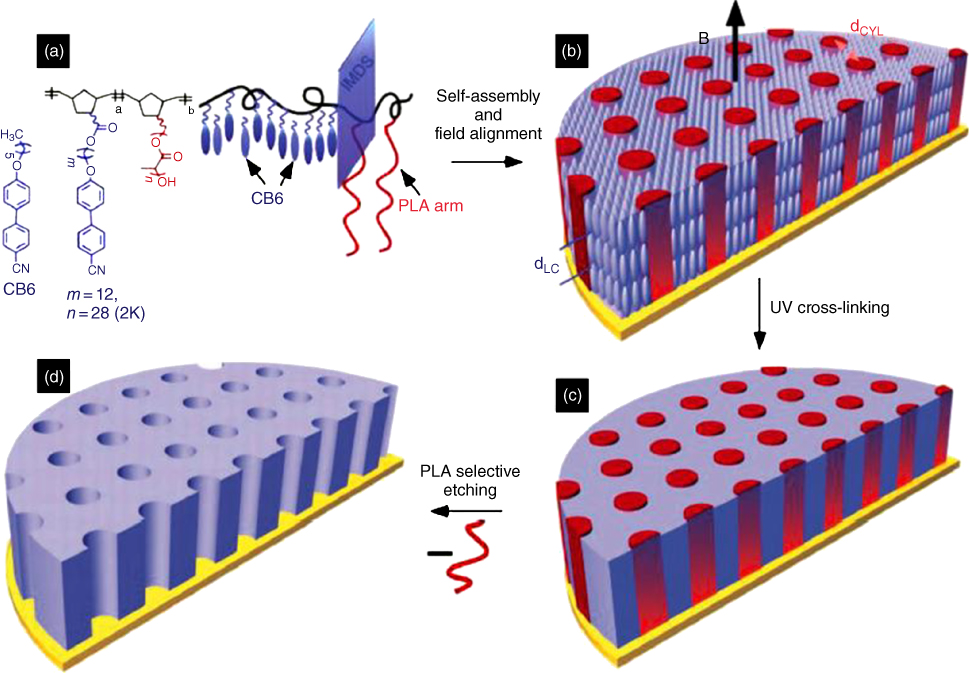
Figure 9.6 (a) Molecular structure of NBCB-b-NBPLA consisting of cyanobiphenyl mesogens (blue rod) and PLA (red), the blue plane is the inter-material dividing surface (IMDS). CB6 is the free mesogen introduced into the system to accelerate the kinetics of magnetic field alignment. (b) Magnetic alignment occurs subject to the positive anisotropy and homogeneous anchoring of the mesogens leading to orientation of cylindrical domains along the field. (c) UV irradiation yielding mechanically robust films. (d) Subsequent PLA etching from the aligned material results in a large-area nonporous membrane over millimeter-scale thicknesses.
(Gopinadhan et al. 2014 [34]. Reproduced with permission of John Wiley & Sons.)
A magnetic field combined with an acoustic field can adjust the type of assembled structure in situ, and therefore drive the assembly to form a large class of structures ranging from discrete clusters to well-ordered crystals. A uniform external magnetic field is used to control the interparticle interactions, and an acoustic standing wave controls the local density of particles. By simultaneously controlling these two fields, a wide range of phase space can be explored in a single experiment. For example, a novel magneto-acoustic assembly platform can direct the assembly of a large class of structures, ranging from discrete colloidal molecules and polymer networks, to well-ordered crystals, by tuning the ratio of the magnetic and acoustic field strengths [36]. The technique can stabilize the assembly structures and transfer them to other substrates, showing promise for future applications in biosensors and photonic devices.
9.6 DNA-Directed Self-Assembly
DNA is a powerful tool that can be attached to nano- and micro-objects and can be used as a programmable “glue” to direct the assembly of mesoscale units. DNA contains four different nucleotide bases, each of which forms a base pair with another complementary base according to a set of canonical rules: adenine with thymine and guanine with cytosine. By simply arranging the sequence of these four nucleotide bases in different DNA strands, a combinatorially large set of binding interactions can be designed as specific hybridizations between complementary DNA strands. Thus, DNA-DSA of fluorophores, proteins, carbon nanotubes, nanoparticles, artificial vesicles, emulsion droplets, and even of living cells was explored recently.
Perhaps the most basic assembly scheme is the following: two nanoparticle solutions are functionalized with two different, but complementary, strands of an oligonucleotide. Hybridization of the complementary DNA strands is the driving force behind the assembly of the nanoparticles (Figure 9.7). This process can produce one-dimensional linear arrays, two-dimensional trimers and tetramers, as well as stable three-dimensional assemblies built up on a substrate [37, 38].
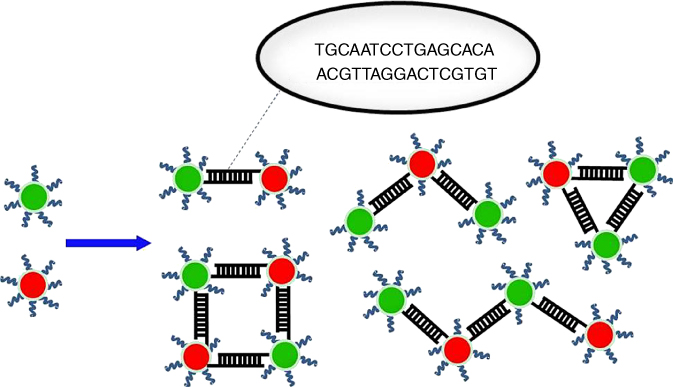
Figure 9.7 Schematic illustration of DNA-directed assembly of nanoparticles by hybridizations between complementary DNA strands.
(Adapted from Barrow et al. 2013 [37] and Zhang et al. 2015 [38].)
This DNA-directed assembly technique can be used to prepare more complicated nanostructures. For example, a simple bottom-up approach was reported for the fabrication of highly surface-enhanced Raman spectroscopy (SERS)-active gold core–satellite nanostructures, using electrostatic and DNA-DSA [39]. In this process, spherical gold nanoparticles (AuNPs) with an average diameter of 30 and 20 nm functionalized with monothiolated DNA (denoted as AuNP/DNA conjugates) were used as core and satellite nanoparticle building blocks, respectively. Firstly, an array of core AuNPs (30-nm diameter) on a silicon substrate was fabricated through electrostatic self-assembly, and then the specific DNA-directed assembly of 20-nm AuNPs onto the core nanoparticles form immobilized 20-nm satellite AuNPs specifically around the core AuNPs. UV-ozone treatment following the successful self-assembly is directed at removing all surface-confined molecules to obtain pristine gold surfaces ready to adsorb analytes for their detection in SERS experiments. It is demonstrated that DNA-DSA provides excellent control over particle distances on the nanoscale. The as-fabricated core–satellite nanostructures exhibit better SERS activities than commercial SERS substrates in signal intensity and reproducibility. This work indicated that DNA-DSA strategies allow for the development of complex assembly processes for the fabrication of complex, well-defined functional nanostructures with future applications well beyond the field of sensing.
Complementary DNA molecules were also reported as glue to direct the self-assembly of hydrogel cubes. Yin and coworkers fabricated hydrogel cubes carrying giant DNA-encoding tandem repeated complementary sequences, and complementary giant DNA was capable of directing the assembly of hydrogel cubes with a wide range of edge lengths (Figure 9.8) [40]. This DNA-DSA of shape-controlled hydrogel modules proved to be highly programmable and controllable, and will open new doors to address the challenge of building complex self-assembled three-dimensional structures for diverse applications in materials sciences and especially in biomaterials. For instance, by encapsulating specific cells inside the hydrogel cubes, the self-assembled structures could be used to build the basic architectures of native tissues.

Figure 9.8 Self-assembly of hydrogel cubes with uniform giant DNA glue modification. (a) Schematic of giant-DNA-directed hydrogel assembly. Giant DNA containing tandem repeats of complementary 48-nt sequences was uniformly amplified on the surface of red and blue hydrogel cubes. Hybridization between the complementary DNA sequences resulted in assembly of hydrogel cubes. (b) Aggregates assembled from red and blue hydrogel cubes carrying complementary giant DNA.
(Qi et al. 2013 [40]. Reproduced with permission of Nature Publishing Group.)
9.7 Directed Self-Assembly of Nanoparticles
Nanoparticles are one of the most promising and prominent components for technological applications due to their unique size-dependent properties including mechanics, electrics, superparamagnetism, sensitivity, chemiluminescence, and catalysis. However, in order to fully harness the potential capabilities of nanoparticles, a high level of direction and control is required. DSA has been identified as a necessary process where the nanoparticles spontaneously organize into useful patterns or other ordered structures by non-covalent interactions [2, 41]. Since DSA of nanoparticles still employs the basic principles of self-assembly, thermodynamic driving forces and other constraints may need to be modulated.
The overall size of nanoparticle aggregates, interparticle spacing between particles, and the collective behavior in nanoparticle ensembles should be carefully constructed, either by direct interactions such as interparticle forces [42–46] or indirectly by means of templates [47–50] and external fields like electric, [51, 52] magnetic, [53, 54] flow [55, 56], and combinations of them [57, 58]. For instance, Figure 9.9 illustrates that under carefully controlled conditions, cubic nanocrystals of magnetite self-assemble into arrays of helical superstructures in a template-free manner with >99% yield [59].
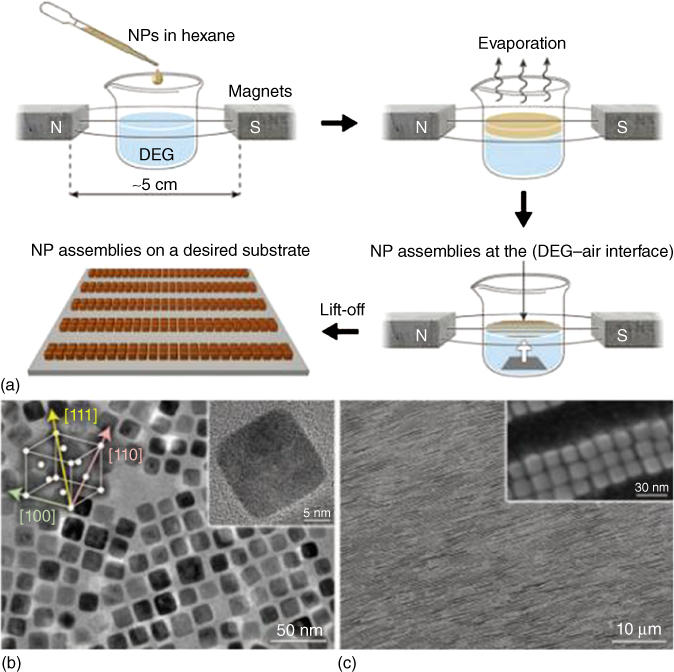
Figure 9.9 (a) Schematic representation of the experimental setup. (b) Transmission electron microscopy and (c) scanning electron microscopy images of the building block and the one-dimensional nanocube belts, respectively.
(Reproduced with permission from [59]. Copyright 2014, Science Publishing Group.)
Moreover, using proper interacting segments, a wide range of different building block combinations is conceivable, from biological origin to inorganic and organic nanoparticles and microparticles. Klajn's group introduced directed co-assembly of a well-ordered mixed mesostructure of high complexity. Three hierarchical levels were bridged up by guiding triblock terpolymers to form soft patchy nanoparticles of different symmetries that, in combination, co-assemble into substructured, compartmentalized materials with predictable and tunable nanoscale periodicities.
One of the major challenges for nanoparticle assembly is the lack of metrology to characterize the kinetics and structures in situ during the assembly process. Nevertheless, DSA opens up avenues for nanoparticles to construct new materials with multifunctions, also a controlled organization over much longer length scales, and satisfies the need for real technological application in smart materials, sensing, photonics, nanolithography, and fascinating artificial nanomachines.
9.8 LB-Technique-Directed Alignment of Nanostructures
The Langmuir–Blodgett (LB) technique is usually used to arrange amphiphiles into ordered packing onto a water surface. Through this technique, amphiphiles can be fabricated into various nanostructures. Now, the LB technique has showed the ability to assemble nanosized building blocks into both closely packed nanosuprastructures and well-defined patterns with high density [60]. The LB assembly process arranging nanostructures is illustrated in Figure 9.10. Nanostructures are first dissolved in a volatile organic solvent and spread onto a water subphase. After solvent evaporation, the nanomaterials are compressed while the surface pressure is monitored and the Langmuir film can be fabricated. Then, the Langmuir film is transferred to a solid substrate by vertical-dipping (LB) or horizontal-lifting (Langmuir–Schaefer, LS) techniques, and then the film device with a certain thickness is fabricated. The density and packing of the film can be adjusted by the compression rate, surface pressure, dipping velocity, temperature, and the initial concentration of nanomaterials. The Yang group firstly reported aligning of the inorganic nanoparticle and nanowire through this procedure [62]. Subsequently, this versatile method was extended to arrange other inorganic nanowires into close-packed films, such as Ge nanowires, VO2 nanowires, Ag nanowires, and PbS nanowires. Recently, this LB technique showed the possibility of large-scale alignment of organic self-assembled nanotubes [61]. A C3-symmetrical glutamic acid derivative self-assembled into hexagonal organic nanotubes and these formed nanotubes can be dispersed into toluene without destroying the assembling nanotubular structures. When spreading the organic nanotube structures on a water subphase, the desired alignment with a large area of dense and well-oriented nanotube array was obtained by the LB technique. Furthermore, both oil-soluble guests, including small molecules and polymers, and water-soluble guests can be encapsulated into nanotubes via the instant gelation method. These guest-contained nanotubes can also be aligned through the same procedures. This study demonstrates that the controlled formation of well-organized nanostructures using the LB method may provide new opportunities for the fabrication of large-scale arrays of functional one-dimensional organic tubular nanostructures.

Figure 9.10 General schematic illustration for preparing nanotube-aligned films by LB technique: First, TMGE nanotubes prepared by gelation upon heating and cooling in toluene and encapsulated with guest molecules using instant gelation method. The formed gel was dispersed in toluene and the tubular structures remained unchanged. Then, spreading the dispersed suspension on the water subphase, using LB technique with repeated compression and expansion procedure, well-aligned nanotube films can be obtained.
(Zhou et al. 2016 [61]. Reproduced with permission of American Chemical Society.)
9.9 Conclusions
Self-assembly has proved its power in organizing small molecular to larger nanostructures and even macroscopic materials. In such a process, the molecular design and the smart utilization of the non-covalent bonds are of utmost importance. Various kinds of molecules such as amphiphiles, π-conjugated molecules, peptides, DNA, polymers, and nanoparticles can serve as building blocks. During the subsequent self-assembly, the external control of the assembly condition and the dynamics are also important in obtaining well-defined nanostructures. With external controls like light and magnetic forces, some molecules like block polymers and nanoparticles can, by DSA, be formed into macroscopic entities and patterns.
Considering the future development of this field, several challenges exist. One is the structural control over a larger scale range. While the control of the nanoscale structures becomes easier, it needs to extend to the macroscopic scale while keeping the unique properties produced in the nanoscale. The second is the robustness of the self-assembly materials. Since many of the self-assembled nanomaterials are composed of the non-covalent bonds, it is needed to further strengthen the mechanical properties of such kind of materials. Third, although we have acquired much knowledge about the non-covalent bonds, structural control, and functional development, many of these are limited to assemblies by one or two components. Since biological and materials systems are generally complicated [63, 64], how to combine different molecules into a complex system in a cooperative or syndetic way remains a challenge. Although there is still a long way to go before we can realize the sophisticated structures such as a cell, a step forward is to expand the supramolecular system to larger nanoarchitectures and beyond.
References
- 1 Ulman, A. (2013) An Introduction to Ultrathin Organic Films: From Langmuir-Blodgett to Self-Assembly, Academic Press.
- 2 Whitesides, G.M. and Grzybowski, B. (2002) Self-assembly at all scales. Science, 295, 2418–2421.
- 3 Lehn, J.M. (1990) Perspectives in supramolecular chemistry-from molecular recognition towards molecular information processing and self-organization. Angew. Chem. Int. Ed., 29, 1304–1319.
- 4 Service, R.F. (2005) How far can we push chemical self-assembly. Science, 309, 95.
- 5 Qin, L., Lv, K., Shen, Z.C., and Liu, M.H. (2015) Self-assembly of organic molecules into nanostructures, in Soft Matter Nanotechnology: From Structure to Function, Wiley-VCH.
- 6 Zhang, L., Wang, T.Y., Shen, Z.C., and Liu, M.H. (2016) Chiral nanoarchitectonics: towards the design, self-assembly, and function of nanoscale chiral twists and helices. Adv. Mater., 28, 1044–1059.
- 7 Elemans, J.A.A.W., Rowan, A.E., and Nolte, R.J.M. (2003) Mastering molecular matter. Supramolecular architectures by hierarchical self-assembly. J. Mater. Chem., 13, 2661–2670.
- 8 Liu, M.H., Zhang, L., and Wang, T.Y. (2015) Supramolecular chirality in self-assembled systems. Chem. Rev., 115, 7304–7397.
- 9 Jeong, S.J., Kim, J.Y., Kim, B.H., Moon, H.S., and Kim, S.O. (2013) Directed self-assembly of block copolymers for next generation nanolithography. Mater. Today, 16, 468–476.
- 10 Aono, M., Bando, Y., and Ariga, K. (2012) Nanoarchitectonics: pioneering a new paradigm for nanotechnology in materials development. Adv. Mater., 24, 150–151.
- 11 Ariga, K., Lee, M.V., Mori, T., Yu, X.Y., and Jonathan, P.H. (2010) Two-dimensional nanoarchitectonics based on self-assembly. Adv. Colloid Interface Sci., 154, 20–29.
- 12 Cui, H., Webber, M.J., and Stupp, S.I. (2010) Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. J. Pept. Sci., 94, 1–18.
- 13 Fuhrhop, J.H. and Wang, T.Y. (2004) Bolaamphiphiles. Chem. Rev., 104, 2901–2938.
- 14 Zhang, L., Wang, X.F., Wang, T.Y., and Liu, M.H. (2015) Tuning soft nanostructures in self-assembled supramolecular gels: from morphology control to morphology-dependent functions. Small, 11, 1025–1038.
- 15 Duan, P.F. and Liu, M.H. (2009) Design and self-assembly of l-glutamate-based aromatic dendrons as ambidextrous gelators of water and organic solvents. Langmuir, 25, 8706–8713.
- 16 Kang, Y.T., Liu, K., and Zhang, X. (2014) Supra-amphiphiles: a new bridge between colloidal science and supramolecular chemistry. Langmuir, 30, 5989–6001.
- 17 Gust, D., Moore, T.A., and Moore, A.L. (1993) Molecular mimicry of photosynthetic energy and electron transfer. Acc. Chem. Res., 26, 198–205.
- 18 Drain, C.M., Varotto, A., and Radivojevic, I. (2009) Self-organized porphyrinic materials. Chem. Rev., 109, 1630–1658.
- 19 Liu, H.B., Xu, J.L., Li, Y.J., and Li, Y.L. (2010) Aggregate nanostructures of organic molecular materials. Acc. Chem. Res., 43, 1496–1508.
- 20 Hu, J.S., Guo, Y.G., Liang, H.P., Wan, L.J., and Jiang, L. (2005) Three-dimensional self-organization of supramolecular self-assembled porphyrin hollow hexagonal nanoprisms. J. Am. Chem. Soc., 127, 17090–17095.
- 21 Qiu, Y.F., Chen, P.L., and Liu, M.H. (2010) Evolution of various porphyrin nanostructures via an oil/aqueous medium: controlled self-assembly, further organization, and supramolecular chirality. J. Am. Chem. Soc., 132, 9644–9652.
- 22 Jelley, E.E. (1936) Spectral absorption and fluorescence of dyes in the molecular state. Nature, 138, 1009–1010.
- 23 Würthner, F., Kaiser, T.E., and Saha-Möller, C.R. (2011) J-aggregates: from serendipitous discovery to supramolecular engineering of functional dye materials. Angew. Chem. Int. Ed., 50, 3376–3410.
- 24 Luo, J.D., Xie, Z.L., Lam, J.W., Cheng, L., Chen, H.Y., Qiu, C.F., Kwok, H.S., Zhan, X.W., Zhu, D.B., and Tang, B.Z. (2001) Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun., 18, 1740–1741.
- 25 Mei, J., Leung, N.L.C., Kwok, R.T.K., Lam, J.W.Y., and Tang, B.Z. (2015) Aggregation-induced emission: together we shine, united we soar!. Chem. Rev., 115, 11718–11940.
- 26 Zhang, S.G. (2003) Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol., 21, 1171–1178.
- 27 De Santis, E. and Ryadnov, M.G. (2015) Peptide self-assembly for nanomaterials: the old new kid on the block. Chem. Soc. Rev., 44, 8288–8300.
- 28 Adams, D.J. (2011) Dipeptide and tripeptide conjugates as low-molecular-weight hydrogelators. Macromol. Biosci., 11, 160–173.
- 29 Marchesan, S., Vargiu, A.V., and Styan, K.E. (2015) The Phe-Phe motif for peptide self-assembly in nanomedicine. Molecules, 20, 19775–19788.
- 30 Duan, P.F., Qin, L., Zhu, X.F., and Liu, M.H. (2011) Hierarchical self-assembly of amphiphilic peptide dendrons: evolution of diverse chiral nanostructures through hydrogel formation over a wide pH range. Chem. Eur. J., 17, 6389–6395.
- 31 Li, W.H. and Müller, M. (2016) Directed self-assembly of block copolymers by chemical or topographical guiding patterns: optimizing molecular architecture, thin-film properties, and kinetics. Prog. Polym. Sci., 54–55, 47–75.
- 32 Stoykovich, M.P., Müller, M., Kim, S.O., Solak, H.H., Edwards, E.W., de Pablo, J.J., and Nealey, P.F. (2005) Directed assembly of block copolymer blends into nonregular device-oriented structures. Science, 308, 1442–1446.
- 33 Stein, A., Wright, G., Yager, K.G., Doerk, G.S., and Black, C.T. (2016) Selective directed self-assembly of coexisting morphologies using block copolymer blends. Nat. Commun., 7, 12366.
- 34 Gopinadhan, M., Deshmukh, P., Choo, Y., Majewski, P.W., Bakajin, O., Elimelech, M., Kasi, R.M., and Osuji, C.O. (2014) Thermally switchable aligned nanopores by magnetic-field directed self-assembly of block copolymers. Adv. Mater., 26, 5148–5154.
- 35 Feng, X.D., Tousley, M.E., Cowan, M.G., Wiesenauer, B.R., Nejati, S., Choo, Y., Noble, R.D., Elimelech, M., Gin, D.L., and Osuji, C.O. (2014) Scalable fabrication of polymer membranes with vertically aligned 1 nm pores by magnetic field directed self-assembly. ACS Nano, 8, 11977–11986.
- 36 Yang, Y., Pham, A.T., Cruz, D., Reyes, C., Wiley, B.J., Lopez, G.P., and Yellen, B.B. (2015) Assembly of colloidal molecules, polymers, and crystals in acoustic and magnetic fields. Adv. Mater., 27, 4725–4731.
- 37 Barrow, S.J., Funston, A.M., Wei, X., and Mulvaney, P. (2013) DNA-directed self-assembly and optical properties of discrete 1D, 2D and 3D plasmonic structures. Nano Today, 8, 138–167.
- 38 Zhang, X., Wang, R., and Xue, G. (2015) Programming macro-materials from DNA-directed self-assembly. Soft Matter, 11, 1862–1870.
- 39 Zheng, Y., Thai, T., Reineck, P., Qiu, L., Guo, Y., and Bach, U. (2013) DNA-directed self-assembly of core-satellite plasmonic nanostructures: a highly sensitive and reproducible near-IR SERS sensor. Adv. Funct. Mater., 23, 1519–1526.
- 40 Qi, H., Ghodousi, M., Du, Y., Grun, C., Bae, H., Yin, P., and Khademhosseini, A. (2013) DNA-directed self-assembly of shape-controlled hydrogels. Nat. Commun., 4, 2275.
- 41 Grzelczak, M., Vermant, J., Furst, E.M., and Liz-Marzán, L.M. (2010) Directed self-assembly of nanoparticles. ACS Nano, 4, 3591–3605.
- 42 Israelachvili, J.N. (2011) Intermolecular and Surface Forces, Academic Press.
- 43 Hoeben, F.J., Jonkheijm, P., Meijer, E., and Schenning, A.P. (2005) About supramolecular assemblies of π-conjugated systems. Chem. Rev., 105, 1491–1546.
- 44 Lim, I.I.S., Mott, D., Ip, W., Njoki, P.N., Pan, Y., Zhou, S., and Zhong, C.J. (2008) Interparticle interactions in glutathione mediated assembly of gold nanoparticles. Langmuir, 24, 8857–8863.
- 45 Giner-Casares, J.J. and Reguera, J. (2016) Directed self-assembly of inorganic nanoparticles at air/liquid interfaces. Nanoscale, 8, 16589–16595.
- 46 Roy, D., Mondal, D., and Goswami, D. (2016) Structure and dynamics of optically directed self-assembly of nanoparticles. Sci. Rep., 6, 23318.
- 47 Nie, Z., Petukhova, A., and Kumacheva, E. (2010) Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotechnol., 5, 15–25.
- 48 Shirman, T., Arad, T., and Van Der Boom, M.E. (2010) Halogen bonding: a supramolecular entry for assembling nanoparticles. Angew. Chem. Int. Ed., 49, 926–929.
- 49 Boal, A.K., Ilhan, F., DeRouchey, J.E., Thurn-Albrecht, T., Russell, T.P., and Rotello, V.M. (2000) Self-assembly of nanoparticles into structured spherical and network aggregates. Nature, 404, 746–748.
- 50 Dong, Z., Asbahi, M., Lin, J., Zhu, D., Wang, Y.M., Hippalgaonkar, K., Chu, H.S., Goh, W.P., Wang, F., Huang, Z., and Yang, J.K. (2015) Second-harmonic generation from sub-5 nm gaps by directed self-assembly of nanoparticles onto template-stripped gold substrates. Nano Lett., 15, 5976–5981.
- 51 Hermanson, K.D., Lumsdon, S.O., Williams, J.P., Kaler, E.W., and Velev, O.D. (2001) Dielectrophoretic assembly of electrically functional microwires from nanoparticle suspensions. Science, 294, 1082–1086.
- 52 Lele, P.P., Mittal, M., and Furst, E.M. (2008) Anomalous particle rotation and resulting microstructure of colloids in AC electric fields. Langmuir, 24, 12842–12848.
- 53 Lee, S.H. and Liddell, C.M. (2009) Anisotropic magnetic colloids: a strategy to form complex structures using nonspherical building blocks. Small, 5, 1957–1962.
- 54 Ding, T., Song, K., Clays, K., and Tung, C.H. (2009) Fabrication of 3D photonic crystals of ellipsoids: convective self-assembly in magnetic field. Adv. Mater., 21, 1936–1940.
- 55 Pasquino, R., Snijkers, F., Grizzuti, N., and Vermant, J. (2010) Directed self-assembly of spheres into a two-dimensional colloidal crystal by viscoelastic stresses. Langmuir, 26, 3016–3019.
- 56 Shereda, L.T., Larson, R.G., and Solomon, M.J. (2008) Local stress control of spatiotemporal ordering of colloidal crystals in complex flows. Phys. Rev. Lett., 101, 038301.
- 57 Mittal, M. and Furst, E.M. (2009) Electric field-directed convective assembly of ellipsoidal colloidal particles to create optically and mechanically anisotropic thin films. Adv. Funct. Mater., 19, 3271–3278.
- 58 Jones, T.B. and Jones, T.B. (2005) Electromechanics of Particles, Cambridge University Press.
- 59 Singh, G., Chan, H., Baskin, A., Gelman, E., Repnin, N., Kral, P., and Klajn, R. (2014) Self-assembly of magnetite nanocubes into helical superstructures. Science, 345, 1149–1153.
- 60 Liu, J.W., Liang, H.W., and Yu, S.H. (2012) Macroscopic-scale assembled nanowire thin films and their functionalities. Chem. Rev., 112, 4770–4799.
- 61 Zhou, X., Cao, H., Yang, D., Zhang, L., Jiang, L., and Liu, M. (2016) Two-dimensional alignment of self-assembled organic nanotubes through Langmuir–Blodgett technique. Langmuir, 32, 13065–13072.
- 62 Yang, P.D. (2003) Nanotechnology: wires on water. Nature, 425, 243–244.
- 63 Bai, C. and Liu, M. (2013) From chemistry to nanoscience: not just a matter of size. Angew. Chem. Int. Ed., 52, 2678–2683.
- 64 Lehn, J.M. (2013) Perspectives in chemistry-steps towards complex matter. Angew. Chem. Int. Ed., 52, 2836–2850.