
Chapter 10
Functional Porous Materials
Watcharop Chaikittisilp
The University of Tokyo, Department of Chemical System Engineering, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
10.1 Introduction
Progress in nanoscience and nanotechnology has accelerated the developments in porous materials during the past decades [1–5]. In addition to the traditional applications of porous materials, that is, adsorption, ion exchange, and catalysis, their uses have been broadened to emerging areas such as sensing, low reflective index (low-n) and low dielectric constant (low-k) materials for electronic devices, and drug delivery. To sustain their developments at a rapid pace, the concept of “nanoarchitectonics” should be applied to further innovate the materials and their applications [6, 7], offering a powerful proposition of making objects and systems not yet envisioned.
Among porous materials, zeolites, crystalline microporous aluminosilicates and other metallosilicates that are constructed from corner-sharing, tetrahedrally coordinated TO4/2 primary units (where T is a tetrahedral atom such as silicon or aluminum), have made the greatest impact to our human lives [1, 8]. However, there is no singular class of materials that can be perfectly used for all applications. This leads to the development of other classes of porous materials, including mesoporous silicas [4], porous organic polymers [3, 5], and metal–organic frameworks (MOFs) [2].
This chapter addresses the overview and the recent developments in the field of “functional” porous materials. It is the intention of the author to provide a fundamental background on the descriptors of porous materials by starting this chapter with the 2015 updated technical report on physisorption of gases summarized by the International Union of Pure and Applied Chemistry (IUPAC) [9]. The functional materials are later described on the basis of their types of functionalities, including porous materials with inorganic, organic, and inorganic–organic frameworks (or pore walls).
10.2 Classification of Porous Materials
Classification of porous materials is done by the IUPAC according to their pore sizes [10]. Pores with widths below about 2 nm are denoted as micropores, those in the range of 2–50 nm are mesopores, and those above about 50 nm are macropores. The term “nanopore” covers these three porous categories, but with an upper limit of about 100 nm [9].
Characterization of porous materials is generally done by means of gas physisorption. In the 1985 IUPAC recommendations [10], physisorption isotherms were grouped into six categories, which were updated in 2015 into eight types based on the recent discoveries of novel porous materials [9]. The 2015 updated classification of sorption isotherms is shown in Figure 10.1. The main revisions are that type I and type IV isotherms are divided into two subtypes, denoted as (a) and (b).
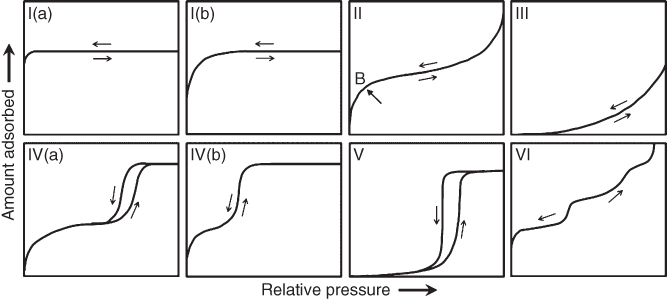
Figure 10.1 2015 IUPAC classification of physisorption isotherms.
Reversible type I isotherms are typically obtained on microporous materials. Depending on the sizes of micropores, uptakes at low relative pressure can be different; type I(a) isotherms are given by microporous materials with pore widths less than about 0.7 nm, whereas type I(b) isotherms are obtained by filling wider micropores and possibly narrow mesopores (<∼2.5 nm). Reversible type II isotherms are found on nonporous or macroporous materials, where mono- and then multilayer adsorption occurs on the surface with unrestricted uptakes up to very high relative pressure. The point B generally indicates the completion of monolayer formation.
Type III isotherms represent very small uptakes at low relative pressure, thereby indicating the weak interactions between the adsorbents and the adsorbates. The absence of the point B suggests that the adsorbed molecules are clustered around the most favorable sites on the surface of nonporous or macroporous materials. In this case, the surface of the materials is not completely wetted as the uptake amount remains finite even at the saturation pressure. Type V isotherms are very similar to type III at low relative pressure, indicating the relatively weak adsorbent–adsorbate interactions. At a higher relative pressure, the clustering adsorption is followed by the pore-filling adsorption. These two types of isotherms are often observed in water adsorption, especially on hydrophobic pore walls [11].
Type IV(a) and type IV(b) isotherms are typically arisen from mesoporous materials; the difference between these subtypes is again related to the pore sizes. In the case of type IV(a) isotherms, capillary condensation is accompanied by hysteresis. This occurs when the pore width is larger than a certain critical width, depending on the adsorbates and sorption temperature (e.g., for nitrogen adsorption in cylindrical pores at 77 K, hysteresis starts to occur for pores wider than about 4 nm). Completely reversible type IV(b) isotherms are therefore given by materials possessing pore sizes below such a critical value.
Reversible stepwise type VI isotherms are seen for the layer-by-layer adsorptions on a highly uniform nonporous surface, where each step represents the formation of a new layer with its height indicating the uptake capacity for each layer.
In the type IV isotherms, mesopores are filled through pore condensation where gas condenses to a liquid-like phase. If mesopores are large enough, hysteresis loops can be observed (i.e., type IV(a)). As is well known, the shape of the hysteresis loops of the type IV(a) isotherms is strongly correlated to the pore structures and sorption mechanisms. As shown in Figure 10.2, the 2015 IUPAC technical report classifies the hysteresis loops into six types [9]. Note that types H1, H2(a), H3, and H4 were identified in the original 1985 recommendation [10].

Figure 10.2 2015 IUPAC classification of hysteresis loops.
In general, the hysteresis loops are associated with capillary condensation in which the vapor–liquid phase transition is delayed owing to the presence of metastable adsorption films and hindered nucleation of liquid. As a result, the adsorption branch of the hysteresis loop is not in thermodynamic equilibrium. On the contrary, because nucleation does not occur during evaporation, the desorption branch is equivalent to an equilibrium liquid–vapor transition. This sorption mechanism is represented by the type H1 loop, which is observed in ordered mesoporous materials with uniform cylindrical pores such as MCM-41 and SBA-15 silicas, ordered three-dimensional pore networks, for instance, MCM-48 and KIT-6 silicas, and some controlled pore glasses.
In materials having ink-bottle-shaped pores, evaporation is also delayed, resulting in types H2(a) and H2(b) hysteresis loops. The desorption branch can be much steeper than the adsorption branch (type H2(a)) because delayed evaporation due to the pore blocking represents a percolation transition. Type H2(a) loop is also given by cavitation-induced evaporation or spontaneous nucleation of bubbles inside the pores in which the pore centers empty while the pore neck remains filled. The cavitation phenomenon occurs in the materials with pore neck diameters smaller than a certain critical size at a given temperature and adsorbates (e.g., a critical neck of 5–6 nm for nitrogen and argon sorption at 77 and 87 K, respectively). Type H2(a) loop is observed in many silica gels, some porous glasses (e.g., vycor), and some ordered mesoporous materials (e.g., SBA-16 and KIT-5 silicas). In the absence of percolation effects and in the case of a wider size distribution of pore necks, for example, mesocellular silica foams, and hydrothermally treated SBA-16 and KIT-5 silicas, H2(b) hysteresis loop is observed.
In type H3 loop, the adsorption branch resembles the type II isotherms, while the desorption branch appears at a critical value of cavitation. This type is found in aggregates of plate-like particles and in materials with macropores that are not completely filled with adsorbates. Type H4 loop is similar to type H3, but the adsorption branch is a combination of the types I and II isotherms, representing materials containing micropores and meso/macropores such as mesoporous zeolites and hierarchical micro- and mesoporous carbons. Type H5 loop also contains the steep desorption step due to cavitation, but another distinct desorption step is also present. This loop is seen in materials containing both open and partially blocked mesopores (e.g., plugged hexagonal templated silicas).
10.3 Functional Frameworks: from Inorganic, through Organic, to Inorganic–Organic
In any porous material, generally, there are four different levels of functionality: (i) the components of pore walls, (ii) ionic species adsorbed on the pore walls via strong ionic interactions, (iii) the organics covalently bound to the pore walls, and (iv) the guest species encapsulated in the porous cavities such as organic molecules and metal clusters (Figure 10.3).

Figure 10.3 Four types of functionality: (a) framework backbones, being inorganic, organic, or inorganic–organic components; (b) ionic (either cationic or anionic) species adsorbed on the pore walls via strong ionic interactions; (c) organics covalently functionalized to the pore walls; and (d) guest species encapsulated in the porous cavities such as organic molecules, enzymes, and metal clusters.
The pore walls can be inorganic, organic, and inorganic–organic compositions. Zeolite is a representative class of inorganic porous materials. Typically, its silica framework is substituted with aluminum, thereby creating anionic aluminosilicate frameworks that constitute Brönsted acid sites when they are counterbalanced by protons. Hydrophobicity/hydrophilicity of zeolites can be controlled by altering the content of aluminum in the frameworks. Zeolites with high Si/Al ratios are hydrophobic, whereas low-silica zeolites are hydrophilic. Besides aluminum, incorporation of other heteroatoms such as gallium, germanium, tin, titanium, and zirconium can yield zeolites with new functionalities that are hard to achieve from conventional aluminosilicate compositions [12–16]. For example, tin-, titanium-, zinc-, and zirconium-containing zeolites show Lewis acidity.
In addition to the framework components, the functionalities of zeolites can be altered depending on the extra-framework cationic species that can be exchanged, making ion-exchanged zeolites capable of being utilized as gas adsorbents, redox catalysts, and optoelectronic devices [17–20]. For example, Cu(II)- and Fe(II)-exchanged zeolites are promising catalysts for selective reduction of NOx [18, 19]. In addition, several metal (metallic, cation, or oxo-complex)-modified zeolites have shown great promise to directly convert methane, a main component in the shale and natural gases, to easily transportable liquid fuels and other useful chemicals [21–24].
In addition to zeolites, inorganic porous materials can be mesoporous silicas, metal oxides, and metals. Functionalities of such mesoporous materials again depend on the chemical compositions of the pore walls. In the case of mesoporous silica, for example, functionalization of silica frameworks has been achieved by incorporation of organic units and other metal atoms into the frameworks [25, 26]. In addition, mesoporous silica can be functionalized by organic moieties on its surface, which has been achieved mostly via four synthetic routes: (i) physical impregnation of monomeric or polymeric organics, (ii) covalent grafting of organosilanes having targeted functional groups, (iii) direct co-condensation of functional group-containing silanes and conventional precursors during synthesis, and (iv) in situ polymerization in the pores of mesoporous silica. These functionalized mesoporous silica-based materials can be used in several areas such as sensing, drug delivery, and CO2 capture [27–29].
Control over pore diameters of functional porous silica is also important. For instance, multiple, orthogonal interactions between structure-directing agents (SDAs), co-SDAs, and silica frameworks can generate porous silica having some periodicity, high surface area, and pore diameters near the boundary between micro- and mesopores using aromatic compounds bearing anionic end groups as SDAs (see Figure 10.4) [30]. By using chiral SDAs, chirality can transfer/transcribe in part to the surface of mesoporous silica and metal, providing enantioselective recognition [31, 32].
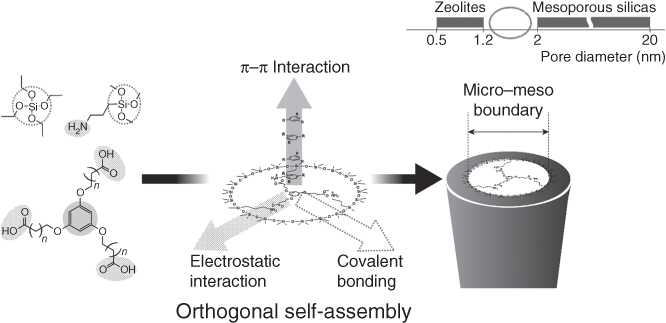
Figure 10.4 Synthesis of nanoporous silica with a pore diameter near the boundary between micro- and mesopores by the orthogonal multiple interactions of SDA–SDA, SDA–silica, and silica–silica.
(Adapted from Muraoka et al. 2015 [30].)
Synthesis of mesoporous materials via a soft-templating method generally employs lyotropic liquid crystals (LLCs) of surfactants as templates. Formation of the targeted materials occurs by condensation or reduction of precursors embedded in the LLC matrix. Alternatively, mesoporous materials can be achieved via a micelle assembly pathway, in which the concentration of surfactants or template molecules is greater than a critical micelle concentration but lower than the point required for the formation of LLC phase. Several mesoporous metal films (e.g., platinum, palladium, and gold) are electrochemically fabricated via this pathway. The resulting films show excellent electrochemical and optical properties [33–35].
In the past decade, synthesis of porous organic frameworks has advanced very rapidly, partly due to urgent needs of low-density porous materials for gas storage [3, 5]. Such frameworks, consisting solely of light elements (hydrogen, boron, carbon, nitrogen, oxygen, etc.), can be constructed by a wide array of standard synthetic strategies in organic chemistry. In an extreme case, covalent organic frameworks (COFs) possessing crystalline structures and high surface area can be constructed under thermodynamic control [36–38]. COFs can be designed and synthesized as two- and three-dimensional frameworks. These materials are composed entirely of covalent bonds between light elements, making them the crystalline material with the lowest density, for example, COF-108: 0.17 g cm−1.
Under carefully chosen reaction conditions, crystalline COFs are obtained under thermodynamic control (i.e., reversibility of the dehydration condensation). For their synthesis, the selection of the right solvent seems to be very important. To foster the formation of a uniform and highly ordered structure, solvents are chosen wherein the reactants are poorly soluble. This approach slows down the reversible condensation. Furthermore, the reactions are often carried out in sealed Pyrex tubes, to ensure the reversible reaction, again to slow down the reversible process, and to minimize defects by self-healing.
As order and crystallinity are not prerequisites for fine control over porosity and functionality of materials, several synthetic porous organic polymers with large surface area and narrow pore size distribution have been reported [3, 5]. In contrast to crystalline COFs, these porous organic polymers are classified as amorphous frameworks. The representatives are polymers with intrinsic microporosity (PIMs) [39, 40], hypercross-linked polymers (HCPs) [41, 42], and conjugated microporous polymers (CMPs) [43, 44] (Figure 10.5). All of them are formed under “kinetic control.”

Figure 10.5 Examples of molecular structures of (a) polymers with intrinsic microporosity (PIMs), (b) hypercross-linked polymers (HCPs), and (c) conjugated microporous polymers (CMPs).
As high rigidity, bulkiness, nonplanarity, and contorted architectures frustrate efficient packing of macromolecules, PIMs can be synthesized through the exploitation of simple and efficient dioxane-forming reactions. The internal surface area of some of these easily accessible polymers is in excess of 1000 m2 g−1 (Brunauer–Emmett–Teller (BET) specific surface area) and micropore size distributions apparently reveal average pore diameters of about 0.7 nm or less. Their microporosity is independent of their processing history, which confirms that the porosity is intrinsic and arises from the rigid macromolecules' inability to completely fill the voids. In contrast to COFs, HCPs, and CMPs, PIMs are solution-processable, making them a candidate material for gas separation membranes [45]. The performance of membranes can be improved by post-synthetic functionalization of organic frameworks and by modification of the geometry of organic backbones [46, 47].
CMPs are mostly synthesized by C−C and C−N coupling chemistry, providing a unique extended conjugation in their backbones. Considering structural chemistry, one may consider that highly rigid monomers should be selected as they can selectively connect together into specific macromolecular networks. By designing the monomers based on the diamond structure, CMPs with BET specific surface areas of about 5000–7000 m2 g−1 can be achieved [48, 49]. From a molecular design viewpoint, the most fascinating property of CMPs is their diversity of π-conjugated skeletons. Therefore, it is synthetically possible to tune the π-conjugation of CMPs systematically and precisely, thereby allowing us to optimize their functionalities. CMPs show great promise in many areas such as chemosensing, light harvesting, and electrochemical energy storage [50–53].
Porous organic–inorganic hybrids are of particular interest as their functionalities can be expanded beyond the typical limits of organic or inorganic components alone because the hybridization of inorganic and organic groups can provide additional synergistic functions arising from specific interactions at the hybrid interfaces. Since the early 1990s, porous MOFs or porous coordination polymers (PCPs) built by metal ions as “joint connectors” and organic “linker” moieties (see Figure 10.6a) became an alternative class of crystalline porous materials and have attracted the close attention of scientists and engineers [2, 5]. Much earlier, however, the syntheses of these materials have been reported in the literatures, for example, Prussian-blue compounds, Werner complexes, and Hofmann clathrates. Considering the abundant surface of MOFs, robust frameworks with potential functions related to zeolites, the so-called second-generation MOFs [54], show unexpected sorption properties. Two prototypes of this generation are [Zn4O(benzenedicarboxylate)3]n, MOF-5 [55], and [Cu(SiF6)(4,4′-bipyridine)2]n [56].

Figure 10.6 (a) Schematic of typical synthesis of MOFs. (b) Synthesis of MOFs in the presence of multiple ligands having different functional groups.
Unlike inorganic crystalline frameworks such as zeolite, interestingly, a number of MOFs possess dynamic/flexible porous frameworks, in which their structures can be transformed responding to guest inclusion/exchange or external stimuli, such as pressure, light, and magnetic and electric fields, the so-called third-generation MOF [54]. This generation of MOFs is known to exhibit unprecedented properties regarding adsorption, for example, guest-selective and gate-opened pressure [2].
Functionalization of MOFs can provide more selective and active sites for specific applications. There are two major ways, namely, introduction of coordinatively unsaturated metal centers [57, 58] and post-synthetic modification of organic linkers [59, 60]. In the former case, there are three different methods employed thus far to introduce exposed metal sites into MOFs. The most common strategy involves the removal of metal-bound volatile guests, mostly the solvent used in their syntheses, which typically function as terminal ligands for the metals embedded within the porous frameworks. The other two methods involve either the incorporation of metal species into organic linkers, for example, as a metallo Schiff base, or the impregnation of frameworks with excess metal cations.
In the latter, MOFs are assembled and then modified with chemicals while preserving the framework structures. Because MOFs are typically obtained by “one-pot” synthesis involving either slow diffusion or direct mixing (generally under solvothermal conditions) of metal ions and organic precursors, there are limitations in the possible functional groups that can be directly introduced into MOFs during their synthesis. Considering that MOFs are sufficiently robust and porous to allow late-stage transformations without compromising overall framework integrity, a wide variety of chemical reactions would be available for modifying the framework components. Thus, modification of organic bridging ligands in a post-synthetic manner offers the ability to tailor the physicochemical environment of the cavities and the pore surface within MOFs, hence allowing the fine-tuning of the interactions with guest species, as well as the stability and reactivity of the frameworks. As a breakthrough in MOF functionalization, MOFs having multiple functional groups with varied contents can be achieved via direct solvothermal synthesis (Figure 10.6b) [61, 62]. Characterizations suggest that these different linking groups are incorporated into the frameworks in a way that mixes the linkers, rather than forming separate domains. The complex arrangements of several functional groups within the pores are expected to provide for properties that are not simply linear sums of those of the pure components.
10.4 Summary and Outlook
As described here, functional porous materials have found their place at the center of nanomaterials. The continuing research and development of porous materials with targeted functionality can offer a platform for nanoarchitectonics toward several potential applications for which other nanomaterials are not appropriate or do not provide satisfactory performances. Considering the continuously increasing demand for materials to be used in new technologies, “ab initio” synthesis of functional porous materials for on-demand applications with the aid of advanced computational and analytical techniques is crucial and should be intensively considered in future research [63–65].
References
- 1 Davis, M.E. (2002) Nature, 417, 813–821.
- 2 Horike, S., Shimomura, S., and Kitagawa, S. (2009) Nat. Chem., 1, 695–704.
- 3 Thomas, A. (2010) Angew. Chem. Int. Ed., 49, 8328–8344.
- 4 Ariga, K., Vinu, A., Yamauchi, Y., Ji, Q., and Hill, J.P. (2012) Bull. Chem. Soc. Jpn., 85, 1–32.
- 5 Slater, A.G. and Cooper, A.I. (2015) Science, 348, aaa8075.
- 6 Ariga, K. and Aono, M. (2016) Jpn. J. Appl. Phys., 55, 1102A6.
- 7 Aono, M. and Ariga, K. (2016) Adv. Mater., 28, 989–992.
- 8 Chaikittisilp, W. and Okubo, T. (2017) Zeolite and zeolite-like materials, in Handbook of Solid State Chemistry, Part 4. Nano and Hybrid Materials (eds R. Dronskowski, S. Kikkawa, and A. Stein), Wiley-VCH Verlag GmbH, Weinheim, pp. 97–119.
- 9 Thommes, M., Kaneko, K., Neimark, A.V., Olivier, J.P., Rodriguez-Reinoso, F., Rouquerol, J., and Sing, K.S.W. (2015) Pure Appl. Chem., 87, 1051–1069.
- 10 Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A., Rouquérol, J., and Siemieniewska, T. (1985) Pure Appl. Chem., 57, 603–619.
- 11 Thommes, M., Morell, J., Cychosz, K.A., and Fröba, M. (2013) Langmuir, 29, 14893–14902.
- 12 Fricke, R., Kosslick, H., Lischke, G., and Richter, M. (2000) Chem. Rev., 100, 2303–2405.
- 13 Notari, B. (1996) Adv. Catal., 41, 253–334.
- 14 Corma, A. and García, H. (2002) Chem. Rev., 102, 3837–3892.
- 15 Román-Leshkov, Y. and Davis, M.E. (2011) ACS Catal., 1, 1566–1580.
- 16 Van de Vyver, S. and Román-Leshkov, Y. (2015) Angew. Chem. Int. Ed., 54, 12554–12561.
- 17 Pham, T.D., Liu, Q., and Lobo, R.F. (2013) Langmuir, 29, 832–839.
- 18 Vanelderen, P., Vancauwenbergh, J., Sels, B.F., and Schoonheydt, R.A. (2013) Coord. Chem. Rev., 257, 483–494.
- 19 Brandenberger, S., Kröcher, O., Tissler, A., and Althoff, R. (2008) Catal. Rev., 50, 492–531.
- 20 Wada, Y., Sato, M., and Tsukahara, Y. (2006) Angew. Chem. Int. Ed., 45, 1925–1928.
- 21 Grundner, S., Markovits, M.A.C., Li, G., Tromp, M., Pidko, E.A., Hensen, E.J.M., Jentys, A., Sanchez-Sanchez, M., and Lercher, J.A. (2015) Nat. Commun., 6, 7546.
- 22 Narsimhan, K., Iyoki, K., Dinh, K., and Román-Leshkov, Y. (2016) ACS Cent. Sci., 2, 424–429.
- 23 Snyder, B.E.R., Vanelderen, P., Bols, M.L., Hallaert, S.D., Böttger, L.H., Ungur, L., Pierloot, K., Schoonheydt, R.A., Sels, B.F., and Solomon, E.I. (2016) Science, 536, 317–321.
- 24 Gerceker, D., Motagamwala, A.H., Rivera-Dones, K.R., Miller, J.B., Huber, G.W., Mavrikakis, M., and Dumesic, J.A. (2017) ACS Catal., 7, 2088–2100.
- 25 Fujita, S. and Inagaki, S. (2008) Chem. Mater., 20, 891–908.
- 26 Hoffmann, F., Cornelius, M., Morell, J., and Fröba, M. (2006) Angew. Chem. Int. Ed., 45, 3216–3251.
- 27 Brunelli, N.A. and Jones, C.W. (2013) J. Catal., 308, 60–72.
- 28 Didas, S.A., Choi, S., Chaikittisilp, W., and Jones, C.W. (2015) Acc. Chem. Res., 48, 2680–2687.
- 29 Tarn, D., Ashley, C.E., Xue, M., Carnes, E.C., Zink, J.I., and Brinker, C.J. (2013) Acc. Chem. Res., 46, 792–801.
- 30 Muraoka, K., Chaikittisilp, W., Yanaba, Y., Yoshikawa, T., and Okubo, T. (2015) Chem. Commun., 51, 10718–10721.
- 31 Bueno-Alejo, C.J., Villaescusa, L.A., and Garcia-Bennett, A.E. (2014) Angew. Chem. Int. Ed., 53, 12106–12110.
- 32 Wattanakit, C., Côme, Y.B.S., Lapeyre, V., Bopp, P.A., Heim, M., Yadnum, S., Nokbin, S., Warakulwit, C., Limtrakul, J., and Kuhn, A. (2014) Nat. Commun., 5, 3325.
- 33 Wang, H., Wang, L., Sato, T., Sakamoto, Y., Tominaka, S., Miyasaka, K., Miyamoto, N., Nemoto, Y., Terasaki, O., and Yamauchi, Y. (2012) Chem. Mater., 24, 1591–1598.
- 34 Wang, H., Ishihara, S., Ariga, K., and Yamauchi, Y. (2012) J. Am. Chem. Soc., 134, 10819–10821.
- 35 Li, C., Dag, Ö., Dao, T.D., Nagao, T., Sakamoto, Y., Kimura, T., Terasaki, O., and Yamauchi, Y. (2015) Nat. Commun., 6, 6608.
- 36 Côté, A.P., Benin, A.I., Ockwig, N.W., O'Keeffe, M., Matzger, A.J., and Yaghi, O.M. (2005) Science, 310, 1166–1170.
- 37 El-Kaderi, H.M., Hunt, J.R., Mendoza-Cortés, J.L., Côté, A.P., Taylor, R.E., O'Keeffe, M., and Yaghi, O.M. (2007) Science, 316, 268–272.
- 38 Diercks, C.S. and Yaghi, O.M. (2017) Science, 355, eaal1585.
- 39 Budd, P.M., Ghanem, B.S., Makhseed, S., McKeown, N.B., Msayib, K.J., and Tattershall, C.E. (2004) Chem. Commun., 230–231.
- 40 McKeown, N.B. and Budd, P.M. (2006) Chem. Soc. Rev., 35, 675–683.
- 41 Germain, J., Hradil, J., Fréchet, J.M.J., and Svec, F. (2006) Chem. Mater., 18, 4430–4435.
- 42 Xu, S., Luo, Y., and Tan, B. (2013) Macromol. Rapid Commun., 34, 471–484.
- 43 Jiang, J.-X., Su, F., Trewin, A., Wood, C.D., Campbell, N.L., Niu, H., Dickinson, C., Ganin, A.Y., Rosseinsky, M.J., Khimyak, Y.Z., and Cooper, A.I. (2007) Angew. Chem. Int. Ed., 46, 8574–8578.
- 44 Xu, Y., Jin, S., Xu, H., Nagai, A., and Jiang, D. (2013) Chem. Soc. Rev., 42, 8012–8031.
- 45 Budd, P.M. and McKeown, N.B. (2010) Polym. Chem., 1, 63–68.
- 46 Du, N., Park, H.B., Robertson, G.P., Dal-Cin, M.M., Visser, T., Scoles, L., and Guiver, M.D. (2011) Nat. Mater., 10, 372–375.
- 47 Carta, M., Malpass-Evans, R., Croad, M., Rogan, Y., Jansen, J.C., Bernardo, P., Bazzarelli, F., and McKeown, N.B. (2013) Science, 339, 303–307.
- 48 Ben, T., Ren, H., Ma, S., Cao, D., Lan, J., Jing, X., Wang, W., Xu, J., Deng, F., Simmons, J.M., Qiu, S., and Zhu, G. (2009) Angew. Chem. Int. Ed., 48, 9457–9460.
- 49 Yuan, D., Lu, W., Zhao, D., and Zhou, H.-C. (2011) Adv. Mater., 23, 3723–3725.
- 50 Kou, Y., Xu, Y., Guo, Z., and Jiang, D. (2011) Angew. Chem. Int. Ed., 50, 8753–8757.
- 51 Liu, X., Xu, Y., and Jiang, D. (2012) J. Am. Chem. Soc., 134, 8738–8741.
- 52 Sprick, R.S., Jiang, J.-X., Bonillo, B., Ren, S., Ratvijitvech, T., Guiglion, P., Zwijnenburg, M.A., Adams, D.J., and Cooper, A.I. (2015) J. Am. Chem. Soc., 137, 3265–3270.
- 53 Sprick, R.S., Bonillo, B., Clowes, R., Guiglion, P., Brownbill, N.J., Slater, B.J., Blanc, F., Zwijnenburg, M.A., Adams, D.J., and Cooper, A.I. (2016) Angew. Chem. Int. Ed., 55, 1792–1796.
- 54 Kitagawa, S., Kitaura, R., and Noro, S.-I. (2004) Angew. Chem. Int. Ed., 43, 2334–2375.
- 55 Li, H., Eddaoudi, M., O'Keeffe, M., and Yaghi, O.M. (1999) Nature, 402, 276–279.
- 56 Noro, S.-I., Kitagawa, S., Kondo, M., and Seki, K. (2000) Angew. Chem. Int. Ed., 39, 2081–2084.
- 57 Kitaura, R., Onoyama, G., Sakamoto, H., Matsuda, R., Noro, S.-I., and Kitagawa, S. (2004) Angew. Chem. Int. Ed., 43, 2684–2687.
- 58 McDonald, T.M., Mason, J.A., Kong, X., Bloch, E.D., Gygi, D., Dani, A., Crocellà, V., Giordanino, F., Odoh, S.O., Drisdell, W.S., Vlaisavljevich, B., Dzubak, A.L., Poloni, R., Schnell, S.K., Planas, N., Lee, K., Pascal, T., Wan, L.F., Prendergast, D., Neaton, J.B., Smit, B., Kortright, J.B., Gagliardi, L., Bordiga, S., Reimer, J.A., and Long, J.R. (2015) Nature, 519, 303–308.
- 59 Cohen, S.M. (2012) Chem. Rev., 112, 970–1000.
- 60 Fei, H. and Cohen, S.M. (2015) J. Am. Chem. Soc., 137, 2191–2194.
- 61 Deng, H., Doonan, C.J., Furukawa, H., Ferreira, R.B., Towne, J., Knobler, C.B., Wang, B., and Yaghi, O.M. (2010) Science, 327, 846–850.
- 62 Kong, X., Deng, H., Yan, F., Kim, J., Swisher, J.A., Smit, B., Yaghi, O.M., and Reimer, J.A. (2013) Science, 341, 882–885.
- 63 Farha, O.K., Yazaydín, A.Ö., Eryazici, I., Malliakas, C.D., Hauser, B.G., Kanatzidis, M.G., Nguyen, S.T., Snurr, R.Q., and Hupp, J.T. (2010) Nat. Chem., 2, 944–948.
- 64 Bai, P., Jeon, M.Y., Ren, L., Knight, C., Deem, M.W., Tsapatsis, M., and Siepmann, J.I. (2015) Nat. Commun., 6, 5912.
- 65 Gallego, E.M., Portilla, M.T., Paris, C., León-Escamilla, A., Boronat, M., Moliner, M., and Corma, A. (2017) Science, 355, 1051–1054.