
Chapter 20
Immunoengineering
Yasuhiro Nakagawa1,2 and Mitsuhiro Ebara1,2,3
1International Research Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, 305-0044, Japan
2University of Tsukuba, Graduate School of Pure and Applied Sciences, 1-1-1, Tennodai, Tsukuba, Ibaraki, 305-8577, Japan
3Tokyo University of Science, Graduate School of Industrial Science and Technology, 6-3-1 Niijuku, Katsushika-ku, Tokyo, 125-8585, Japan
20.1 Introduction
The immune system provides a vital cellular response that protects the body against bacterial and/or viral infections. Infections from microorganisms, toxic cells (e.g., cancer cells), metabolized substances, and foreign substances are disposed by the immune system to maintain homeostasis of the human body. Although essential, the immune system attacks foreign materials such as medical implants and rejects them, making it difficult to embed such materials in the body. To overcome this immunological problem, materials used in vivo called biomaterials were developed on the basis of the strategy of reducing interactions with biomolecules (e.g., cells, proteins, lipids, nucleic acids, hormones, sugars, and amino acids). A variety of medical equipment uses this strategy as life-saving and life-improving options for countless patients. This concept has also been important in improving the fields of drug delivery system (DDS) and regenerative medicine.
However, many researchers have been recently interested in developing the next generation of biomaterials that promote or inhibit immune responses. This is driven by the brilliant development of regenerative therapies and DDS research, which strongly favor the fabrication of new immunomodulatory biomaterials by combining research in materials, immunology, and molecular biology. Revealing how cells, cytokines, hormones, and gene expressions are involved in specific biological functions is very important to optimize these biomaterials and medical devices. Research reports regarding immunomodulatory biomaterials have largely increased in the academic literature. However, controlling the immune system with biomaterials is still a challenge due to their methodological complexity. As shown in Figure 20.1, antitumor activity is expected when immune systems are activated; excess activation can cause unexpected symptoms such as allergies. Suppressing of the immune system lowers inflammation but also leads to diminished resistance against infections. Therefore, to obtain immunotherapy with a biomaterial, it is necessary to perfectly understand the biological reaction induced by this material when inserted into the body and to adequately design the shape, physical properties, and chemical properties of the material.
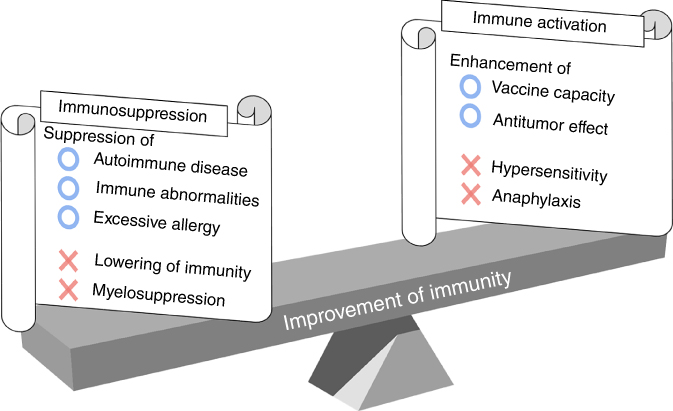
Figure 20.1 The trade-off relationship between immune activation and immunosuppression.
Biomaterials can be classified into three categories according to their association with the immune system (Figure 20.2). The first category is called “immunoevasive biomaterials” developed under the strategy of reducing interactions with biomolecules, as described earlier. The second type is called “immune-activating biomaterials” designed to exhibit antitumor and drug-responsive properties. Finally, the third kind is known as “immunosuppressive biomaterials” that modulate unnecessary uncontrollable inflammation and inhibit rejection responses. Throughout this chapter, we describe these three biomaterial strategies in detail.

Figure 20.2 Biological reactions and expected effects of immunoevasive, immune-activating, and immunosuppressive biomaterials. iDC, immature dendritic cell; mDC, mature dendritic cell; tDC, tolerogenic dendritic cell; Th1, T helper 1 cell; Th2, T helper 2 cell; CTL, cytotoxic T lymphocyte; Mφ, macrophage; Breg, regulatory B cell; Treg, regulatory T cell; Tr1, type 1 regulatory T cell.
20.2 Immunoevasive Biomaterials
In the early days of biomedical research, several materials used in the host body were chosen without considering biocompatibility and many proved to be pathogenic or toxic. Over time, the general understanding of immune systems deepened, and the first generation of biomaterials was developed to mimic the physical properties of replaced tissues with a minimal toxic response in the host [1]. Since then, countless more types of immunoevasive biomaterials have been developed and utilized in clinical trials.
Poly(ethylene glycol) (PEG) is one of most famous nonionic immunoevasive biomaterials. PEG is an industrially manufactured polyether compound and is widely used in surfactants, lubricants, medicaments, and cosmetics [2]. PEG shows perfect compatibility against water through hydrogen bonding between H2O molecules and oxygen in the PEG backbone. Research of bio (blood)-compatible materials using PEG-modified surfaces started in the early 1980s [3]. The surface of the linear PEG molecule was modified with one fixed branch, while the other chain end was able to move freely. These hydrophilic polymer brushes have been shown to exhibit non-fouling properties against biomolecules. For example, Osterberg et al. reported that PEG brush modifications and adsorption onto a polystyrene surface reduced protein adsorption effectively [4]. More recently, Nagasaki et al. found that surfaces modified with PEG of different molecular weights (6 : 2.5 kDa = 9 : 1 w/w) could strongly suppress nonspecific protein adsorption [5]. As shown in Figure 20.3, this superior antithrombotic property comes from the suppression of biomolecule contact due to higher density PEG graft chains. Even though only hydrophilicity was considered for biocompatibility, this finding shows that interfacial energy and exclusion volume considerably affects non-fouling surface properties.

Figure 20.3 Relationship between protein size and nonspecific adsorption ability on various surfaces. Surfaces modified with PEG at two different molecular weights exhibited superior antifouling properties [5].
While PEG is a nonionic material, ionic immunoevasive biomaterials have also been developed. Ionic immunoevasive biomaterials can be classified as cationic, anionic, and amphoteric. Amphoteric materials can be either copolymers of cationic and anionic monomers or polymers of zwitterionic monomers. Generally, amphoteric materials have better immunoevasive abilities than anionic and cationic materials. Kitano et al. reported that copolymers of methacrylic acid (anionic monomer) and N,N-dimethylaminopropyl methacryl amide (cationic monomer) with a 1 : 1 composition are more biocompatible than other copolymers with the same monomer copolymerization but different ratios [6]. They performed polarized Raman spectroscopy studies, which revealed that despite being an electrolyte, the amphoteric polymer does not disturb hydrogen bonds in the water [7]. Polarized Raman spectroscopy evaluation could detect the deficiency in hydrogen bonds between water molecules by measuring the intensity of OH vibrations. This study indicates that mono-ionic materials disrupted hydrogen bonds in water, whereas zwitterionic materials left these bonds intact. As for the other type of ionic biocompatible material, excluding copolymer of nonionic monomers, commonly used polymers of zwitterionic monomers including sulfobetaines such as sulfobetaine methacrylate (SBMA) [8], phosphobetaines such as poly(2-methacryloyloxyethyl phosphorylcholine) (MPC) [9], and carboxybetaines such as carboxybetaine methacrylate (CBMA) [10], are well known (Table 20.1).
Table 20.1 Ionic biocompatible polymers
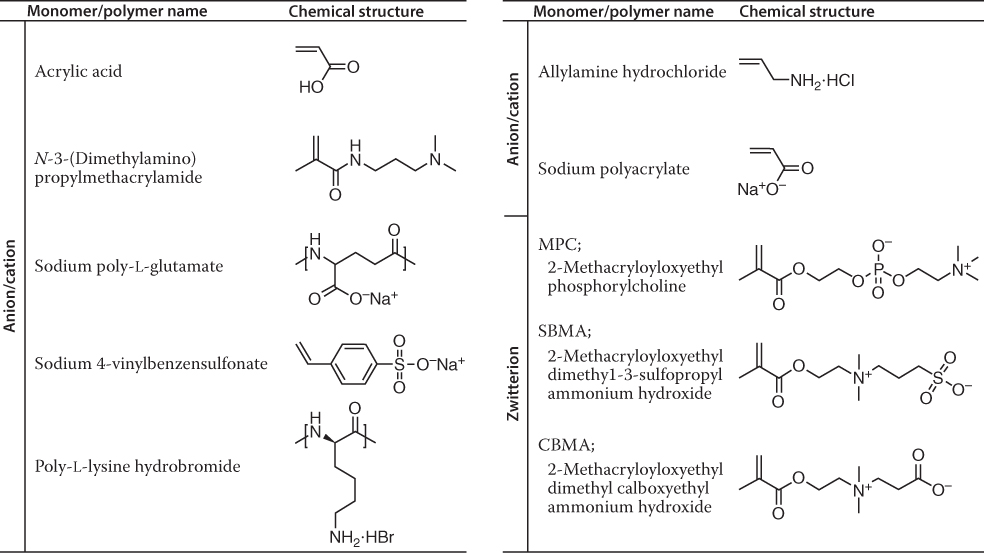 |
In particular, MPC polymers developed by Ishihara and coworkers have been extensively applied to various medical devices, including an artificial joint (Aquala®, Japan Medical Materials), cardiovascular stent (Endeavor®: Medtronic), left ventricular assist device (EVAHEART®, Sun Medical), oxygenator (PrimO2x®: Sorin), and soft contact lenses (Proclear®: Cooper Vision). MPC has a phosphate anion site and quaternary amine cation site that is designed to mimic phosphatidylcholine, which is present in the cell membrane (lipid bilayer membrane) in vivo. In addition to the medical field, MPC is also widely used in cosmetics, shampoos, and textile processing agents. In general, to commercialize biomaterials, cooperation between academic researchers and industries is essential to ensure a stable supply on an industrial scale. In addition, it is necessary for a medical device manufacturer to successfully manufacture a device using this biomaterial and ultimately to evaluate its safety and functionality and be allowed clinical use. That process is extremely difficult; however, MPC is known as the model that broke through these hurdles.
20.3 Immune-Activating Biomaterials
In recent years, new biomaterials have been developed to exploit the immune response of hosts to provide or improve therapeutic effects. The first application of this concept was the use of nonbiological adjuvant materials that increased the host immune response to vaccines. Many adjuvants have been approved for use in clinical treatments; calcium hydroxide and aluminum phosphate have been approved by the Food and Drug Administration (FDA) in the United States, as they strongly activate cellular and humoral immunity. Recent biomaterials have built on this background and aim to improve upon the adjuvant design. For example, adjuvants using biodegradable materials such as γ-polyglutamic acid (γ-PGA) [11], poly(lactic-co-glycolic acid) (PLGA) [12], and poly(ϵ-caprolactone) (PCL) [13] have been reported. This concept is also expected to be effective in antitumor therapies. Generally, antigen delivery to antigen-presenting cells (APCs), protection of antigens, and activation of natural immunity are important mechanisms that affect antitumor immune responses. When designing physically, chemically, and biologically stimuli-responsive materials, it is important to consider spatiotemporal control of antigens in vivo for improving antitumor effects.
Generally, it is important to avoid the induction of danger signals when antigens are delivered to APCs. For example, while major histocompatibility complex (MHC) class I present antigens that originate from the cytoplasm, MHC class II present antigens that originate extracellularly and are exposed in the endolysosomes. This is also observed for toll-like receptor (TLR) proteins, wherein TLR4 and TLR5 function at the cell membrane but TLR7 and TLR9 operate in the endosomes. Generally, the internal pH of endosomes (pH 5.0–6.0) is acidic than biological extracellular fluids (pH 7.4). Although biodegradable materials such as PCL and PLGA break down at internal endosomal pH, the degradation rate is generally not fast enough for effective drug deliveries. Fréchet et al. overcame this problem by using pH-cleavable cross-linkers on anti-DEC205 (dendritic and epithelial cells, 205 kDa) antibodies containing hydroxyethyl acrylamide microparticles [14]. These particles are rapidly hydrolyzed under lysosomal acidic conditions (pH 5) and release antigens into dendritic cells (DCs). Released antigens are recognized by MHC I glycoproteins exposed on DC surfaces and finally induce differentiation of naïve cluster of differentiation (CD) 8+ T cells to cytotoxic T lymphocytes (CTLs) (Figure 20.4a). By contrast, Stayton et al. proposed antigen delivery strategies that mimic virus infection mechanisms. Viruses infect cells by binding to receptors on host cell surfaces and fusing with cell membranes. This lipid membrane fusion phenomenon is caused by hydrophobic structural changes of membrane fusion proteins induced by endosomal pH. After this process, viruses enter the host cytosol and begin self-replication. Stayton et al. focused on polypropylic acid (PPAA), which has a pKa around pH 6.0–6.5, making it hydrophilic at biological pH while dramatically hydrophobic at endosomal pH due to the protonation of carboxyl groups along its backbone [15]. Because of the polymer phase transition, PPAA disrupted lipid bilayer membranes and antigens tethered to PPAA by disulfide bonds were released under endosomal reductive conditions. Exposure of MHC I antigens and activation of CTLs were successful due to the synergetic effect of lysosomal destruction and antigen release (Figure 20.4b) [16]. In addition, targeting to B cells and cytosol delivery were achieved by tethering of anti-CD22 antigen to PPAA [17].
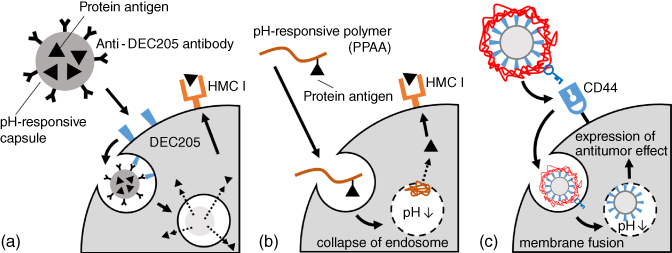
Figure 20.4 (a) Targeting to dendritic cells and release of protein antigen in cells. Particles cross-linked with a pH-responsive cross-linking agent selectively disintegrate and release antigens in the acidic environment of a lysosome. (b) Polypropylic acid (PPAA)is able to disrupt the endosomal membrane due to abrupt protonation at endosomal pH because it has a pKa around pH 6.0–6.5. (c) Antitumor effect of hyaluronic acid (HA) immobilized on HVJ-E. The HA layer improves stability in bloodstream and works as a ligand for CD44, which is overexpressed on cancer cells. In addition, as this layer diffuses in endosomal pH, HVJ-E can fuse with the endosomal membrane due to revealed fusion proteins and induce strict tumor toxicity.
Next, we introduce an example of antitumor immunity induced by virus envelopes. Murine parainfluenza virus type 1 (also known as hemagglutinating virus of Japan (HVJ)) has high cell fusion ability and a 15-kDa RNA genome. Recently, inactivated HVJ has been used as a novel envelope (HVJ-envelope: HVJ-E) [18]. This virus is promising for nucleic acid, antibody, and anti-cancer drug delivery. Kaneda et al. revealed that HVJ-E shows antitumor activity itself. Reported antitumor mechanisms include the activation of natural killer (NK) cells, induction of CTLs in tumor tissues, and the inhibition of regulatory T (Treg) cells [19]. For example, direct injection of HVJ-E into tumor tissues of tumor-bearing mice induce the upregulation of chemokines from tumor tissues, causing the maturation of DCs and activation of NK cells, CD4+ T cells, and CD8+ T cells. This phenomenon is caused by the induction of viral RNA to DCs and the parallel induction of IL6 and IFN-β. To improve its antitumor activity, Okada et al. functionalized HVJ-E with hyaluronic acid (HA) to act as a ligand for CD44 (Figure 20.4c) [20]. The HVJ-E surface was modified using layer-by-layer (LBL) deposition that built up alternating layers of cationic glycol chitosan and anionic HA. These piled polymer layers improve retention time in the bloodstream by inhibiting interactions between HVJ-E and red blood cells and can selectively degrade in cancer tissues under endosomal pH and hyaluronan-targeting enzymes.
It is important to consider various characteristics such as size, shape, hardness, and surface properties when designing biomaterials for adjuvants and DDS carriers as these properties greatly affect their kinetics and biological responsiveness. For example, particles with a size of 25 nm or less administered into the blood are effectively transported to the lymph nodes, but the delivery efficiency of 100 nm particles was only one-tenth lower than that of 25 nm particles (Figure 20.5a) [21]. The effect of particle size on immunity has also been reported. Aluminum salts are commonly used as adjuvants owing to their ability to form submicron aggregates with protein antigens that promote the production of IL1β through the activation of NAKP3 inflammasomes. Sharp et al. investigated various sizes of PCL and PLGA particles that were taken up by dendritic cells and found an upregulation of IL1β for particles smaller than 1 µm [22]. It has also been shown that the shape of particles affects the activity of APCs. Champion et al. fabricated polystyrene particles of various shapes (spheres, oblate ellipsoids, long spheres, elliptical disks, rectangular disks, and UFO type) and investigated the phagocytic response of macrophages [23]. When macrophages in the presence of elliptical disks with a major axis length of 14 µm and a minor axis length of 3 µm adhered to the tip of the long axis, they quickly took up particles within 6 min. However, when the initial adhesion was made on the flat surface of the short axis, the disks were not taken up by the cells (Figure 20.5b). Remodeling of the cytoskeletal protein actin seems to contribute significantly to these phenomena. Biological responsiveness of biomaterials also greatly depends on the physical properties of their surfaces (charge, hydrophobicity, ligand, etc.). For example, cationic nanoparticles can enter cells by transiently puncturing the cell membrane, thereby causing cytotoxicity. It is also known that the degree of hydrophobicity is one of the major triggers in the recognition of biomaterials by TLRs. Verma et al. fabricated gold nanoparticles as small as 5 nm and coated their surfaces with hydrophobic and anionic domains [24]. They prepared particles in which these domains are either randomly or regularly arranged and compared the uptake of these particles by dendritic cells. Particles with regularly arranged domains were taken up three times more than those with randomly arranged ones (Figure 20.5c). Moreover, particles with randomly arranged domains were trapped in endosomes, whereas regularly arranged particles were released into the cytoplasm without going through endocytosis. These results are similar to the cell membrane penetration mechanism of cell-penetrating peptides (CPPs), suggesting the possibility of designing new materials that mimic CPPs.
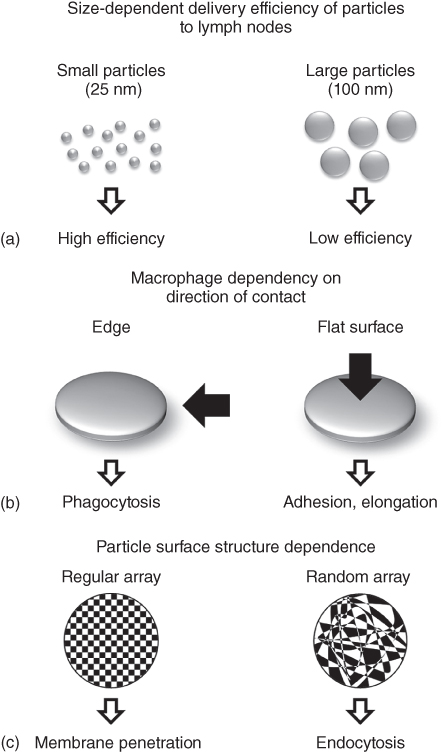
Figure 20.5 Immune response differences in physical, geometrical, and physicochemical properties of particles. (a) Size-dependent delivery efficiency of particles to target. (b) Macrophages show phagocytosis when they adhere to the tip of elliptical particles, but they exhibit adhesion/extension behavior when they adhere to flat surfaces of elliptical particles. (c) For gold nanoparticles modified with hydrophobic and anionic domains, nanoparticles with regularly arranged surface induces cell membrane permeability, whereas nanoparticles with randomly distributed surface are endocytosed by cells.
20.4 Immunosuppressive Biomaterials
In addition to activating immunity, recent biomaterials have been developed to induce immune tolerance for the treatment of transplant rejections and autoimmune diseases. In general, the immune system effectively eliminates foreign substances such as pathogenic microorganisms, but does not respond to self-biomolecules, symbiotic microorganisms, and environmental substances. However, in autoimmune diseases, the immune system misidentifies some molecules as foreign and elicits a response against them. In this case, the immune system can also excessively respond to pathogens, causing tissue injury and immunopathological diseases. For example, chronic inflammatory disorders such as inflammatory bowel disease, allergic reactions, cancer, obesity, diabetes, collagen disease, and Alzheimer's disease are all caused by the impaired maintenance of immune homeostasis. As the tolerance of the immune system is maintained by Treg cells, immunosuppressive biomaterials should induce Treg cells for the treatment of autoimmune diseases caused by excessive, chronic, and uncontrollable immune reactions. In this section, such immunosuppressive biomaterials are discussed.
To induce immune tolerance, researchers have developed strategies for the delivery of adjuvants, antigens, and drugs via various biomaterials. For example, Phillips et al. succeeded in inhibiting immune responses using PLGA particles that contained costimulatory antisense oligonucleotides for DCs and T cells [25]. After recognition and engulfment of these particles, immature DCs (iDCs) presented biologically degraded antigens on their surfaces. These presented antigens were recognized by naive CD4+ T cells and enhanced immune tolerance by suspending autoimmune responses. In addition, this immune tolerance antigen delivery system was used to treat type 1 diabetes (non-obesity diabetes) in mice by reducing APC antigens. In another example, PLGA particles modified with anti-DEC205 antibodies, anti-CD1c antibodies, and P-2 peptides were also developed [26]. This example showed the application of DDS to immunological tolerance, as well as the delivery of various adjuvants and antibodies based on a molecular targeting approach.
Previous immunosuppressive biomaterials discussed earlier simply operate as delivery carriers, and the materials themselves do not have any anti-inflammatory properties. However, in recent years, immunosuppressive materials have been developed, and a few specific examples are introduced here. Stable organic free radicals such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) are often used as catalysts for oxidation reactions in organic synthesis and can act as radical scavengers that detect radical generation reacting systems. Samuni et al. focused on the radical scavenging ability of TEMPO for removing reactive oxygen species (ROS) at inflammatory regions [27]. Normally, large amounts of ROS are produced during inflammation, and exposure to this oxidative stress can cause various diseases such as arteriosclerosis. However, reactive oxygen must be selectively removed as it plays an important role during intracellular aerobic respiration in mitochondria. Therefore, low-molecular-weight substances such as TEMPO diffuse into mitochondria by concentration gradient and work as a toxin. However, Nagasaki and coworkers overcame this issue by tethering TEMPO molecules to PEG polymer chains [28]. It prevented TEMPO from permeating through the cell membrane and enabled the removal of unnecessary ROS in the extracellular environment selectively by PEG tethering. This redox active polymer proved useful in many fields, including medicines, medical devices, cosmetics, luxury goods, and health foods. For example, Nagasaki and coworkers succeeded in improving the cognitive function of mice that model Alzheimer's disease by orally administering TEMPO-functionalized PEG [29]. In addition, TEMPO-functionalized PEG microparticles showed inhibition of nonspecific tissue readhesion after surgery that far exceeds regular PEG [30].
While inflammation suppression by low-molecular drugs is the most common treatment method for inflammatory diseases, new therapies focusing on inflammatory cytokines have recently been attracting attention. For example, anti-IL6 receptor antibodies have shown great achievements in clinical treatments of autoimmune diseases such as rheumatism and Castleman's disease [31]. However, these antibody drugs are extremely expensive; hence, the development of novel cytokine therapies without antibodies was requested by countless patients. These requests motivated several researchers to investigate the anti-inflammatory effects of apoptotic cells as a treatment strategy in place of antibodies. Voll et al. reported that apoptosis induced leukocytes to downregulate inflammatory cytokine production and upregulate anti-inflammatory cytokines for the production of immune cells [32]. Apoptosis is a genetically programmed cell death mechanism and is necessary to regulate embolic development and maintain homeostasis. Many studies also revealed that phospholipid phosphatidylserine (PtdSer) acts as a trigger molecule for these anti-inflammatory effects [33]. This is especially intriguing as most apoptotic cells are simply engulfed by macrophages and cleared by noninflammatory mechanisms [34]. Normally, cell membranes have a lipid bilayer constructed by phospholipids such as electrically neutral sphingomyelin (Sph) and phosphatidylcholine (PtdCho) that occupy the outside membrane and anionically charged phosphatidylethanolamine (PtdEA), PtdSer, and phosphatidylinositol (PtdIno) that are located on the cytosol side [35]. Cell membranes keep this heterogeneous structure by active transportation at inner sites by scramblase [36], and the cytoskeleton [37] is therefore stabilized by anionic phospholipids. However, apoptosis induces the destruction of these highly maintained asymmetrical structures, forcing PtdSer to be exposed on the surface of apoptotic cell membranes, which are then recognized by immune cells [38] (Figure 20.6). Several therapeutic effects of PtdSer liposomes have been reported for chronic heart failure [39], Alzheimer's disease [40], and osteoporosis [41]. These therapeutic effects are based on the conditioning of intracellular signaling factors using only biological responses of immune cells and have been reported to be physiologically and highly effective. Excellent therapeutic effects in vivo have also been shown for many inflammatory diseases, making PtdSer a promising candidate for highly efficient treatments. This therapy is also very cost-effective as it uses inexpensive PtdSer liposomes rather than molecular targeted drugs.

Figure 20.6 Major phospholipids constituting the cell membrane and collapse of asymmetrical distribution of the phospholipid bilayer due to the progression of apoptosis. Sph, sphingomyelin; PtdCho, phosphatidylcholine; PtdEA, phosphatidylethanolamine; PtdSer, phosphatidylserine; PtdIno, phosphatidylinositol.
Pioneering in this field has mostly been based on the use of low-molecular drugs, but more methods to imitate and utilize immune mechanisms and biomolecules have been developed as they became more deeply understood. In addition, new materials conforming to biological mechanisms have shown more effective therapeutic effects, and further development of this field is expected in the near future.
20.5 Conclusions
Improved understanding of regenerative medicine, DDS, and molecular biology served as the foundation for developing novel functional biomaterials. In fact, successful development of synthetic materials that imitate specific cellular functions will strongly influence further advancements in the medical field. For example, the greatest advantage of synthetic materials is their ease of use (e.g., cheapness, stability, safety, adaptability). Even if particle-type biomaterials are stored in a dry state, they are able to disperse promptly when dissolved in water, just before use. Furthermore, the therapeutic effect of biomaterials is not transient unlike those of conventional drug therapies; they can control long-term healing. This may enable complete treatment of a disease and lead to considerable social contribution.
References
- 1 Hench, L. (1980) Science, 208, 826.
- 2 Bailey, F.E. and Koleske, J.V. (1990) Alkylene Oxides and Their Polymers, Medical Dekker, Inc., New York.
- 3 Harris, J.M. (1992) in Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications (ed. J.M. Harris), Springer, Boston, MA.
- 4 Osterberg, E., Bergstrom, K., Holmberg, K., Schuman, T.P., Riggs, J.A., Burns, N.L., Van Alstine, J.M., and Harris, J.M. (1995) J. Biomed. Mater. Res., 29, 741.
- 5 Nagasaki, Y., Kobayashi, H., Katsuyama, Y., Jomura, T., and Sakura, T. (2007) J. Colloid Interface Sci., 309, 524.
- 6 Kitano, H., Nagaoka, K., Tada, S., Gemmei-Ide, M., and Tanaka, M. (2008) Macromol. Biosci., 8, 77.
- 7 Kitano, H., Sudo, K., Ichikawa, K., Ide, M., and Ishihara, K. (2000) J. Phys. Chem. B, 104, 11425.
- 8 Kitano, H., Mori, T., Takeuchi, Y., Tada, S., Gemmei-Ide, M., Yokoyama, Y., and Tanaka, M. (2005) Macromol. Biosci., 5, 314.
- 9 Ishihara, K., Aragaki, R., Ueda, T., Watenabe, A., and Nakabayashi, N. (1990) J. Biomed. Mater. Res., 24, 1069.
- 10 Carr, L.R., Xue, H., and Jiang, S. (2011) Biomaterials, 32, 961.
- 11 Nilsson, J.S., Broos, S., Akagi, T., Akashi, M., Hermansson, A., Cayé-Thomasen, P., Lindstedt, M., and Greiff, L. (2014) Acta Otolaryngol., 134, 1034.
- 12 Jiang, W., Gupta, R.K., Deshpande, M.C., and Schwendeman, S.P. (2005) Adv. Drug Delivery Rev., 57, 391.
- 13 Benoit, M., Baras, B., and Gillard, J. (1999) Int. J. Pharm., 184, 73–84.
- 14 Kwon, Y.J., James, E., Shastri, N., and Fréchet, J.M.J. (2005) Proc. Natl. Acad. Sci. U S A, 102(51): p. 18264.
- 15 Murthy, N., Robichaud, J.R., Tirrell, D.A., Stayton, P.S., and Hoffman, A.S. (1999) J. Controlled Release, 61, 137.
- 16 Yin, X., Hoffman, A.S., and Stayton, P.S. (2006) Biomacromolecules, 7, 1381.
- 17 Berguig, G.Y., Convertine, A.J., Shi, J., Palanca-Wessels, M.C., Duvall, C.L., Pun, S.H., Press, O.W., and Stayton, P.S. (2012) Mol. Pharmaceutics, 9, 3506.
- 18 Kaneda, Y., Yamamoto, S., and Nakajima, T. (2005) Advances in Genetics, vol. 53, Academic Press, p. 307.
- 19 Kurooka, M. and Kaneda, Y. (2007) Cancer Res., 67, 227.
- 20 Okada, T., Uto, K., Sasai, M., Lee, C.M., Ebara, M., and Aoyagi, T. (2013) Langmuir, 29, 7384.
- 21 Reddy, S.T., van der Vlies, A.J., Simeoni, E., Angeli, V., Randolph, G.J., O'Neil, C.P., Lee, L.K., Swartz, M.A., and Hubbell, J.A. (2007) Nat. Biotechnol., 25, 1159.
- 22 Sharp, F.A., Ruane, D., Claass, B., Creagh, E., Harris, J., Malyala, P., Singh, M., O'Hagan, D.T., Pétrilli, V., Tschopp, J., O'Neill, L.A.J., and Lavelle, E.C. (2009) Proc. Natl. Acad. Sci. U.S.A., 106, 870.
- 23 Champion, J.A. and Mitragotri, S. (2006) Proc. Natl. Acad. Sci. U.S.A., 103, 4930.
- 24 Verma, A., Uzun, O., Hu, Y., Hu, Y., Han, H.-S., Watson, N., Chen, S., Irvine, D.J., and Stellacci, F. (2008) Nat. Mater., 7, 588.
- 25 Phillips, B., Nylander, K., Harnaha, J., Machen, J., Lakomy, R., Styche, A., Gillis, K., Brown, L., Lafreniere, D., Gallo, M., Knox, J., Hogeland, K., Trucco, M., and Giannoukakis, N. (2008) Diabetes, 57, 1544.
- 26 Lewis, J.S., Zaveri, T.D., Crooks Ii, C.P., and Keselowsky, B.G. (2012) Biomaterials, 33, 7221.
- 27 Samuni, A., Krishna, C.M., Mitchell, J.B., Collins, C.R., and Russo, A. (1990) Free Radical Res. Commun., 9, 241.
- 28 Toh, K., Yoshitomi, T., Ikeda, Y., and Nagasaki, Y. (2011) Sci. Technol. Adv. Mater., 12, 065001.
- 29 Chonpathompikunlert, P., Yoshitomi, T., Vong, L.B., Imaizumi, N., Ozaki, Y., and Nagasaki, Y. (2015) PLoS One, 10, e0126013.
- 30 Yoshitomi, T. and Nagasaki, Y. (2015) Biomater. Sci., 3, 810.
- 31 Nishimoto, N., Kishimoto, T., and Yoshizaki, K. (2000) Ann. Rheum. Dis., 59, i21.
- 32 Voll, R.E., Herrmann, M., Roth, E.A., Stach, C., Kalden, J.R., and Girkontaite, I. (1997) Nature, 390, 350.
- 33 Gaipl, U.S., Beyer, T.D., Baumann, I., Voll, R.E., Stach, C.M., Heyder, P., Kalden, J.R., Manfredi, A., and Herrmann, M. (2003) Immunobiology, 207, 73.
- 34 Bagalkot, V., Deiuliis, J.A., Rajagopalan, S., and Maiseyeu, A. (2016) Adv. Drug Delivery Rev., 99, Part A, 2.
- 35 Verkleij, A. and Post, J. (2000) J. Membr. Biol., 178, 1.
- 36 Tanaka, K., Fujimura-Kamada, K., and Yamamoto, T. (2011) J. Biochem., 149, 131.
- 37 Vanags, D.M., Pörn-Ares, M.I., Coppola, S., Burgess, D.H., and Orrenius, S. (1996) J. Biol. Chem., 271, 31075.
- 38 Somersan, S. and Bhardwaj, N. (2001) J. Cell Biol., 155, 501.
- 39 Harel-Adar, T., Mordechai, T.B., Amsalem, Y., Feinberg, M.S., Leor, J., and Cohen, S. (2011) Proc. Natl. Acad. Sci. U.S.A., 108, 1827.
- 40 Hashioka, S., Han, Y.-H., Fujii, S., Kato, T., Monji, A., Utsumi, H., Sawada, M., Nakanishi, H., and Kanba, S. (2007) Free Radical Biol. Med., 42, 945.
- 41 Wu, Z., Ma, H.M., Kukita, T., Nakanishi, Y., and Nakanishi, H. (2010) J. Immunol., 184, 3191.