
Utilizing open source software to facilitate communication of chemistry at RSC
Abstract:
The Royal Society of Chemistry is one of the world’s premier chemistry publishers and has an established reputation for the development of award-winning platforms such as Prospect and ChemSpider. Using a small but agile in-house development team, we have combined commercial and open source software tools to develop the platforms necessary to deliver capabilities to our community of users. This book chapter will review the systems that have been developed in-house, what they deliver to the community, the challenges encountered in developing our systems and utilizing open source code, and how we have extended available code to make it fit-for-purpose.
3.1 Introduction
The Royal Society of Chemistry (RSC) is the largest organization in Europe with the specific mission of advancing the chemical sciences. Supported by a worldwide network of 47 000 members and an international publishing business, our activities span education, conferences, science policy and the promotion of chemistry to the public. The information-handling requirements of the publishing division have always consumed the largest proportion of the available software development resources, traditionally dedicated to enterprise systems to develop robust and well-defined systems to deliver published content to customers. Internal adoption of open source solutions was initiated with the development of Project Prospect [1], and then extended with the acquisition of ChemSpider [2]. ChemSpider delivered both a platform incorporating much open source software, staff expertise in cheminformatics, as well as new and innovative functionality. The small but agile in-house development team have combined commercial and free/open source software tools to develop the platforms necessary to deliver capabilities to the user community. This book chapter will review the systems that have been developed in-house, what they will deliver to the community, the challenges encountered in utilizing these tools and how they have been extended to make them fit-for-purpose.
3.2 Project Prospect and open ontologies
RSC began exploring the semantic markup of chemistry articles, together with a number of other publishers in 2002, providing support for a number of summer student projects at the Unilever Centre in Cambridge University. This work led to an open source Experimental Data Checker [3], which parsed the text of experimental data paragraphs and performed validation checks on the extracted and formatted results. This collaboration led to RSC involvement, as well as collaboration with Nature Publishing Group [4] and the International Union of Crystallography [5], in the SciBorg project [6]. The resulting development of OSCAR [7] (Open Source Chemistry Analysis Routines) as a means of marking up chemical text and linking concepts and chemicals with other resources, was then explored and was ultimately used as the text mining service underpinning the award-winning ‘Project Prospect [1]’ (see Figure 3.1).

Figure 3.1 A ‘prospected’ article from RSC. Chemical terms link out to ontology definitions and related articles as appropriate and chemical names link out to ChemSpider (vide infra)
It was essential to develop both a flexible and cost-effective solution during this project. Software development was started from scratch, using standards where possible, but still facing numerous unknowns. Licensing a commercial product for semantic markup would have been difficult to justify and also risked both inflexibility and potential limitations in terms of rapid development. As a result, it was decided to work alongside members of the OSCAR development team, contributing back to the open source end product and providing a real business case to drive improvements in OSCAR. This enabled the creation of a parallel live production system, and RSC became the first publisher to semantically markup journal articles, this resulted in the ALPSP Publishing Innovation award in 2007 [8]. More importantly, the chosen path provided a springboard to innovation within the fields of publishing chemistry and the chemical sciences. What follows is a summary of the technical approach that was taken to deliver Project Prospect.
If the architecture for a software project is designed in the right way then in many cases it is possible to set up the system using open source software such as scaffolding [9] and migrate parts of it, as necessary, to more advanced modules, either open or closed source software, as it is determined what the requirements are. For Project Prospect the following needs were identified:
1. a means of extracting chemical names from text and converting them to electronic structure formats;
2. a means of displaying the resulting electronic structure diagrams in an interface for the users;
3. a means of storing those chemicals separately from the article XML;
4. a means of finding non-structural chemical and biomedical terms in the text.
Chemical names commonly contain punctuation, for example [2-({4-[(4-fluorobenzyl)oxy]phenyl}sulfonyl)-1,2,3,4-tetrahydroisoquinolin-3-yl](oxo)acetic acid, or spaces, like diethyl methyl bismuth, or both, and hence cause significant problems for natural language processing code that has been written to handle newswire text or biomedical articles. For this reason, the Sciborg project [6] required code that would identify chemical names so that they would not interfere with further downstream processing of text. Fortunately, a method for extracting chemical structures out of text was already available. The OSCAR software provided a collection of open source code components to meet the explicitly chemical requirements of the Sciborg project. It delivered components that determined whether text was chemical or not, RESTful web services for the Chemistry Development Kit (CDK) [10], routines for training language models, and, importantly, the OPSIN parser [11], which lexes candidate strings of text and generates the corresponding chemical structures. The original version of OPSIN produced in 2006 had numerous gaps but was still powerful enough to identify many chemicals. We also used the ChEBI database [12] as the basis of a chemical dictionary.
In order to display extracted chemical structures, the CDK was used via OSCAR. Although the relevant routines were not entirely reliable and, specifically, did not handle stereochemistry, they were good enough to demonstrate the principle. Following the introduction of the International Chemical Identifier (InChI) [13], and clear interest by various members of the publishing industry and software vendors in supporting the new standard, it was decided to store the connection tables as InChIs. The InChI code is controlled open source, open but presently only developed as a single trunk of code by one development team. The structures were stored as InChIs both in the article XML and in a SQL Server database. For the non-structural chemical and biomedical terms contained within the text, resources that were accessible to the casual reader were identified as being most appropriate. This application was launched with the IUPAC Gold Book [14], which had recently been converted to XML, and with the Gene Ontology [15]. A more detailed account of the integration is given elsewhere [16]. Unfortunately, the Gold Book turned out to have too broad a scope and too narrow coverage to be particularly useful for this work. Initially attempts were made to markup text with all of the entries in the Gold Book, but too many of them, such as cis- and trans-, were about nomenclature and parts of names, so only hand-picked selections from the resource were used.
Named reactions and analytical methods were additional obvious areas to select for the further development of ontologies applicable to chemistry publishing. RXNO [17] represents named reactions, for example the Diels-Alder reaction, which are particularly easy to identify and a method was established to determine under which classification a reaction falls. The Chemical Methods Ontology (CMO) [18] was initially based on the 600 or so terms contained within the IUPAC Orange Book [19] and then extended based on our experience with text-mining. It now contains well over 2500 terms and covers physical chemistry as well as analytical chemistry. RXNO and CMO have been provided as open source and are available from Google Code where we have trackers and mailing lists [17] [18]. When ChemSpider was acquired in 2009 (vide infra). a number of processes were changed to make use of many of the tools and interfaces available within the system. The processes associated with (1), (2) and (3) listed above have been changed but (4), the method by which non-structural chemical and biomedical terms are found in the text, remains the same. At present we utilize OSCAR3, a greatly improved version of OPSIN [11] and ACD/Labs’ commercial name to structure software [20]. The large assortment of batch scripts and XSLT transforms has now been replaced by a single program, written in C#, for bulk markup of documents, and it memorizes the results of the name to structure transformations. The key difference for structure rendering is that ChemSpider stores the 2D layouts in addition to the InChIs in the database and as a result the rendering process is now a lot simpler. The ChemSpider image renderer is also used in place of the original CDK, providing significant improvements in structure handling and aesthetics. The original RSC Publishing SQL Server database serving the Prospect Project has now been replaced by integration with ChemSpider, meaning that substructure searching is now available as well as cross-referencing to journal information from other publishers that has been deposited in ChemSpider.
New approaches are being investigated to enhance the semantic markup of RSC publications and to roll out new capabilities as appropriate. These now include the delivery of our semantically enriched articles to the Utopia platform [21], see Chapter 15 by Pettifer and collegues for more details on this innovative scientific literature tool. The development of the RSC semantic markup platform owes much of its success to the availability of the open source software components, developed by a team of innovative scientists and software developers, and these are now used in parallel with both in-house and commercial closed source software to deliver the best capabilities.
3.3 ChemSpider
ChemSpider [22] was initially developed on a shoestring budget as a hobby project, by a small team, simply to contribute a free resource to the chemistry community. Released at the American Chemical Society (ACS) Spring meeting in Chicago in March 2007, it was seeded with just over 10 million chemicals sourced from the PubChem database [23]. Following a two-year period of expanding the database content to over 20 million chemicals, adding new functionality to the system to facilitate database curation and crowd-sourced depositions of data, as well as the development of a series of related projects, ChemSpider was acquired by the Royal Society of Chemistry [2]. The original strategic vision of providing a structure-centric community for chemistry was expanded to become the world’s foremost free access chemistry database and to make subsets of the data available as open data.
The database content in ChemSpider (see Figure 3.2), now over 26 million structures aggregated from over 400 data sources, has been developed as a result of contributions and depositions from chemical vendors, commercial database vendors, government databases, publishers, members of the Open Notebook Science community and individual scientists. The database can be queried using structure/ substructure searching and alphanumeric text searching of chemical names and both intrinsic, as well as predicted, molecular properties. Various searches have been added to the system to cater to various user personae including, for example, mass spectrometrists and medicinal chemists. ChemSpider is very flexible in its applications and nature of available searches.
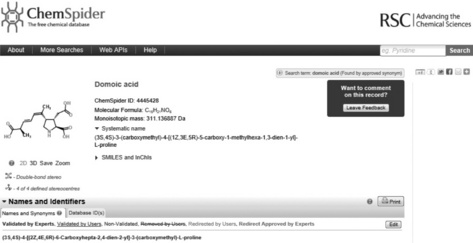
Figure 3.2 The header of the chemical record for domoic acid (http://www.chemspider.com/4445428) in ChemSpider. The entire record spans multiple pages including links to patents and publications, pre-calculated and experimental properties and links to many data external data sources and informational web sites
The primary ChemSpider architecture is built on commercial software using a Microsoft technology platform of ASP.NET and SQL Server 2005/2008 as at inception, it allowed for ease of implementation, projected longevity and made best use of available skill sets. Early attempts to use SQLite as the database were limited by performance issues. The structure databasing model was completely developed in-house. The InChI library is the basis of the ChemSpider registration system as is the exact searching in ChemSpider (which uses InChI layer separation and comparison). As a result, ChemSpider is highly dependent on the availability of the open source InChI library for InChI generation. The choice between using InChI identifiers versus alternative chemical structure hashing algorithms (e.g. CACTVS hash codes [24]) was largely based on community adoption. Attempts were made to develop our own version of hash codes early on but were abandoned quickly as the standard InChI library was already out of beta and increasingly used in the chemical community. No modifications to the InChI source code have been made except for small changes to the libraries allowing multiple versions of the InChI code to coexist in one process address space.
The GGA Bingo toolkit (SQL Server version) [25] is used for substructure and similarity searches in ChemSpider. The open source library GGA software is developed by a small team of geographically co-located developers and, to the best of our knowledge, they do not allow the source code to be modified outside of their organization. This platform was chosen over other possible solutions for ChemSpider as the team was knowledgeable, professional and agile. The original version of Bingo was made available only on SQL Server 2008 while ChemSpider was running on SQL Server 2005 at that time. As the software was available as open source, we recompiled the source code to work on SQL Server 2005 and the GGA team fixed version discrepancies quickly and provided a working version of Bingo for SQL Server 2005 within a one-day turnaround. This is a testament to the skills of the software team supporting this open source product.
In order to perform both structure searching and substructure searching, a manner by which to introduce a chemical structure drawing is required. We provide access to a number of structure drawing tools, both Java and JavaScript. Two of these structure editors are open source (GGA’s Ketcher [26] and JChemPaint [27]) and we implemented them without modification.
There are various needs on the ChemSpider system for the conversion of chemical file formats and we utilize the open source OpenBabel package for this purpose [28]. Although the software code was functional, we have identified a number of general issues with the code including inversion of stereo centers and loss of other chemical information. We believe that OpenBabel is a significant contribution to the cheminformatics community that will continue to improve in quality.
We generate 3D conformers on the fly from the 2D layouts in the database. We chose the freely available Balloon optimizer [29], primarily because it is free and fast; and it is a command line tool and was relatively easy to integrate. Balloon is not, however, open source, and cannot be modified. We use Jmol [30] to visualize the resulting optimized 3D molecular structures as well as crystallographic files (CIFs) where available. We do not use the Java-based JMol to visualize regular 2D images as it can add a significant load to browsers.
Literature linking from ChemSpider to open internet services has been established in an automated fashion taking advantage of freely available application programming interfaces to such web sites as PubMed [31], Google Scholar [32] and Google Patents [33]. Validated chemical names are used as the basis of a search against the PubMed database searching only against the title and the abstract. In this way, a search on cholesterol, for example, would only retrieve those articles with cholesterol in the title and abstract rather than the many tens of thousands of articles likely to mention cholesterol in the body of the article. A similar approach has been taken to integrate to Google Scholar and Google Patents. It should be noted that the Application Programming Interfaces (APIs) are free to access but are not open source. PubMed (through Entrez) [34] has both SOAP and RESTful APIs. The Entrez API is both extensive and robust, providing access to most of the NCBI/NLM [35] electronic databases. Google now provides RESTful APIs and has deprecated the SOAP services that it once supported. This probably reflects the trend to support only lightweight protocols for modern web applications. All of these APIs are called in a similar way: a list of approved synonyms associated with a particular ChemSpider record is listed, sorted by ‘relevance’ (which is calculated based on the length of the synonym as well as its clarity), then used to call against the API. The result (whether SOAP, XML or HTML) is then processed by an ‘adapter’, transformed into an intermediate XML representation and passed through XSLT to produce the final HTML shown in the ChemSpider records.
The value of analytical data is as reference data for comparing against other lab-generated data. Acquisition of a spectrum and comparison against a validated reference spectrum speeds up the process of sample verification without the arduous process of full data analysis. As a result of this general utility, ChemSpider has provided the ability to upload spectral data of various forms against a chemical record such that an individual chemical can have an aggregated set of analytical data to assist in structure verification. As a result of contributions from scientists supporting the vision of ChemSpider as a valuable centralizing community-based resource for chemical data for chemists, over 2000 spectra have been added to ChemSpider in the past 2 years with additional data being added regularly. These data include infrared, Raman, mass spectrometric and NMR spectra, with the majority being 1H and 13C spectra. Spectral data can be submitted in JCAMP format [36] and displayed in an open source interactive applet, JSpecView [37], allowing zooming and expansion. JSpecView is open source but the code seems to lack a clearly defined architecture and boundaries. JSpecView has been modified to visualize range selection (inverting a region’s color while dragging a mouse cursor). One of the main problems faced with supporting JSpecView is that it understands only one of the many flavors of JCAMP produced by spectroscopy vendors. This is not the fault of JSpecView but rather the poor adherence to the official JCAMP standard by the spectroscopy vendors. An alternative spectral display interface is the ChemDoodle spectral web component [38], which is a ‘Spectrum Canvas’ and renders a JCAMP spectrum in a web page along with controls to interact with it – for example to zoom in on a particular area of interest. However, it relies on HTML5, which limits its usage to modern standards compliant browsers that support HTML5 (for example Google Chrome and Firefox) and, as described earlier, limits its use in most versions of Internet Explorer unless the Google Chrome Frame plug-in is installed. This form of spectral display has not yet been implemented in the ChemSpider web interface but has been installed to support the SpectralGame [39] [40] on mobile devices.
Although ChemSpider is not an open source project per se, depending for its delivery on a Microsoft ASP.NET platform and SQL Server database, it should be clear that the project does take advantage of many open source components to deliver much of the functionality including file format conversion and visualization. In particular the InChI identifier, a fully open source project, has been a pivotal technology in the foundation of ChemSpider and has become essential in linking the platform out to other databases on the internet using InChIs.
3.4 ChemDraw Digester
The ChemDraw Digester is an informatics project bridging the previous two topics discussed – it is a tool that uses the structure manipulation programs contained in ChemSpider’s code to help enhance RSC articles. In the first section we saw that if the most important chemical compounds in a paper can be identified and deposited to ChemSpider then the article can be enhanced with links to provide readers with more compound information. These compounds were generated by using name to structure algorithms after extraction of the chemical identifier. However, more often than defining a compound by name, chemistry authors refer to and define compounds in their paper by figures in the manuscript where the molecular structures are being discussed (see Figure 3.3) [41].
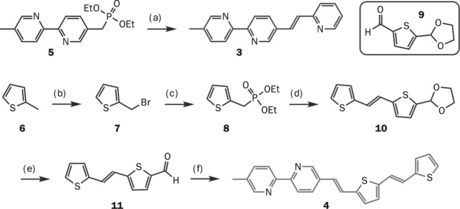
Figure 3.3 Example of figure in article defining compounds (Reproduced by permission of The Royal Society of Chemistry)
Where the images accompanying a manuscript have been generated using the structure drawing package ChemDraw [42], the RSC requests that authors supply these images not only as image files, but also in their original ChemDraw format (with the file extension ‘.cdx’), as these files preserve the chemical information of the structures within them. As the ChemDraw file format can also incorporate graphical objects and text, the files often contain labels (reference numbers or text) that correspond to the references of the compound in the corresponding manuscript, as in the example figure. Therefore, by ‘digesting’ a ChemDraw file we could potentially decorate these occurrences of the compounds’ identifying labels in the manuscript with a link to its chemical structure in ChemSpider – this is the basic aim of the ChemDraw Digester.
The most crucial part of this digestion process is to find each compound in the original ChemDraw file, match it up with its corresponding label, and then convert its 2D molecular structure into the MDL MOLfile format [43] (with extension.mol). The conversion from ChemDraw to mol format is required so that the files can be concatenated to make a MDL SDF file (with extension.sdf) suitable for deposition to ChemSpider. This SDF file [44] is also supplemented with article publication details in its associated data fields, which are used during deposition to create links from the new and existing compound pages in ChemSpider back to the source RSC article. Once deposited to ChemSpider, the related IDs of each compound can be retrieved and used to markup their names and references in the source article with reverse links to the ChemSpider compounds. The ChemDraw format is, unfortunately, not an open standard and it is not straightforward to digest in order to extract and convert the chemical structures and their associated labels. It is a binary file format, and although there is good documentation [45], deciphering it is a painstaking process and this would require considerable effort.
Fortunately, as discussed previously there is an existing routine to convert ChemDraw files to SDF using the ‘convert’ function of OpenBabel [46]. The ChemDraw digester was written using a Visual Studio, and .NET framework as a C# service with an ASPX/C# web front-end so that ultimately it can be reintegrated with the main ChemSpider web site. As a result, it could reference the native C++ OpenBabel library in the same way as the main ChemSpider code – via a wrapper managed C++ assembly (OBNET), which only exposed functionality required for the Digester and ChemSpider. The real advantage of OpenBabel being open source is that source code can be adjusted and the assembly recompiled, allowing adjustments required to deal with the real ChemDraw files from authors. These adjustments primarily involved adding new functionality, such as a new ‘splitter’ function to split ChemDraw files that contain multiple ‘fragment’ objects (molecules) into separate ChemDraw files so that each could be processed separately. Another issue is that the ChemDraw format supports more features than MOL, so some information is lost in this conversion. As a result, the ChemDraw reader had to be adjusted to read in this information and store it in the associated data fields in the SDF file generated – for example special bond types are represented by the PubChem notation. The other more important example of data lost from the original ChemDraw file is that of text labels associated with molecules. The difficulty in this case is to define how to match up a structure with its label. As a first step, OpenBabel was adjusted to recognize text labels that had been specifically grouped with a particular structure by the author. However, it became clear that in practice authors rarely used this grouping feature for this purpose, so that the vast majority of labels in the figures would be lost.
The ChemDraw Digester incorporates a review step where the digested information can be reviewed in an editable web page as shown in Figure 3.4. If a label is wrong or absent it can be amended but this is a time-consuming process, and the ultimate aim for the ChemDraw Digester is that it could be run as a fully automated process that does not require human intervention.
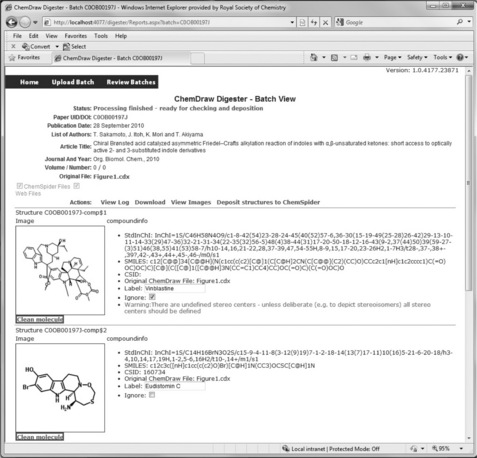
As an alternative to manual correction, the OpenBabel source code was modified to return labels for structures based on proximity, as well as grouping. A function was added which was called when a ‘fragment’ object (molecule or atom) was found. The function calculates the distance between the fragment object and all of the ‘text’ objects in the file (based on their 2D coordinates), so that the closest label to it could be identified. If the distance between the fragment and its closest text was less than the distance between that same text and any other fragment, then the value of the ‘text’ property of the text object (the text in the label) was associated with the structure and returned in the SDF file produced. Certain checks were also built in to ignore labels that do not contain any alphanumeric characters (e.g. ‘+’).
The ChemDraw Digester is presently in its final stages of development and testing, all of the processed structures in the SDF file will be reviewed for mistakes and compared with those in the figures of the article so that all discrepancies are identified. We are already aware of some areas that will need attention. Some can be dealt with by post-processing the structures in the SDF file after digestion – for example, it is common for authors to draw boxes in the ChemDraw files for aesthetic reasons, and to draw these molecules by simply drawing four straight line bonds. As a result, we have added a filter which by default ignores these rogue cyclobutane molecules, and similarly ethane molecules, which are commonly used to draw straight lines. When a structure is flagged to be ignored for any reason it will not be deposited into ChemSpider or marked up in the original article, but at the reviewing stage any automatically assigned ‘ignore’ flags can be overridden (see the Ignore checkbox in Figure 3.4). Molecules are also flagged to be ignored based on some basic checks of their chemistry such as having a non-zero overall charge, possessing atoms with unusual atom valences, undefined stereochemistry, etc. Another issue encountered was one in which the molecule is not drawn out explicitly but, for example, is represented by a single ‘node’ which is labeled, e.g. ‘FMoc’. These groups are automatically expanded but the placement of atoms in these expanded groups is sometimes peculiar and leads to ugly 2D depictions of the molecules. This can be addressed by allowing the ability to apply a cleaning algorithm to the relevant MOL structure in the SDF file to tidy up and standardize the bond lengths and angles to prevent atom overlap and very long bonds.
These are examples that can be dealt with by post-processing the structures in the SDF file. However, as OpenBabel source code availability allows customization, then issues that cannot be fixed with postprocessing can be dealt with during the initial ChemDraw to SDF conversion. One such issue is that authors may use artistic license to overlay another ChemDraw object onto a molecule – for example to only draw part of a larger structure. The objects can be lines to indicate dangling bonds (even more problems are caused when these are not drawn as graphical objects but instead as various variations of ethane molecules as in Figure 3.5(a)), graphical pictures (e.g. circles to indicate beads as in Figure 3.5(b)) or brackets (e.g. commonly used to indicate repeat units polymers as in Figure 3.5(c)). The objects are usually overlaid onto an unlabeled carbon to give the appearance of a bond from the drawn molecule to these objects. The current OpenBabel algorithm would interpret the ChemDraw by simply identifying a carbon atom, and treating any objects overlaid on it as separate entities rather than bonded in any way, and it would not be possible to detect any error in the final molecule that was output. Although it is difficult to envisage any way that we could fully interpret such molecules, we could modify the OpenBabel convert function to return a warning when a chemical structure overlaps any other ChemDraw object so that these can be ignored by default, rather than processed incorrectly. Another very common problem which is difficult to find a solution for, is dealing with Markush structures – see Figure 3.5(d). Authors commonly save valuable space in the figures of their articles by representing multiple, similar compounds by defining part of the structure with a place holder, for example the label ‘R’ and supplying a label (usually elsewhere in the ChemDraw file) defining the different groups that could be substituted for R. This would require quite an extensive alteration to OpenBabel to deal with it correctly, but it is at least conceivable.
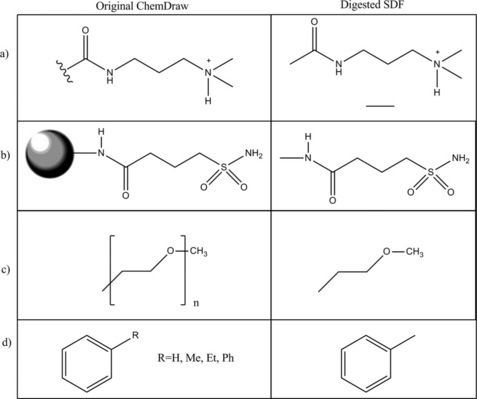
Figure 3.5 Examples of ChemDraw molecules which are not converted correctly to MOL files by OpenBabel
The long-term aim of the ChemDraw Digester is for it to process all ChemDraw files supplied with RSC articles automatically. In fact, with some extra effort, it may be possible to extract embedded ChemDraw file objects from Microsoft Word files and digest them, and this may allow even more structures to be identified, even when authors do not send ChemDraw files. However, rather than simply using the various checks on molecules to filter out and omit problem structures automatically, it would be more useful to be able to feed these warnings back to the authors to give them the opportunity to revise their ChemDraw files and images, so that they could be used as intended. For this reason, another long-term aim of the ChemDraw Digester is for it to be made available as part of the ChemSpider web site as an author tool. If this were possible then authors could upload their own ChemDraw files when writing their articles, and view the Review web page (as in Figure 3.4) and see a clear indicator of whether the structures drawn in their figures do indeed adequately define the molecules to which they are referring, and if not what issues need to be fixed. Poorly drawn structures are more common than one would expect in academic papers so the ability to raise the standard of chemistry in RSC articles would benefit both authors and readers. The Digester is presently in a testing phase with the RSC editors and we expect to roll it out to general usage within the organization shortly.
3.5 Learn Chemistry Wiki
The RSC’s objective is to advance the chemical sciences, not only at a research level but also to provide tools to train the next generation of chemists. The RSC’s LearnChemistry platform [47] is currently being developed to provide a central access point and search facility to make it easier to access the various different chemistry resources that it provides. ChemSpider contains a lot of useful information for students learning chemistry but there is also a lot of information which is not relevant to their studies, which might be confusing and distracting. As a result, the RSC is developing a teaching resource, which will belong to LearnChemistry, for students in their last years of school, and first years of university (ages 16–19), which restricts the compounds and the properties, spectra and links displayed for each, to those relevant to their studies. However, students do not just need compound information in isolation – it is most useful when linked to and from study handouts and laboratory exercises. In addition, this resource is not just intended to be read and browsed, but interactive – allowing students to answer a variety of quiz questions, and allowing chemical educators to contribute to the content – so this resource is to be called the Learn Chemistry Wiki (see Figure 3.6 [48]). The platform on which this community web site is built is required to support the ability for multiple users to contribute collaboratively and to be easily customizable both in terms of appearance and functionality.
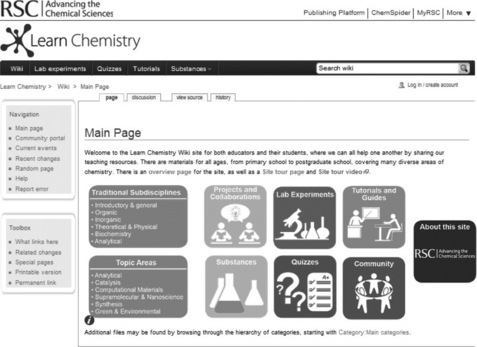
As such, an obvious start point was MediaWiki open source software [49]. It is easy to administrate and customize in terms of initial setup, managing user logins, tracking who is making changes and reverting these when necessary. It is also easy for untrained users to add pages and edit existing ones as many people are familiar with the same editing interface as Wikipedia [50]. It is also easy to program enhancements to the basic functionality as not only is all of the PHP code itself open source, but it has also been designed to allow extensions (programs called when the wiki pages are loaded) to be built into it. There are many extensions already available [51] and as these are also open source, it is straightforward to learn how they work and write new extensions. For all of these levels of MediaWiki use, there is plenty of documentation readily available by performing internet searches. An Industrial application of Mediawiki for knowledge management and collaboration is described in Chapter 13 by Gardner and Revell.
The basic setup of the wiki is straightforward – anyone can view the web site, but a login is required to edit or add pages. Anyone can register for a login and make changes, but this functionality is primarily aimed at teachers. Changes to the site will be monitored by administrators, and can be crowd-curated by other teachers. The content of the web site is separated into different namespaces. The ‘Lab’, ‘TeacherExpt’ and ‘Expt’ sections contain traditional HTML content – a mixture of formatted text, pictures and links which describe experiments (an overview, teachers’ notes and students’ notes, respectively).
Each page in the ‘Substance’ section contains compound information. Most of this information is dynamically retrieved from a corresponding ChemSpider compound page – images of its structure and a summary of its properties (molecular formula, mass, IUPAC name, appearance, melting and boiling points, solubility, etc.), and links to view safety sheets and spectra. Where there is a link to a Wikipedia page from the linked ChemSpider compound, the lead section of the Wikipedia article is shown in the page with a link to it. It is also possible to add extra information, for example references to the pages using the regular MediaWiki editing interface. There are currently approximately 2000 of these substance pages that correspond to simple compounds that would commonly be encountered during the last years of school and first years of university.
Creating these substance pages posed a variety of technical challenges. The decision to retrieve as much information as possible from ChemSpider and Wikipedia rather than to store it in the Learn Chemistry Wiki was taken in the interests of maintainability, as both of these sources will potentially undergo continuous curation. The issue of retrieving compound images from ChemSpider’s web site and incorporating them into the wiki pages was easily addressed by using the ‘EnableImageWhitelist’ option [52] in the local settings configuration. However, to retrieve text from the ChemSpider server required installation of the ‘web service’ extension [53]. This is very easy to install in the same way as the majority of MediaWiki extensions – simply by copying the source PHP file into the extensions directory of the MediaWiki setup files, and referencing this file in the main local settings file of the MediaWiki installation.
In the LearnChemistry wiki, the main identifier for each compound is its name, which appears in the title of the compound page. However, in ChemSpider the main identifier for each compound with a particular structure is its ChemSpider ID and it is more future-proof to call information from its web services by querying this rather than its name, which is potentially subject to curation. This mapping between the wiki page name and ChemSpider ID is crucial, and was painstakingly curated before the LearnChemistry pages were created. This also needs to be easily maintainable in case a change is needed for whatever reason in the future. The ‘Data’ extension [54] is used to manage this mapping. Each substance page contains hidden text, which uses the extension to set a mapping of the compound name to the ChemSpider ID. The extension can then be used to retrieve this mapped information whenever the ChemSpider ID needs to be used (e.g. in a web service call), in the substance page but also any other in the wiki, rather than ‘hardcoding’ the ChemSpider ID into many places.
The substance pages themselves were created by writing and running a MediaWiki ‘bot’ – a PHP script that accesses the MediaWiki API to login to the wiki, read information from it, or edit pages in it. There is a lot of information on the internet describing the MediaWiki API [55], and examples of bot scripts to get started [56]. In two chapters in this book, Alquier (Chapter 16) and also Harland and co-workers (Chapter 17) describe the benefits and application of semantic MediaWiki. For each batch of substance pages to be created, an input file was made containing the basic inputs required to populate the page. The bot script firstly retrieves the login page of the wiki and supplies it with user credentials, then logs in and retrieves a token to be used when accessing other pages on the wiki. The script then recurses through the input file, constructs a URL for each new substance page in edit mode, posts the new content, and then saves the changes made. The Snoopy open source PHP class [57] played the crucial role of effectively simulating a web browser in this process – it was very well documented and straightforward to implement.
Discoverability is also important for these substance pages. An important objective was to make the substance pages searchable by structure (as ChemSpider is). An easy way to do this from outside the site is to use the ‘Add HTML Meta And Title’ extension [58], which was used to set the meta-data keywords and description on each substance page for search engines to use, and making sure that the InChI key was included in the meta-data. Structure searching within the wiki (not just from internet search engines) is also necessary, so that students or teachers can draw a molecule using a chemical drawing package embedded within a wiki page. When they click on a Search button in the page, the InChI key of the drawn structure is compared with that of all the substance pages in the wiki, and any matches are returned. This is rather a specialized requirement and required the development of a new extension. Developing a new extension was made easier by investigating the range of extensions that are already available for MediaWiki and reviewing the code behind them which is enabled by the fact that the MediaWiki hooks and handlers are all open source, well documented, and transparent. The functionality of this structure search was split so as to create two new MediaWiki extensions rather than one: the first embedded a structure drawer into a wiki page and the second added a Search button, which when clicked would display the search results. The reason for splitting the functionality into two separate extensions was that various other applications of the structure drawer had been suggested (which will be described shortly) and by this design the first extension could be used for various other applications without duplicating code.
Although a new MediaWiki extension needed to be developed to add a structure editor to a wiki page, it was not necessary to start from scratch as various open source structure editors already exist that can be embedded into web pages. The GGA Ketcher [26] structure editor available in ChemSpider was chosen as the structure editor of choice and implemented in the structure drawing extension because it is easy to use and is based on Javascript so does not require any extra additional add-ins or Flash support to be installed (which could be a problem in a school environment). It was also very easy to integrate into a MediaWiki extension. To incorporate a Ketcher drawing frame into a HTML page, it was simply necessary to download the Javascript and CSS files that comprise the Ketcher code, reference these in the head section of the HTML of a wiki, add the Ketcher frame, table and buttons to the body of the HTML, and add an onload attribute to the page to initialize the Ketcher frame. The only part of these steps which was not immediately straightforward for a version 1.16.0 MediaWiki extension to add to the web page in which it was called, was the step of adding an onload attribute to the page, but a workaround was used that involved adding a Javascript function to the HTML head, which was called at the window’s onload event. The resulting extension was called the KetcherDrawer extension.
The accompanying extension would add a Search button and would need to perform several actions when clicked. The first action is to take the MOL depiction of the molecule that has been drawn (which is easily retrieved via a call to the Ketcher Javascript functions) and convert it into an InChI key so that this can be searched on. This conversion is done using the IUPAC InChI code [59], and any warnings that are returned are displayed in the wiki page, for example if stereochemistry is undefined or any atom has an unusual valence. The next action is to post this InChIKey to a search of the wiki – this was done by using the MediaWiki API to silently retrieve the results of this search. If one matching substance page was found then the page would redirect to view it. If no match for the full InChIKey was found, then a second search was submitted to the MediaWiki API to find any matches for just the first half of the InChIKey. This roughly equates to broadening the search to find matches for the molecule’s skeleton. Any results from this search are listed in the wiki page itself, with a warning that no exact match could be found for the molecule but that these are similar molecules. After these two extensions had been written, it was then possible to add the functionality to perform a structure search within the wiki just by calling the KetcherDrawer and KetcherQuizAnswer extensions in the page.
A DisplaySpectrum extension was also written to add an interactive spectrum to a wiki page. As explained earlier, we use two possible display tools for spectra: JspecView and the ChemDoodle spectral display. Approximately two-thirds of current viewers of the RSC educational web sites use a web browser that does not support canvases, and in most school environments the installation of plug-ins is not an option. To make the best of both worlds, the DisplaySpectrum extension automatically tests the browser being used: if it supports canvases then it displays the spectrum using the ChemDoodle spectral viewer [38], and if not it uses the JspecView applet [37].
This chapter has demonstrated how a simplified version of both the information in, and functionality of ChemSpider has been integrated into the LearnChemistry educational web site, using the collaborative aspects of MediaWiki to allow these and other related pages, such as quizzes and descriptions of experiments to then be built up. The system was pieced together from many different open source programs and libraries, which would not have been possible without the flexibility of the MediaWiki platform on which the platform is based.
3.6 Conclusion
RSC has embraced the use of free/open source cheminformatics and Wiki tools in order to deliver multiple systems to the chemistry community that facilitate learning, data sharing and access to data and information of various types. By utilizing open source code where appropriate, and by integrating with other commercial platforms, we have been able to deliver a rich tapestry of functionality that could not otherwise have been achieved without significantly higher investment. In choosing our commercial vendor for our substructure search engine, we also opted for an open source platform with the GGA software.
Our experiences of using free/open source software are generally very positive. In a number of cases we have been able to take the software components as are and drop them into our applications to be used without any recoding and using the existing software interfaces as delivered. In most cases, our involvement with the code developers has either been negligible or has required significant dialog to resolve issues. In the cheminformatics domain of open source software, we have found commercial open source software to be of excellent quality and rigorously tested and well supported. For open source software of a more academic nature, we have found that small teams (where the software is supported by one group, for example) are highly responsive and effective in addressing identified issues, whereas applications with a broad development base are less so. In certain cases we have had to invest significant resources in optimizing the software for our purposes and knitting it into our applications. We generally find that documentation suffices for our needs, or that our development staff can understand the code even without complete documentation.
The true collaborative benefits of platforms such as ChemSpider will be felt as the multitude of online resources are integrated into federated searches and semantic web linking in a manner that single queries can be distributed across the myriad of resources to provide answers through a single interface. There is a clear trend in life sciences towards more open access to chemistry data. In the near future this may provide additional pre-competitive data allowing the development of federated systems such as the Open PHACTS [60], using ChemSpider as an integral part of the chemistry database and search engine. The Open PHACTS platform will allow pharmaceutical companies to link data across the abundance of life science databases that are already and will increasingly become available. ChemSpider is likely to become one of the foundations of the semantic web for chemistry and, with an ongoing focus for enabling collaboration and integration for life sciences, will be an essential resource for future generations.
3.7 Acknowledgments
ChemSpider is the result of the aggregate work of many contributors. All core ChemSpider development is led by Valery Tkachenko (Chief Technology Officer) and we are indebted to our colleagues involved in the development of much of the software discussed in this chapter. These include Sergey Shevelev, Jonathan Steele and Alexey Pshenichnov. Our RSC platforms are supported by a dedicated team of IT specialists that is second to none. The authors acknowledge the support of the open source community, the commercial software vendors (specifically Accelrys, ACD/Labs, GGA Software Inc., OpenEye Software Inc., Dotmatics Limited), many data providers, curators and users for their contributions to the development of the data content in terms of breadth and quality.
3.8 References
[1] Project Prospect. [Accessed September 2011]; Available from: http://www.rsc.org/Publishing/Journals/ProjectProspect/FAQ.asp
[2] Royal Society of Chemistry acquires ChemSpider. [Accessed September 22nd 2011]; Available from: http://www.rsc.org/AboutUs/News/PressReleases/2009/ChemSpider.asp
[3] Adams, S.E., et al. Experimental data checker: better information for organic chemists. Org Biomol Chem. 2004; 2(21):3067–3070.
[4] Nature Publishing Group. [Accessed September 2011]; Available from: http://www.nature.com/npg_/company_info/index.html.
[5] International Union of Crystallography. Available from: http://www.iucr.org/.
[6] Sciborg Project. [Accessed September 2011]; Available from: http://www.cl.cam.ac.uk/research/nl/sciborg/www/.
[7] OSCAR on Sourceforge. [Accessed September 2011]; Available from: http://sourceforge.net/projects/oscar3-chem/.
[8] Project Prospect wins ALPSP award. [Accessed September 2011]; Available from: http://www.rsc.org/Publishing/Journals/News/ALPSP_2007_award.asp.
[9] Scaffolding. [Accessed September 2011]; Available from: http://depth-first.com/articles/2006/12/21/scaffolding/.
[10] Steinbeck, C., et al. The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J Chem Inf Comput Sci. 2003; 43(2):493–500.
[11] Lowe, D.M., et al. Chemical name to structure: OPSIN, an open source solution. J Chem Inf Model. 2011; 51(3):739–753.
[12] de Matos, P., et al. Chemical Entities of Biological Interest: an update. Nucleic Acids Res. 2010; 38:D249–D254. [(Database issue)].
[13] The IUPAC International Chemical Identifier (InChI). [Accessed September 2011]; Available from: http://www.iupac.org/inchi/.
[14] IUPAC Gold Book. [Accessed September 2011]; Available from: http://goldbook.iupac.org/.
[15] The Gene Ontology. [Accessed September 2011]; Available from: http://www.geneontology.org/.
[16] Batchelor, C.R., Corbett, P.T., Semantic enrichment of journal articles using chemical named entity recognition. ACL ‘07 Proceedings of the 45th Annual Meeting of the ACL on Interactive Poster and Demonstration Sessions. 2007:45–48.
[17] The RXNO Reaction Ontology. [Accessed September 2011]; Available from: http://code.google.com/p/rxno/.
[18] The Chemical Methods Ontology. [Accessed September 2011]; Available from: http://code.google.com/p/rsc-cmo/.
[19] Inczedy, J., Lengyel, T., Ure, A.M., Compendium of Analytical Nomenclature (definitive rules 1997) – The Orange Book, 3rd edition, 1998.
[20] ACD/Labs Name to Structure batch. Available from: http://www.acdlabs.com/products/draw_nom/nom/name/.
[21] Hull, D., Pettifer, S.R., Kell, D.B. Defrosting the digital library: bibliographic tools for the next generation web. PLoS Comput Biol. 2008; 4(10):e1000204.
[22] ChemSpider. [Accessed September 2011]; Available from: http://www.chemspider.com.
[23] The PubChem Database. [Accessed September 2011]; Available from: http://pubchem.ncbi.nlm.nih.gov/.
[24] Gregori-Puigjane, E., Garriga-Sust, R., Mestres, J. I ndexing molecules with chemical graph identifiers. J Comput Chem. 2011; 32(12):2638–2646.
[25] The GGA Software Bingo Toolkit. [Accessed September 2011]; Available from: http://ggasoftware.com/opensource/bingo.
[26] The GGA Ketcher Structure Drawer. [Accessed September 2011]; Available from: http://ggasoftware.com/opensource/ketcher.
[27] JChemPaint Sourceforge Page. [Accessed September 2011]; Available from: http://sourceforge.net/apps/mediawiki/cdk/index.php?title=JChemPaint.
[28] OpenBabel Wiki Page. [Accessed September 2011]; Available from: http://openbabel.org/wiki/Main_Page.
[29] The Balloon 3D Optimizer. [Accessed September 2011]; Available from: http://users.abo.fi/mivainio/balloon/.
[30] Jmol: An Open Source Java viewer for chemical structures in 3D. [Accessed September 2011]; Available from: http://jmol.sourceforge.net/.
[31] PubMed. [Accessed September 2011]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/.
[32] Google Scholar. [Accessed September 2011]; Available from: http://scholar.google.com/.
[33] Google Patents. [Accessed September 2011]; Available from: http://www.google.com/patents.
[34] Entrez, the Life Sciences Search Engine. [Accessed September 2011]; Available from: http://www.ncbi.nlm.nih.gov/sites/gquery.
[35] NCBI, the national Center for Biotechnology Information. [Accessed September 2011]; Available from: http://www.ncbi.nlm.nih.gov/.
[36] Published JCAMP-DX Protocols. [Accessed September 2011]; Available from: http://www.jcamp-dx.org/protocols.html.
[37] Lancashire, R.J. The JSpecView Project: an Open Source Java viewer and converter for JCAMP-DX, and XML spectral data files. Chem Cent J. 2007; 1:31.
[38] ChemDoodle web components [Accessed September 2011]; Available from: http://web.chemdoodle.com/.
[39] Bradley, J.C., et al. The Spectral Game: leveraging Open Data and crowdsourcing for education. J Cheminform. 2009; 1(1):9.
[40] The SpectralGame. [Accessed September 2011]; Available from: http://www.spectralgame.com/.
[41] Younes, A.H., et al. Electronic structural dependence of the photophysical properties of fluorescent heteroditopic ligands – implications in designing molecular fluorescent indicators. Org Biomol Chem. 2010; 8(23):5431–5441.
[42] Cambridgesoft ChemDraw. [Accessed September 2011]; Available from: http://www.cambridgesoft.com/software/chemdraw/.
[43] The Molfile Format. [Accessed September 2011]; Available from: http://goldbook.iupac.org/MT06966.html.
[44] The SDF file format. [Accessed September 2011]; Available from: http:// www.epa.gov/ncct/dsstox/MoreonSDF.html#Details.
[45] CDX File format specification. [Accessed September 2011]; Available from: http://www.cambridgesoft.com/services/documentation/sdk/chemdraw/cdx/ index.htm.
[46] Guha, R., et al. The Blue Obelisk-interoperability in chemical informatics. J Chem Inf Model. 2006; 46(3):991–998.
[47] Learn Chemistry. [Accessed September 2011]; Available from: http://www.rsc.org/learn-chemistry.
[48] Learn Chemistry Wiki. [Accessed September 2011]; Available from: http:// www.rsc.org/learn-chemistry/wiki.
[49] MediaWiki. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/MediaWiki.
[50] Wikipedia. [Accessed September 2011]; Available from: http://www.wikipedia.org/.
[51] Mediawiki Extensions. [Accessed September 2011]; Available from: http:// www.mediawiki.org/wiki/Category:All_extensions.
[52] MediaWiki EnableImageWhitelist extension. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/Manual:$wgEnableImageWhitelist.
[53] MediaWiki Webservice extension. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/Extension-.Webservice.
[54] Mediawiki Data extension. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/Extension:Data.
[55] MediaWiki API. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/API:Main_page.
[56] MediaWiki Bot to make pages. [Accessed September 2011]; Available from: http://meta.wikimedia.org/wiki/MediaWiki_Bulk_Page_Creator.
[57] Snoopy PHP class. [Accessed September 2011]; Available from: http://sourceforge.net/projects/snoopy/.
[58] MediaWiki Add HTML Meta and title extension. [Accessed September 2011]; Available from: http://www.mediawiki.org/wiki/Extension:Add_ HTML_Meta_and_Title.
[59] IUPAC InChI v1.03. [Accessed September 2011]; Available from: http://www.iupac.org/inchi/release103.html.
[60] Open PHACTS. [Accessed September 2011]; Available from: http://www.openphacts.org/.