
Chapter 17
Materials Nanoarchitectonics: Drug Delivery System
Yohei Kotsuchibashi
Shizuoka Institute of Science and Technology, Faculty of Science and Technology, Department of Materials and Life Science, 2200-2 Toyosawa, Fukuroi, Shizuoka, 437-8555, Japan
17.1 Introduction
The origin of the controlled drug delivery system (DDS) using polymeric biomaterials dates back to the 1960s, with materials sizing continuously decreasing from macro (1970s) to micro (1980s), and nano (1990s to present) [1, 2]. Using the Ringsdorf model for polymer drugs [3], various types of nanoparticles were produced in the 2000s. The development of nanoparticles was supported by three key technologies: (i) PEGylation; (ii) passive targeting, that is, solid tumor accumulation via the enhanced permeability and retention (EPR) effect; (iii) active targeting, that is, specific cell interaction by ligand-conjugated polymeric materials. PEGylation is the covalent attachment between poly(ethylene glycol) (PEG) and another substance; examples include small molecules, peptides, proteins, antibodies, and their fragments, and oligonucleotides [4–9]. This coupling decreases immunogenicity and increases blood-circulation time [10]. The EPR effect is a phenomenon whereby nanoparticles tend to accumulate in tumor tissue because of the gaps in blood vasculature around the tumor that are caused by the rapid growth of cancer cells and their underdeveloped lymphatic vasculature [11]. For even more effective accumulation, nanocarriers with a superficial affinity site that interacts with target cancer cells have been developed. To achieve controlled drug delivery, precisely designed materials are required. In this chapter, the focus is on the most recent materials developed for DDS. The chapter is divided into three parts: (i) diagnosis from tissues to the organelles using nanomaterials, (ii) current thermoresponsive drug carriers, and (iii) smart nanocarriers for specific drugs derived from benzoxaborole.
17.1.1 Diagnosis from Tissues to the Organelles Using Nanomaterials
The diagnosis of disease in its early stage is very useful from a preventative and cost-efficiency point. The biomaterials used in the field of diagnosis are required to have high stability and brightness [12]. Clinically, there are many different types of diagnostic systems such as optical imaging, magnetic resonance imaging (MRI), computed tomography (CT), ultrasound (US), positron emission tomography (PET), and single photon emission computed tomography (SPECT). They all have their own unique advantages, but also intrinsic limitations [13, 14]. The application of novel imaging materials to existing medical equipment offers a clear advantage when considering the practical applications of these biomaterials. Nanomaterials with an affinity for target cells can recognize disease tissues. For example, the receptor of folic acid (Mw = 441.40) is expressed on the cell surface in various types of malignancies of the ovary, brain, kidney, breast, lung, and myeloid cell. Therefore, folic acid molecules combined with drug carriers (e.g., copolymers, micelles, and liposomes) can engage in active targeting and result in cancer accumulation [15]. Interestingly, the shape of the material can also affect its accumulation in organs. Decuzzi et al. prepared silica materials with different shapes (spherical, hemispherical, discoidal, and cylindrical) to investigate their accumulation in organs by injection via the tail vein; a large accumulation of discoidal particles was observed in the majority of organs, especially the brain, spleen, lungs, and heart. It is hypothesized that this specialized form interacts strongly with vessel walls [16]. Systemic, local, and oral administration of dosage forms can also be optimized for drug delivery [17]. The oral route of administration is particularly advantageous, as this form of treatment is minimally invasive. The floating particles of quantum dots (QDs) in the air can also be adsorbed via the respiratory tracts, with their excretive routes dependent on particle size. As QDs are only 34 nm in diameter, they are immediately transferred to the lymph nodes through the alveoli. Those with a diameter less than 6 nm were excluded from the kidney via the blood stream [18]. The dosage routes for DDS will be discussed actively in the future. Wilhelm et al. analyzed the in vivo accumulation of administered nanoparticles in tumor tissues using over a hundred articles from the last decade [19]. According to their results, a median of 0.7% of the injected dose of nanoparticles was delivered to the targeted tumor tissues, meaning that only 7 out of 1000 nanoparticles accumulated in the solid tumor. In addition, the delivery efficiency was affected by various characteristics, including particle diameter (10–100 nm: 0.6%; >200 nm: 0.4%), zeta potential (negative: 0.5%; neutral: 0.7%; positive: 0.6%), shape (spherical: 0.7%; rod: 0.8%), cancer type, and tumor model. Median delivery efficiency has not improved in the past 10 years. Therefore, precise material design will likely be the breakthrough in this field; multidimensional data such as the simulation of particle pharmacokinetics and analysis of big-data on the biomaterials will be required with the help of supercomputers. Recently, with the development of fluorescent materials, accumulation techniques have been utilized to image/analyze local information on the organelles of a single living cell. A temperature-responsive polymer with fluorescence units was used to monitor the local temperature in living cells, with differences in fluorescence intensity observed depending on organelle [20]. There was a difference of 0.96 °C in the average temperature between the nucleus and cytoplasm, while localized high temperatures were observed near the mitochondria. Tracking the activities of living mitochondria is significant because of their important roles in cell functioning, including energy production, lipid metabolism, innate immunity, aging, and cell death. Hu et al. focused on phenomena that often occur during imbalanced reactive oxygen species (ROS) production in dysfunctional mitochondria, using a probe that could detect endogenous superoxide (O2•−) in living cells [21]. The prepared fluorescent probe was also used for O2•− detection in zebrafish embryos in vivo. A series of mitochondria-selective probes were also reported. Another example involves a quantitative probe prepared for assessing adenosine triphosphate (ATP) levels in the organelles of living cells [22]. The probe was an ϵ-subunit (ATP-binding protein) of the bacterial F0F1-ATP synthase sandwiched between cyan- and yellow-fluorescent proteins, which could detect ATP concentrations (7.4 µM–3.3 mM) in the cells as a fluorescence resonance energy transfer (FRET), resulting in structural changes via interactions with ATP. Interestingly, the mitochondrial matrix in HeLa cells had a significantly lower ATP level than the cytoplasm and nucleus, even though the mitochondrion is the cell's ATP-producing organelle. This low ATP level in mitochondria was also observed in non-cancer NIH 3T3 cells. It was hypothesized that this phenomenon was caused by or related to adenine nucleotide translocator pumps. Chang et al. prepared pH-responsive fluorescent hybrid nanoparticles for real-time cell labeling and endocytosis tracking [23]. The hybrid nanoparticles were composed of amphiphilic block copolymers, a silica-rich shell, rapid cell-penetrating peptides (HIV-1 TAT), and an encapsulated pH-responsive fluorophore. The local fluorescence intensity increased in acidic lysosomes in human breast cancer cells (MCF-7) as incubation time increased (1, 4, and 8 h). These unique fluorescent probes provided insight into the intracellular environment and biological interactions occurring at the scale of a single living cell.
17.1.2 Current Thermoresponsive Drug Carriers
Previously, drug release from nanoparticles was mainly triggered by drug diffusion. To achieve switchable “on–off” drug release, nanoparticles have been combined with stimuli-responsive (or smart) polymers. Stimuli-responsive polymers can recognize a slight change in the temperature, pH, light, magnetic field, or molecular concentration of their external environment [24, 25]. Among them, temperature is particularly useful because it is possible to use body temperature (including local variable temperatures in the tissues and cells) as the trigger (Figure 17.1). It is also relatively easy to control the local body temperature externally (e.g., hyperthermia). There have been excellent reviews on thermoresponsive drug carriers that can encapsulate various types of drugs, including low-molecular-weight compounds, synthetic polymers, proteins, nucleic acids, and inorganic materials [26]. The focus here is on recent thermoresponsive carriers. Thermoresponsive polymers are mainly classified into two types based on their thermoresponsive behavior: lower critical solution temperature (LCST) and upper critical solution temperature (UCST) types [27]. The LCST types of thermoresponsive polymers can dissolve in an aqueous solution at a temperature lower than the LCST; above the LCST, these polymers can become hydrophobic because of accelerated polymeric intra- and intermolecular hydrophobic interactions. This reversible solubility change has been widely applied in the development of model proteins, triggers for self-assemblies, “on–off” switches for protein activities, cell sheet technologies, drug carriers, column chromatography, and sensors, among others [28, 29]. In contrast, UCST copolymers can dissolve in a solvent above the UCST and are insoluble below the UCST. This phenomenon occurs via specific interactions such as hydrogen bonding and electrostatic interactions [27, 30]. Recently, dual thermoresponsive copolymers that have two LCST and/or UCTS have been reported [31]. Thermoresponsive copolymers have been actively investigated in preclinical studies. For example, Li et al. prepared an immune-micelle with poly(N-isopropyl acrylamide) (PNIPAAm) in the shell for the treatment of gastric cancer [32]. Anti-Her2 antibody Fab fragments were anchored in the micelle. The immune-micelle was injected intravenously into Balb/c nude mice with gastric cancer; consequently, the tumor volume after experimentation was five times less than that of the control. Feng et al. applied a thermoresponsive rodlike micelle to in vivo gene therapy for nucleus pulposus regeneration [33]. The rodlike micelle was loaded with hemooxygenase-1 plasmid DNA and directly injected into the rat disk tissue space for gene therapy; the therapy produced a high healing effect by reducing inflammatory responses and increasing the glycosaminoglycan content. Thermoresponsive supramaterials that are connected via non-covalent bonding, such as hydrogen bonds and electrostatic interactions, have also been used as drug carriers. Chi et al. prepared polymeric vesicles consisting of supra-amphiphilic macromolecules, which release the encapsulated model drug calcein by cooling to 25 °C or heating to 60 °C [34]. The thermoresponsive copolymers that are especially used as drug carriers are required to have not only biocompatibility but also rapid response, small hysteresis during the heating/cooling process, and a nondependent response with regard to concentrations and ionic strengths. The ethylene glycol types of thermoresponsive copolymers satisfy the abovementioned conditions [35]. Body temperature is constantly maintained via homeostasis, but it is known that some tissues such as tumors and sites of infections have naturally elevated temperatures compared with that of normal tissues [26]. There are also temperature differences among different organs. For example, the temperature inside the liver is usually around 38 °C, which is higher than that inside the intestine. Using this temperature difference, efficient drug delivery to the intestine was achieved using polymeric carriers transported via the bile duct, as the temperature of the bile duct is similar to that of the surrounding liver tissue [36]. Nanoparticle formation is usually affected by heating processes, whether they be direct heating (10 → 40 °C) or gradual heating (10 → 20 → 30 → 40 °C, with a 15-min interval at each temperature). Our ethylene glycol–based block copolymers did not demonstrate a large size difference between heating processes, but the dissolution of viscous polymers in PBS still requires several tens of minutes [37]. In the future, polymeric nanoparticles will be required to not only have the ability to be stored in solid (highly viscous) state but also rapid dissolution/preparation as biomaterials [38]. Thermoresponsive materials also have an advantage in that heat is relatively easy to apply to specific tissues from outside the body. For example, a heat source of iron particles excited via magnetic field has been incorporated into a system of thermoresponsive organic/inorganic hybrid materials and applied to drug carriers, diagnosis, and separation systems [39]. Examples of other heat sources include light energy that is converted into heat through metals (Au, Ag, Pt, Ge, etc.) [40], dyes [41], carbon materials [42], and semiconductors [43]. Among them, near-infrared (NIR) light (700–1100 nm) is a useful photothermal therapy with regard to DDS because of its noninvasive and relatively deep-tissue penetration. Gold nanomaterials are one of the most commonly used materials with NIR light via the strong surface plasmon resonance (SPR) and scatter, as a high photothermal effect can be produced [44]. It is possible to induce cell death (apoptosis and necrosis) via NIR light, resulting in the heating of Au nanomaterials [45]. NIR light has been applied to modified red blood cells (RBCs) as the trigger for encapsulated drug release [46]. The RBC is an excellent drug carrier because of its high biocompatibility, long circulating half-life (120 days in humans), membrane flexibility/stability, absence of nucleus/organelles/DNA, high surface-to-volume ratio, and easy modification of its cell membrane [47]. The photothermal nanocomplex indocyanine green-bovine serum albumin and doxorubicin (DOX) were encapsulated into RBCs modified with the RGD (Arg-Gly-Asp) peptide for tumor targeting via angiogenic endothelium αγβ3-integrin. Eighty percent of DOX was released after 5 min under NIR light irradiation. Thus, it can be easily seen why thermoresponsive drug carriers and their heating processes will continue to attract research as biomaterials.
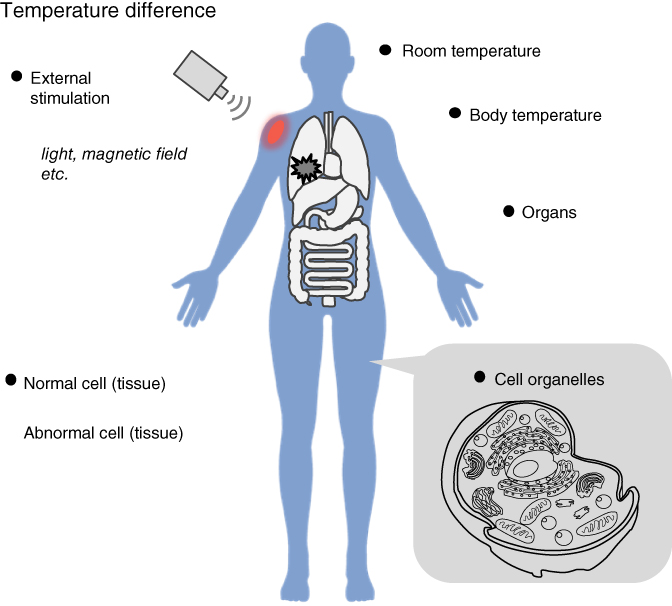
Figure 17.1 Temperature difference in a living body.
17.1.3 Smart Nanocarriers for Benzoxaborole-Based Drugs
An optimized carrier for specific drugs can be designed using the concept of nanoarchitectonics [48]. Recently, benzoxaborole and its boron-containing derivatives have attracted attention for their possible use as therapeutic drugs for various types of diseases [49]. The first report on benzoxaborole was published in 1957 [50]. In 2006, benzoxaborole began to garner attention as a compound that can strongly interact with cis-diols as compared to the typical phenyl boronic acid (PBA) [51]. The Ka (M−1) values of benzoxaborole with saccharides in pH 7.4 were 2.6 (d-fructose), 5.6 (d-glucose), and 3.3 (NeuSAc) times higher than PBA [52]. This strong interaction is achieved via a strain structure that is induced by the compound's five-membered oxaborole ring, which results in a lower pKa [53]. AN2690 is benzoxaborole derivative that contains a fluorine, and was reported as an effective treatment for onychomycosis in 2007; it can obstruct the protein synthesis of the fungus via interacting with fungal fungous leucyl tRNA [54]. AN2690 was approved by the Food and Drug Administration (US FDA) in 2014, and was marketed as the drug tavaborole. Other examples of benzoxaborole derivatives include AN2728 (crisaborole), which has been complemented in a phase III clinical trial as an anti-inflammatory drug. Benzoxaborole-based drug candidates have also been reported to have the following effects: antibacterial (gram-negative or positive), antiparasitic, anti-inflammatory, anti-malarial, and anti-African sleeping sickness [49]. While almost all arylboronic acid derivatives show low mammalian cell toxicity, there are reports that these derivatives show mutagenicity against bacteria in the Ames test (benzoxaborole was nonmutagenic) [55, 56]. The underlying explanation for this difference has not yet become clear. The relationship between mutagenicity and arylboronic acids was investigated via the chemical shifts of 11B nuclear magnetic resonance (NMR) spectra [57]. According to the report, the mutagenicity was observed in arylboronic acids with a 11B NMR peak at 29.5–28 ppm. Research on benzoxaborole derivatives has mainly been focused on its efficacy as a medicine, with few reports on how these low-molecular-weight drugs can be transported to their intended target. Therefore, carriers of benzoxaborole derivatives based on DDS concepts will be required in the future. From the point of view of encapsulation in drug carriers, the reversible covalent bond of the benzoxaborole derivatives with diol groups is an advantage when designing complex drug release profiles (Figure 17.2). There are reports of different approaches to drug carriers for benzoxaborole derivatives, such as modifications, encapsulations, prodrugs, and organic–inorganic hybrids. Benzoxaborole and tavaborole (AN2690) were simply encapsulated in poly-l-lactic acid (PLLA) films, which were applied to the MDA-MB-231 cancer cells to investigate their effects on cell migration, growth, and morphology [58]. The diffusion profiles of the encapsulated benzoxaborole drugs from the films fit the Fickian diffusional model. There are few papers on benzoxaborole derivatives as anticancer drugs; PBA, for example, has been reported to be able to reduce the migration of breast and prostate cancer cells [64, 65]. Cancer cells tended toward the apoptotic state after treatment with free AN2690. The PLLA films loading the benzoxaborole were floated in the media in order to avoid direct interaction with the cancer cells, and the results demonstrated inhibition of cell migration, an effect similar to that of the free drug. Inorganic carriers for benzoxaborole via the coordination network were reported by the same group [59]. Mg–Al-layered double hydroxide was selected as the biodegradable inorganic matrix, and almost 100% of the encapsulated benzoxaborole was observed to be released after 100 min, which was a more rapid release than that of the PLLA films. We synthesized benzoxaborole-based methacrylamide monomers and their copolymers [61–63]. Reversible covalent bonding with diol groups was inherited from the parent benzoxaborole group in these copolymers. Using this property, benzoxaborole-based polymeric gels were achieved at pH 7.4 by mixing the diol with glyco-polymers [61]. The gel morphology was controlled from simple bulks to nanoparticles via polymer concentrations. The functionality of benzoxaborole-based polymers (BOP) could even be applied to mixed gels; three different functionalities, that is, temperature, pH, and glucose, were successfully added to control gel formation/collapse. The mixed BOP and glyco-polymer gel system was combined with photoinduced proton transfer chemistry (photoacid generators (PAGs): o-nitrobenzaldehyde) for local and remote control of gel disintegration [62]. PAGs can rapidly release protons by UV irradiation and produce local pH change in the solution/gel. The pH decrease led to the breakage of diol-benxozaborole bonding. The BOP principle was also applied to the functionalization of poly(ethylene-co-vinyl alcohol) (EVOH), which is used in a wide range of fields, such as food packaging, funnel tanks, and medical applications because of its high gas barrier and biocompatibility [66, 67]. The functionalized EVOH nanofiber meshes with BOP, with its pH-responsive COOH groups prepared by electrospinning. The modified EVOH nanofiber meshes showed a high stability in acidic or alkaline aqueous solutions. After a 24 h immersion test, over 97% of EVOH and BOP remained within the nanofiber mesh. The anionic-charged nanofiber mesh was strongly dyed by a model drug of methylene blue via electrostatic interaction at pH 7.4. The dye was rapidly released from the fibers under acidic conditions [63]. Using BOPs, Mahalingam et al. proposed a HIV neutralization system targeting the mannose groups of gp120 protein on the virus [60]. The BOP-glycopolymer nanoparticle was prepared with an antigen to trigger activated T cells for cancer immunotherapy and vaccine development [68]. The interaction of benzoxaborole derivatives with diol groups is strongly affected not only by the derivatives but also by the structures of the diol compounds. The stereoisomerism of the sugars and benzoxaborole was investigated using BOPs and several types of sugar polymers [69]. The interaction force was found to be the greatest in mannose, followed by galactose and glucose; the weak interaction of glucose was due to the presence of a 4,6-diol without a cis-diol group. The benzoxaborole derivatives require release from the polymer chains to display their original efficacy as drugs, that is, prodrug. Ke et al. prepared a doxorubicin (DOX) hanging polycarbonate micelle by incorporating a pH-breakable interaction between the NH2 and aldehyde groups [70]. The fixed DOX in the micelle was enhanced for intracellular release, which is effective in the treatment of cancer with multidrug resistance. There are also benzoxaborole derivatives with NH2 groups, allowing for the application of these breakable groups to the prodrug design of benzoxaborole derivatives. In this way, benzoxaborole derivatives can be combined with DDS, and their reversible covalent bonds applied to the design of effective loading/release systems.

Figure 17.2 Benzoxaborole derivatives loading or bearing materials for drug delivery system. (A) Chemical structures of (a) benzoxaborole and (b) its derivatives. (B) PLLA film.
(Sene et al. 2016 [58]. Reproduced with permission of Royal Society of Chemistry.) and (C) Mg–Al-layered double hydroxide.
(Sene et al. 2015 [59]. Reproduced with permission of American Chemical Society.) with benzoxaborole derivatives. (D) HIV neutralization.
(Mahalingam et al. 2011 [60]. Reproduced with permission of American Chemical Society.). Polymeric benzoxaborole-based (E) nanogel.
(Kotsuchibashi et al. 2013 [61]. Reproduced with permission of American Chemical Society.), (F) gels.
(Kotsuchibashi et al. 2015 [62]. Reproduced with permission of American Chemical Society.), and (G) nanofiber.
(Adapted from Kotsuchibashi and Ebara 2016 [63].)
17.2 Conclusion and Future Trends
This chapter focused on materials used to create DDS. With the development of nanotechnology, the targets of drug delivery have been getting smaller and smaller, from organs to cell organelles. Now, controlling material sizing, shapes, surface properties, and functionalities is relatively easy, and we can embody the materials from creative intracerebral designs. High accumulation of nanomaterials in the targeted tissues and cells in vivo will be possible with these excellent designs. Nanocarriers with multiple stimuli-responsive properties will play an important role in not only targeted drug delivery but also in controlled drug loading/release. In addition, data on exclusive nanocarriers for specific drugs will continue to be actively reported (e.g., benzoxaborole derivatives using reversible covalent bonds). And, these revolutionary nanomaterials are certain to signal the beginning of a new stage in the design of DDS.
References
- 1 Hoffman, A.J. (2008) Controlled Release, 132, 153–163.
- 2 Nishiyama, N. and Kataoka, K. (2006) Adv. Polym. Sci., 193, 67–101.
- 3 Ringsdorf, H.J. (1975) Polym. Sci., 51, 135–153.
- 4 Zhao, H., Rubio, B., Sapra, P., Wu, D., Reddy, P., Sai, P., Martinez, A., Gao, Y., Lozanguiez, Y., Longley, C., Greenberger, L.M., and Horak, I.D. (2008) Bioconjugate Chem., 19, 849–859.
- 5 Pasut, G. and Veronese, F.M. (2007) Prog. Polym. Sci., 32, 933–961.
- 6 Woodburn, K.W., Holmes, C.P., Wilson, S.D., Fong, K.L., Press, R.J., Moriya, Y., and Tagawa, Y. (2012) Xenobiotica, 42, 660–670.
- 7 Wang, Y.S., Youngster, S., Grace, M., Bausch, J., Bordens, R., and Wyss, D.F. (2002) Adv. Drug Delivery Rev., 54, 547–570.
- 8 Chapman, A.P. (2002) Adv. Drug Delivery Rev., 54, 531–545.
- 9 Ng, E.W., Shima, D.T., Calias, P., Cunningham, E.T. Jr., Guyer, D.R., and Adamis, A.P. (2006) Nat. Rev. Drug Discovery, 5, 123–132.
- 10 Ikeda, Y. and Nagasaki, Y. (2014) J. Appl. Polym. Sci., 131, 40293.
- 11 Maeda, H., Wu, J., Sawa, T., Matsumura, Y., and Hori, K. (2000) J. Controlled Release, 65, 271–284.
- 12 Burns, A., Ow, H., and Wiesner, H. (2006) Chem. Soc. Rev., 35, 1028–1042.
- 13 Willmann, J.K., van Bruggen, N., Dinkelborg, L.M., and Gambhir, S.S. (2008) Nat. Rev. Drug Discovery, 7, 591–607.
- 14 Lee, D.-E., Koo, H., Sun, I.-C., Ryu, J.H., Kim, K., and Kwon, I.C. (2012) Chem. Soc. Rev., 41, 2656–2672.
- 15 Bae, Y. and Kataoka, K. (2009) Adv. Drug Delivery Rev., 61, 768–784.
- 16 Decuzzi, P., Godin, B., Tanaka, T., Lee, S.-Y., Chiappini, C., Liu, X., and Ferrari, M. (2010) J. Controlled Release, 141, 320–327.
- 17 Tibbitt, M.W., Dahlman, J.E., and Langer, R. (2016) J. Am. Chem. Soc., 138, 704–717.
- 18 Choi, H.S., Ashitake, Y., Lee, J.H., Kim, S.H., Matsui, A., Insin, N., Bawendi, M.G., Semmler-Behnke, M., Frangioni, J.V., and Tsuda, A. (2010) Nat. Biotechnol., 28, 1300–1303.
- 19 Wilhelm, S., Tavares, A.J., Dai, Q., Ohta, S., Audet, J., Dvorak, H.F., and Chan, W.C.W. (2016) Nat. Rev. Mater., 1, 16014.
- 20 Okabe, K., Inada, N., Gota, C., Harada, Y., Funatsu, T., and Uchiyama, S. (2012) Nat. Commun., 3, 705.
- 21 Hu, J.J., Wong, N.K., Ye, S., Chen, X., Lu, M.Y., Zhao, A.Q., Guo, Y., Ma, A.C.H., Leung, A.Y.H., Shen, J., and Yang, D. (2015) J. Am. Chem. Soc., 137, 6837–6843.
- 22 Imamura, H., Nhat, K.P.H., Togawa, H., Saito, K., Iino, R., Kato-Yamada, Y., Nagai, T., and Noji, H. (2009) Proc. Natl. Acad. Sci. U.S.A., 106, 15651–15656.
- 23 Chang, S., Wu, X., Li, Y., Niu, D., Gao, Y., Ma, Z., Gu, J., Zhao, W., Zhu, W., Tian, H., and Shi, J. (2013) Biomaterials, 34, 10182–10190.
- 24 Stuart, M.A.C., Huck, W.T.S., Genzer, J., Müller, M., Ober, C., Stamm, M., Sukhorukov, G.B., Szleifer, I., Tsukruk, V.V., Urban, M., Winnik, F., Zauscher, S., Luzinov, I., and Minko, S. (2010) Nat. Mater., 9, 101–113.
- 25 Elsabahy, M. and Wooley, K.L. (2012) Chem. Soc. Rev., 41, 2545–2561.
- 26 Karimi, M., Zangabad, P.S., Ghasemi, A., Amiri, M., Bahrami, M., Malekzad, H., Asl, H.G., Mahdieh, Z., Bozorgomid, M., Ghasemi, A., Boyuk, M.R.R.T., and Hamblin, M.R. (2016) ACS Appl. Mater. Interfaces, 8, 21107–21133.
- 27 Roy, D., Brooks, W.L.A., and Sumerlin, B.S. (2013) Chem. Soc. Rev., 42, 7214–7243.
- 28 Akimoto, J., Nakayama, M., and Okano, T. (2014) J. Controlled Release, 193, 2–8.
- 29 Kikuchi, A. and Okano, T. (2002) Prog. Polym. Sci., 27, 1165–1193.
- 30 Seuring, J. and Agarwal, S. (2012) Macromol. Rapid Commun., 33, 1898–1920.
- 31 Kotsuchibashi, Y., Ebara, M., Aoyagi, T., and Narain, R. (2016) Polymers, 8, 380.
- 32 Li, W., Zhao, H., Qian, W., Li, H., Zhang, L., Ye, Z., Zhang, G., Xia, M., Li, J., Gao, J., Li, B., Kou, G., Dai, J., Wang, H., and Guo, Y. (2012) Biomaterials, 33, 5349–5362.
- 33 Feng, G., Chen, H., Li, J., Huang, Q., Gupte, M.J., Liu, H., Song, Y., and Ge, Z. (2015) Biomaterials, 52, 1–13.
- 34 Chi, X., Yu, G., Shao, L., Chen, J., and Huang, F. (2016) J. Am. Chem. Soc., 138, 3168–3174.
- 35 Lutz, J.-F., Akdemir, Ö., and Hoth, A. (2006) J. Am. Chem. Soc., 128, 13046–13047.
- 36 Wang, X., Li, S., Wan, Z., Quan, Z., and Tan, Q. (2014) Int. J. Pharm., 463, 81–88.
- 37 Kotsuchibashi, Y., Ebara, M., Hoffman, A.S., Narain, R., and Aoyagi, T. (2015) Polym. Chem., 6, 1693–1697.
- 38 Yoshida, Y., Takahashi, A., Kuzuya, A., and Ohya, Y. (2014) Polym. J., 46, 632–635.
- 39 Gupta, A.K. and Gupta, M. (2005) Biomaterials, 26, 3995–4021.
- 40 Zhao, W. and Karp, J.M. (2009) Nat. Mater., 8, 453–454.
- 41 Zan, M., Li, J., Huang, M., Lin, S., Luo, D., Luo, S., and Ge, Z. (2015) Biomater. Sci., 3, 1147–1156.
- 42 Levi-Polyachenko, N.H., Merkel, E.J., Jones, B.T., Carroll, D.L., and Stewart, J.H. IV (2009) Mol. Pharmaceutics, 6, 1092–1099.
- 43 Li, W., Zamani, R., Rivera Gil, P., Pelaz, B., Ibáñez, M., Cadavid, D., Shavel, A., Alvarez-Puebla, R.A., Parak, W.J., Arbiol, J., and Cabot, A. (2013) J. Am. Chem. Soc., 135, 7098–7101.
- 44 Ahmad, R., Fu, J., He, N., and Li, S. (2016) J. Nanosci. Nanotechnol., 16, 67–80.
- 45 Ali, M.R.K., Ali, H.R., Rankin, C.R., and El-Sayed, M.A. (2016) Biomaterials, 102, 1–8.
- 46 Sun, X., Wang, C., Gao, M., Hu, A., and Liu, Z. (2015) Adv. Funct. Mater., 25, 2386–2394.
- 47 Villa, C.H., Anselmo, A.C., Mitragotri, S., and Muzykantov, V. (2016) Adv. Drug Delivery Rev., 106, 88–103.
- 48 Ariga, K., Kawakami, K., Ebara, M., Kotsuchibashi, Y., Ji, Q., and Hill, J.P. (2014) New J. Chem., 38, 5149–5163.
- 49 Adamczyk-Woźniak, A., Borys, K.M., and Sporzyński, A. (2015) Chem. Rev., 115, 5224–5247.
- 50 Torssell, K. (1957) Ark. Kemi, 10, 507–511.
- 51 Dowlut, M. and Hall, D.G. (2006) J. Am. Chem. Soc., 128, 4226–4227.
- 52 Ellis, G.A., Palte, M.J., and Raines, R.T. (2012) J. Am. Chem. Soc., 134, 3631–3634.
- 53 Zhdankin, V.V., Persichini, P.J., Zhang, L., Fix, S., and Kiprof, P. (1999) Tetrahedron Lett., 40, 6705–6708.
- 54 Rock, F.L., Mao, W., Yaremchuk, A., Tukalo, M., Crepin, T., Zhou, H., Zhang, Y.K., Hernandez, V., Akama, T., Baker, S.J., Plattner, J.J., Shapiro, L., Martinis, S.A., Benkovic, S.J., Cusack, S., and Alley, M.R. (2007) Science, 316, 1759–1761.
- 55 Scott, H. and Walmsley, R.M. (2015) Mutat. Res., 777, 68–72.
- 56 Ciaravino, V., Plattner, J., and Chanda, S. (2013) Environ. Mol. Mutagen., 54, 338–346.
- 57 Pellizzaro, M.L., Covey-Crump, E.M., Fisher, J., Werner, A.-L.D., and Williams, R.V. (2015) Chem. Res. Toxicol., 28, 1422–1426.
- 58 Sene, S., McLane, J., Schaub, N., Bégu, S., Mutin, P.H., Ligon, L., Gilbert, R.J., and Laurencin, D. (2016) J. Mater. Chem. B, 4, 257–272.
- 59 Sene, S., Bégu, S., Gervais, C., Renaudin, G., Mesbah, A., Smith, M.E., Mutin, P.H., Lee, A., Nedelec, J.-M., Bonhomme, C., and Laurencin, D. (2015) Chem. Mater., 27, 1242–1254.
- 60 Mahalingam, A., Geonnotti, A.R., Balzarini, J., and Kiser, P.F. (2011) Mol. Pharmaceutics, 8, 2465–2475.
- 61 Kotsuchibashi, Y., Agustin, R.V.C., Lu, J.-Y., Hall, D.G., and Narain, R. (2013) ACS Macro Lett., 2, 260–264.
- 62 Kotsuchibashi, Y., Ebara, M., Sato, T., Wang, Y., Rajender, R., Hall, D.G., Narain, R., and Aoyagi, T. (2015) J. Phys. Chem. B, 119, 2323–2329.
- 63 Kotsuchibashi, Y. and Ebara, M. (2016) Polymers, 8, 41.
- 64 McAuley, E.M., Bradke, T.A., and Plopper, G.E. (2011) Cell Adh. Migr., 5, 382–386.
- 65 Bradke, T.M., Hall, C., Carper, S.W., and Plopper, G.E. (2008) Cell Adh. Migr., 2, 153–160.
- 66 Mokwena, K.K. and Tang, J. (2012) Crit. Rev. Food Sci. Nutr., 52, 640–650.
- 67 Itoh, S., Suzuki, C., and Tsuji, T. (2006) J. Biomed. Mater. Res. A, 77, 294–303.
- 68 Lin, M., Zhang, Y., Chen, G., and Jiang, M. (2015) Small, 11, 6065–6070.
- 69 Lin, M., Sun, P., Chen, G., and Jiang, M. (2014) Chem. Commun., 50, 9779–9782.
- 70 Ke, X., Coady, D.J., Yang, C., Engler, A.C., Hedrick, J.L., and Yang, Y.Y. (2014) Polym. Chem., 5, 2621–2628.