
Chem2Bio2RDF: a semantic resource for systems chemical biology and drug discovery
Abstract:
This chapter describes an integrated semantic resource of drug discovery information called Chem2Bio2RDF. Heterogeneous public data sets pertaining to compounds, targets, genes, diseases, pathways and side effects were integrated into a single resource, which is freely available at chem2bio2rdf.org. A number of tools that use the data are described, along with the implementation challenges that derived from the project including details of where data is stored, how to organize it, and how to address data quality and equivalence.
18.1 The need for integrated, semantic resources in drug discovery
'How do we find the needles in the haystacks?' is a question that has been in the minds of pharmaceutical researchers since the early 1990s when high-throughput methods (screening, microarray analyses, and so on) began producing huge volumes of data about compounds, targets, genes, and pathways, and the interactions between them. The question is predicated on the assumption that somewhere in these vast haystacks of data can be found 'needles' – key pieces of knowledge or insight that could help find new drugs or new understandings of disease processes.
Unfortunately, producing the haystacks of data has turned out more straightforward than knowing how to sift through them, or even knowing if the needles are there or how to identify them when they are found. The 2000s saw this question move beyond pharmaceutical companies into the public arena, as huge volumes of data, particularly about chemical compounds and their biological activities, became available in the public domain, including such resources as PubChem, ChemSpider, and ChEMBL.
The needle-in-a-haystack analogy hides a subtler issue: that although most public data sets are centered on chemical or biological entities (compounds, genes, and so on), the most useful insights often lie in the relationships between these entities. Although some sets, such as ChEMBL, do represent a constrained set of relationships (e.g. between compounds and targets), these are not networked to other kinds of relationships, and so wider patterns cannot be seen. Yet it is these patterns that must be key to understanding the systematic effects of drugs on the body. A more appropriate analogy than haystacks is the Ishihara color blindness test, in which to find the hidden patterns one has to look at the whole picture with the right set of lenses.
It is with this in mind, that a research project at Indiana was developed to prototype new ways of representing publicly available entities and relationships as large-scale integrated sets, and new ways of data-mining them (new lenses in the analogy) to reveal the hidden patterns. Since this research began in 2005, many new relevant technologies have come to the fore (particularly in the area of the Semantic Web), but the problems have remained the same: finding ways to integrate public data sets intelligently; providing a common access and computation interface; developing tools that can find patterns across data sets; and developing new methodologies that make these tools applicable in real drug discovery problems.
Our initial work involved the development of ChemBioGrid [1], an open infrastructure of web services and computational tools for drug discovery, operating at the interface of cheminformatics and bioinformatics. This infrastructure allowed the quick development of new kinds of tools that integrated both cheminformatics and bioinformatics applications. However, this did not address the problem of data integration, which is addressed by the topic of this chapter, Chem2Bio2RDF. Data integration is a difficult problem [2, 3], as by definition it involves heterogeneous data sets that have often been developed from different disciplines and groups, each with their own terminology and ways of representing data. Traditionally, integration has been achieved using relational databases, a tortuous manual process that involves complex, formalized relational schema. The new technology that has made this process much more feasible is RDF (Resource Description Framework), a simple language that allows the representation of pairs of entities and the relationships between them (RDF triples). These take the form of Subject–Predicate–Object. For example, we could simply represent a fact about the author using the triple < David Wild > < Is_ a > < Author >. Each RDF triple can be considered as two nodes of a network connected by an edge. In aggregate, the RDF triples describe a network of entities and relationships between them.
18.2 The Semantic Web in drug discovery
The power of RDF lies in its simplicity, but also in the advent of three other Semantic Web technologies which unleash its power: triple stores for efficient storage, access and searching of RDF; SPARQL, a language for querying RDF stores, and ontologies for the standardization of the content of RDF triples. More details on the RDF and semantic web approach are discussed in Chapter 19, where Bildtsen and co-authors describe the 'Triple map' application, and how it is used for semantic searching and knowledge collaboration in biomedical research. These technologies have only recently reached a point of maturity where they are practically useful, and in some ways the Semantic Web was over-hyped before it was mature enough, leaving an impression in some quarters that it is not technically up to the job of data integration. It is the opinion of the author that the technologies have reached a point where they are not only technically capable, but also are the only current technologies capable of tackling this problem. Indeed, as the chapters by Bildtsen and colleagues (Chapter 19), Harland and colleagues (Chapter 17) and Alquier (Chapter 16) show, this technology is now gaining traction within industry.
Recently, there has been an escalation in activity in the application of Semantic Web methods in drug discovery as a means of integration, including the well-funded EU OpenPhacts project [4], the W3 Semantic Web in Health Care and Life Sciences Interest Group [5], CSHALS [6] – a conference on Semantics in Health Care and Life Sciences, and Linked Open Drug Data (LODD) – a central repository for RDF related to drugs [7]. More widely in the biological sciences, the Semantic Web is having an impact through efforts such as Bio2RDF [8]. Bio2RDF covers a large number of genetic, pathway, protein, and enzyme sets, but does not cover chemical sets, except the KEGG ligand database. It thus overlaps with Chem2Bio2RDF, but the key differentiator of the latter is that chemical sets are mapped fully into genetic, protein, and pathway sets.
The Semantic Web provides a technical infrastructure for the large-scale integrated data-mining that is necessary, but advances have also been made in the data-mining techniques themselves, particularly in the fields of chemogenomics (the study of the relationships between chemical compounds – or drugs – and genes) and systems chemical biology [9] (a new term relating to the integrated application of cheminformatics and bioinformatics techniques to uncover new understanding of the systematic effects of chemical compound on the body). Recent research in chemogenomics has included the development of generalizable algorithms for predicting new compound–target interactions [10], creation of predictive networks in domain areas such as Kinases [11], and combination approaches that can be used to predict off-target interactions and new therapeutic uses for drugs [12]. However, application of these methods beyond the original research has been limited by lack of integrated access to public data. Little research has been done in Systems Chemical Biology outside of chemogenomics, for instance relating compounds to side effects, pathways, or disease states, primarily due to the lack of available tools and resources for relating compounds to entities other than targets or genes [9].
Chem2Bio2RDF was designed to address these gaps in data accessibility by providing access to a wide range of data sets covering compounds, drugs, targets, genes, assays, diseases, side effects, and pathways in a single, integrated format.
18.3 Implementation challenges
The main issues of implementing Chem2Bio2RDF were those that plague any implementation that uses emerging technologies: which particular technologies to employ and which to reject (at least initially). For Chem2Bio2RDF, this boiled down to four questions: (1) how should the data be stored and accessed; (2) where should the data be stored; (3) how to organize the data, and whether an OWL ontology should be used to semantically annotate the data; and (4) how to address data quality and equivalence across sets.
18.3.1 Data storage and access
Prior to our Semantic Web implementation, we stored a variety of public data sets in a PostgreSQL relational database enhanced with cheminformatics search functionality using the gNova CHORD cartridge [13], with access directly through JDBC or ODBC database connections, or via web service interfaces (in our ChemBioGrid architecture). All of the data and services resided on the same machine, making access easier. Our initial version of Chem2Bio2RDF preserved this architecture, keeping the data in a relational database and using the D2R tool to provide an RDF SPARQL interface to the relational data set. However, this proved to have limitations that severely restricted the utility of Chem2Bio2RDF: namely (1) cross-data set searching is difficult to implement; and (2) it is hard to embed an ontology. So after initial testing we moved to use Virtuoso Triple Store as our basic representation, which is a true RDF-triple store, thus allowing all the data to be treated as a graph, enabling easy cross-data set searching, and permitting the later development of a Chem2Bio2RDF ontology. For migration, we used D2R to generate RDF for all the relations in PostgreSQL, and then exported them into the triple store. We also found that Virtuoso was much more efficient than the PostgreSQL/D2R implementation: as D2R eventually searches the data by SQL, speed of searching is highly dependent on the structure of the tables and associated indices. Additionally, Virtuoso provides a web interface for data management, and provides a REST web service allowing the data to be searched from other endpoints.
Using either D2R or Triple Store provides access to searching using SPARQL, but this has domain limitations, most notably it does not provide cheminformatics- or bioinformatics-specific searching capabilities such as similarity searching, substructure searching, or protein similarity searching. We thus had to extend SPARQL to allow such queries. This was done using the open source Jena ARQ [14] with cheminformatics functionality from the Chemistry Development Kit (CDK), ChemBioGrid, and bioinformatics functionality from BioJava.
18.3.2 Where data is stored
We opted to use a single medium-performance server (a Dell R510 with four quad-core Xenon processors, 1 TB storage) to store all of the data in our triple store and also to provide searching capabilities. Thus far, this has provided good real-time access to the data assuming no more than two searches are being performed at the same time. Keeping all of the data in one location (versus federating searches out) has proved useful in permitting query security (i.e. queries are not broadcast outside our servers) but a concern is that if we expand Chem2Bio2RDF to new, much larger data sources such as from Genome Wide Association Searches or patient data records, or if demand on our Chem2Bio2RDF server increases dramatically, we may not be able to scale searching to meet the needs. There is thus a likely future need to permit searches to be intelligently distributed in parallel, perhaps using cloud technologies.
18.3.3 How to organize the data
We decided to organize our data sets into six categories based on the kinds of biological and chemical concepts they contain. These categories are: chemical & drug (drug is a subclass of chemical), protein & gene, chemogenomics (i.e. relating compounds to genes, through interaction with proteins or changes in expression levels), systems (i.e. PPI and pathway), phenotype (i.e. disease and side effect), and literature. However, we did not initially develop an OWL ontology, instead depending on 'same-as' relationships between data sets (e.g. PubChem Compound X is the same as Drugbank Drug Y). This decision was made due to the difficulty in defining a scope for an ontology before we had a good idea how Chem2Bio2RDF was to be used. We subsequently developed a set of use-cases that allowed us to describe a constrained, implementable ontology for Chem2Bio2RDF. As we already had access to some other ontologies (such as Gene Ontology), this really boiled down to a chemogenomic ontology for describing the relationship between compounds and biological entities. This was aligned with other related ontologies, submitted to NCBO BioPortal, and will be described in an upcoming publication.
18.3.4 Data quality and equivalence
Addressing quality is fraught with numerous complexities in details – for example is a PubChem BioAssay IC5 0 result comparable with one in CheMBL or from an internal assay? Is an experimental result always more significant than a predicted result or an association extracted from a journal article? What happens when we get so many links between things that we cannot separate the signal from the noise? We are clearly constrained by the inherent quality of the data sources available. For Chem2Bio2RDF we decided on two principles: (1) we would not constrain users from making their own quality decisions (e.g. by excluding or including data sets or data types); and (2) we would not make judgments about equivalence beyond the very basic (two compounds equivalent in two data sets, etc.). Thus we pushed addressing primarily to the tool level, allowing users to select which data sets they are comfortable using, and understanding the caveats in doing so.
18.4 Chem2Bio2RDF architecture
Chem2Bio2RDF is available as a triple store with a SPARQL endpoint, can be accessed indirectly through a variety of tools, and all of the data can be downloaded in RDF format from the Chem2Bio2RDF website [15]. Our Chem2Bio2OWL ontology is also freely available for download.
As previously described, our data sets are organized into six categories based on the kinds of biological and chemical concepts they contain. Some data sources are listed in multiple categories. Some of the data used were previously employed in relational database format in our prior work and in this case they were simply converted into RDF/XML via the D2R server. For the rest of the data sets, we acquired the raw data set (by downloading from web sites), and converted the data into our relational database using customized scripts. These are then published as RDF in the Virtuoso Triple Store. The data can be queried via a SPARQL endpoint.
A list of data sets included in Chem2Bio2RDF is shown in Table 18.1, along with the number of RDF triples for each set. We have developed a streamlined process for the addition of new data sets. We adopted PubChem Compound ID (CID) as the identifier for compounds, and UniProt ID for protein targets. The compounds represented by other data formats (e.g. SMILES, InChi and SDF) were mapped onto the compound ID via InChi keys. All the triples are stored together and the whole set is called the Chem2Bio2RDF data set. Initially, we developed a schema to classify the concepts and the RDF resources in Chem2Bio2RDF. The RDF data can be explored and queried on our web site (www.chem2bio2rdf.org). Chem2Bio2RDF and its related tools rely heavily on open source software: a list of open source software used in Chem2Bio2RDF along with links for where the software can be downloaded is given in Table 18.2.
Table 18.1
Data sets included in Chem2Bio2RDF ordered by number of RDF triples
| Data set | Triples |
| ChEMB | 57 795 793 |
| PubChem Bioassay | 5 908 479 |
| Comparative Toxicogenomics Data set (CTD) | 4 933 484 |
| Miscellaneous Chemogenomics Sets | 4 526 267 |
| ChEBI | 2 906 076 |
| Database of Interacting Proteins (DIP) | 1 113 871 |
| BindingDB | 1 027 034 |
| HUGO HGNC (genes) | 860 350 |
| KiDB (CWRU) | 745 026 |
| UniProt | 596 274 |
| PharmGKB | 512 361 |
| Human Protein Reference Database (HPRD) | 477 697 |
| KEGG (Pathways) | 477 697 |
| MATADOR (Chemogenomics) | 269 656 |
| BindingMOAD | 255 257 |
| DrugBank | 189 957 |
| Sider Side Effects Database | 127 755 |
| Toxicogenomics Tracking Database (TTD) | 116 767 |
| Miscellaneous QSAR sets | 32 206 |
| Drug Combination Database (DCDB) | 20 891 |
| OMIM | 17 251 |
| Reactome (Pathways) | 15 849 |
Table 18.2
Open source software used in Chem2Bio2RDF
| Software | Purpose | Where to find it |
| Virtuoso Open Source Edition | RDF Triple Store | http://www.openlinksw.com/wiki/main/Main |
| D2R | RDF interface to relational database | http://www4.wiwiss.fu-berlin.de/bizer/d2r-server/ |
| Chemistry Development Kit (CDK) | Cheminformatics functionality in SPARQL search | http://sourceforge.net/projects/cdk/ |
| BioJava | Bioinformatics functionality in SPARQL search | http://biojava.org/wiki/Main_Page |
| Jena ARQ | Adding functionality to SPARQL | http://jena.sourceforge.net/ARQ/ |
| Cytoscape | Visualization of query results | http://www.cytoscape.org/ |
| Protégé | Ontology Editing | http://protege.stanford.edu/ |
| SIMILE | Data visualization in web pages | http://www.simile-widgets.org/exhibit/ |
18.5 Tools and methodologies that use Chem2Bio2RDF
We have developed a variety of tools and algorithms that employ the Chem2Bio2RDF architecture. How some of these relate to Chem2Bio2RDF is shown in Figure 18.2. All of the tools are freely available for public use, and where possible the code has been submitted into open source repositories.

Figure 18.2 Tools and algorithms that employ Chem2Bio2RDF. More details and access to the tools can be obtained at http://djwild.info.
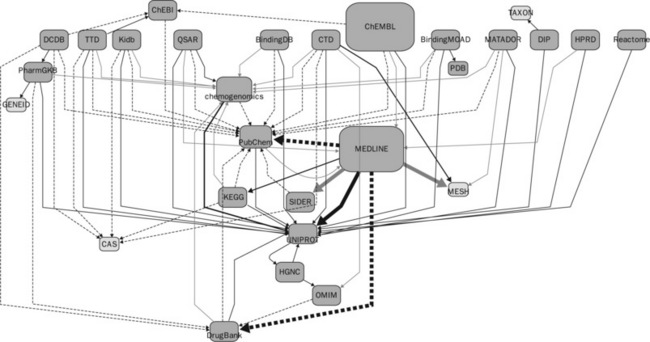
Figure 18.1 Chem2Bio2RDF organization, showing data sets and the links between them. Compound-related links are shown as dashed lines, protein/gene-related links are shown in dark gray, and other links are shown in light gray
Graph theory is well established for the analysis and mining of networked data, and lends itself naturally to application to RDF networks. We implemented an algorithm for computing semantic associations previously applied to social networks [16] for finding multiple shortest or otherwise meaningful paths between any two entities in a network, to enable all of the network paths within a given path length range between any pair of Chem2Bio2RDF entities to be identified. We have recently combined this with the BioLDA algorithm [17] described below into an association search tool that shows, for any pair of entities, the network paths between them that have the highest level of literature support. This has proven useful particularly for suggesting gene associations that can account for a drug's side effects or interactions with a disease, which has led us to develop a modification of the algorithm that allows the search to be restricted to only those paths that contain a particular type of entity (such as a gene).
We adapted a second algorithm from the social networking community to bring scholarly publications into our networks. A database of recent PubMed abstracts (for the last four years) was analyzed to identify Bioterms, that is terms that can be associated with entities in our existing network – for example, the name of a side effect, gene, compound, or drug. These Bioterms constitute an association between a PubMed article and an entry in one of our databases, producing an RDF association that can be mined. These Bioterms were further applied to a modified Latent Dirichlet Allocation algorithm (a method of identifying latent topics – or clusters – in a set of documents based on their word frequency) to identify latent topics in the PubMed literature. Publications and entities can then be probabilistically associated with these topics, and the product of multiple associations over a path used to create a measure of distance between entities (via topics) known as KL-divergence. The automatically identified topics along with their associated Bioterms show a surprising correlation with real areas of study, such as psychiatric disorders. Our resultant BioLDA algorithm [17] can be used for a variety of purposes, including identifying previously unknown Bioterm connections between research areas, constraining other searches to topic areas, and ranking of association paths by literature support as implemented in our association search tool.
We are currently taking the association search and literature-based methods a step further to provide quantitative measures of the association between any two items. We have developed a Semantic Link Association Prediction (SLAP) algorithm to provide such a quantitative measure based on the semantics and topology of the network. We developed an associated tool to provide both quantitative assessments of association strength along with graphical descriptions of the association paths. Initial studies, which will be published shortly, indicate a high rate of success in missed-link predictions (essentially leave-one-out studies using the network). Thus methods such as SLAP appear to be useful in predicting associations that might not already be known in the scientific literature or databases.
A large part of drug discovery involves the creation of new chemical compounds for possible therapeutic use, and new compounds can be integrated into the networks by chemical similarity (i.e. associating them with known compounds with similar chemical structures). This is the basis of our tool called WENDI [18] that embeds new compounds into our networks and permits examination of the potential therapeutic applications of the compounds. A recent development of WENDI, called Chemogenomic Explorer, extends the methods to create 'evidence paths' between compounds and diseases that constitute different paths through the network (via genes, pathways, and so on). These are generated using RDF inference tools, and in aggregate represent a cluster of independent or semi-independent evidence linking a compound to a disease. We think this evidence clustering is important as a way of mitigating the risks of errors in data, as well as the known propensity for individual pieces of published medical research to be later proved incorrect [19].
18.6 Conclusions
Our experience developing Chem2Bio2RDF has shown that Semantic Web technologies are becoming capable of meeting, at least to a reasonable degree, the challenges of data integration, and that the increasing availability of data management tools (including open source) make this process much easier. We consider Chem2Bio2RDF as a working prototype, but hope that it demonstrates that such complex, integrative networks can be built and exploited effectively for drug discovery applications. For robust, commercial applications, issues such as scalability and reliability will come into play, and it remains to be seen how well the technologies address these concerns.
18.7 References
[1] Dong, X., Gilbert, K.E., Guha, R., et al. Web service infrastructure for chemoinformatics. Journal of Chemical Information and Modeling. 2007; 47(4):1303–1307.
[2] Wild, D.J. Mining large heterogeneous datasets in drug discovery. Expert Opinion on Drug Discovery. 2009; 4(10):995–1004.
[3] Slater, T., et al. Beyond Data Integration. Drug Discovery Today. 2008; 13:584–589.
[5] http://www.w3.org/2001/sw/hcls/.
[6] http://en.wikipedia.org/wiki/CSHALS.
[7] Samwald, M., et al. Linked open drug data for pharmaceutical research and development. Journal of Cheminformatics. 2011; 3:19.
[8] Belleau, F., et al. Bio2RDF: towards a mashup to build bioinformatics knowledge systems. Journal of Biomedical Informatics. 2008; 41(5):706–716.
[9] Oprea, T.I., et al. Systems Chemical Biology. Nature Chemical Biology. 2007; 3:447–450.
[10] Keiser, M.J., et al. Predicting new molecular targets for known drugs. Nature. 2009; 462:175–181.
[11] Metz, J.T., et al. Navigating the Kinome. Nature Chemical Biology. 2011. [Web publication date 20 February 2011.].
[12] Xie, L., et al. Structure-based systems biology for analyzing off-target binding. Current Opinion in Structural Biology. 2011. [Web publication date 1 February 2011.].
[13] http://www.gnova.com.
[14] http://jena.sourceforge.net/ARQ.
[15] http://www.chem2bio2rdf.org.
[16] Tang J, Zhang J, Yao L, Li J, Zhang L and Su Z. ArnetMiner: Extraction and Mining of Academic Social Networks. In Proceedings of the Fourteenth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (SIGKDD'2008), pp. 990–8.
[17] Wang, H., et al. Finding complex biological relationships in recent PubMed articles using Bio-LDA. PLoS One. 2011; 6(3):e17243.
[18] Zhu, Q., et al. WENDI: A tool for finding non-obvious relationships between compounds and biological properties, genes, diseases and scholarly publications. Journal of Cheminformatics. 2010; 2:6.
[19] Ioannidis, J. Why most published research findings are false. PLoS Med. 2005; 2(8):e124.