
3.6. Heat of Reaction/Heat of Formation
Heat of reaction is an important parameter in estimating reactive hazards. If the end product composition is known then heat of reaction can be determined by the difference between chemical energy of products and reactants. Put differently:
![]()
Typically heat of reaction in a weight basis is more commonly used as opposed to mole basis for reactivity assessment (Medica, 1995).
Present approaches involve estimating heat of reaction using calorimetric techniques (Hsu et al., 2012; Preston et al.). However, assessing reactivity through calorimetric techniques is resource consuming and thus only practical for a limited set of compounds (Saraf et al., 2003). As far as modeling approaches go, Dow Chemical has been a pioneer in using computational chemistry to estimate thermochemical properties, including heat of reaction. Quoting from (Westmoreland, 2002):
Dave Frurip, a member of the laboratory involved in producing the JANAF tables of thermochemical properties (Dow, 1971), expressed the opinion that computational chemistry for the calculation of change in enthalpy of reaction, a crucial property in safety analysis, has reached “maintenance mode.”
Significant cost advantage has already been proven in using computational chemistry to estimate the heat of reaction (Westmoreland, 2002). In 1996, at a conference, a speaker from Dow Chemical showed that the cost of G2 or Gaussian-2 quantum chemical calculation for calculating heat of formation ($20,000) is less than 30% of the cost ($70,000) incurred to obtain the same through calorimetric measurements (Westmoreland, 2002). The advancement of powerful computers, at the beginning of the year 2000, made the comparison looked paler, that is, $2000 for a G3 or Gaussian-3 quantum calculation to $100,000 for a calorimetric measurement. As a side note, Gaussian-n theories are known as quantum chemistry composite methods intending for high accuracy calculations through combining results of several calculations. Several works (Curtiss et al., 1990, 1991, 1998, 2007; Pople et al., 1989; Curtiss and Raghavachari, 1993) exists on Gaussian-n theories and their implementation, details of which is beyond the scope of this book. However, we will look into various methods of estimating heat of reaction. Note that, computational techniques for estimation of heat of reaction are also applicable toward estimation of heat of combustion and heat of formation.
3.6.1. Macroscale Modeling Approaches
CART or calculated adiabatic reaction temperature–based approach to assess reactivity hazards employs Gibbs free energy minimization technique for heat of reaction estimation. The methodology requires the estimation of the adiabatic temperature increase due to the reaction from estimating the heat of reaction and average specific heat information of the reaction mixture. Adiabatic reaction temperature can be calculated as
![]()
A 1400 K value of the temperature rise is defined as the cut-off value for a possible explosion scenario (Saraf et al., 2003). Lower values such as 1200 K have been recommended (Melhem and Shanley, 1996) for a conservative approach.
As mentioned above, minimization of Gibbs free energy can be implemented to estimate adiabatic reaction temperature. Employing Gibbs free minimization of the reactants and possible products, one can find the mixture composition at thermodynamic equilibrium. While both kinetics and thermodynamics of the system is important, for instantaneous or fast reactions such as combustion, a system will achieve thermodynamic equilibrium in a very short span of time, given the reactant with stoichiometric proportions are available. Process Simulation software such as ASPEN Technology has module (RGibbs) built in it to help user find equilibrium composition through minimization of Gibbs free energy for a multicomponent multiphase (solid–liquid–gas) system. Other computer programs, COCO (van Baten, 2010) and APMonitor Optimization Suite, also have the ability to perform same calculations. It has been also illustrated that using Microsoft Excel's solver functionality; such a problem can be solved (Lwin, 2000). Through the process of Gibbs energy minimization, one is able to estimate the heat of reaction and average value of specific heat to leading to calculation of adiabatic reaction temperature.
As an example, let's go through briefly over the steps that are recommended for Gibbs free energy minimization (Lwin, 2000) for a gas phase reaction:
1. Define total Gibbs free energy of the system with n moles as 
2. Define fugacity coefficient. For gas phase reaction, it can be defined as: 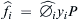
3. Substitute standard state (denoted by superscript “0”) property values for each component
4. Substitute fugacity coefficient equation for gas phase reaction into Gibbs free energy equation
5. Minimize the total Gibbs free energy expression subject to the elemental mass balance over the whole system
Implementation of the above mentioned steps are available in the computer program APMonitor Optimization Suite (Hedengren, 2008). Owing to complexities involved, mathematical approaches implemented toward minimization of Gibbs free energy for a multicomponent and multiphase system are various. The set includes linear programming (Rossi et al., 2009), nonlinear programming (Castier et al., 1989; Castillo and Grossmann, 1981; Mather, 1986), global optimization technique (McDonald and Floudas, 1995), interval method (Lin and Stadtherr, 2004), and several other approaches along with variations of the methods mentioned here. Details related to mathematical algorithms of these methods are skipped here.
Problem:
Liquid pentane is burned with 20% excess air. The initial temperature of both the reactants is 298 K. Assuming complete combustion, find the adiabatic flame temperature:
1. Assuming constant Cp for each component
2. Assuming Cp takes the form A + BT for each component
The ability to determine multiphase equilibrium allows one to define the final state of a given system. For scenarios with complete combustion.
3.6.2. Molecular Modeling Approaches
3.6.2.1. Molecular group activity
Amount of energy or heat released from decomposition or reaction correlate strongly to the potential intensity of reactive hazards. CHETAH (Frurip et al., 1989; Davies et al., 1985), an ASTM computational program, which stands for Chemical Thermodynamic and Energy Release Program, has the ability to assess chemical reactivity hazard. It is based on implementation of Benson's method of group activity (Benson et al., 1969; Benson and Buss, 1958). The proposed law can be defined as: “If 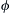 is a molecular property, then for the disproportional reaction:
is a molecular property, then for the disproportional reaction: 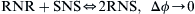 as the separation between R and S becomes large compared to their dimensions.” The basis for the law is analogous to the fact that in infinitely dilute multicomponent systems, molecular properties can be reasonably and accurately estimated by adding contribution from individual atoms toward these properties. This is possible because of the short-range intermolecular forces in the absence of substantial Coulombic interaction, such as in the case of hydrocarbons. However, with the presence of long-range Coulombic forces, such as for polar molecules, such an approach will lead to erroneous molecular property estimation. While CHETAH's principal aim is to predict deflagration/detonation potential from molecular structure, it is as well a useful tool to assess chemical reactivity hazard owing to its ability to estimate heat of reaction and thermodynamic properties of individual substances through the same principles. It identifies and estimates the greatest energy release scenario of reactants decomposing into different product species. Benson group increment theory (Benson and Buss, 1958; Benson et al., 1969), developed additivity rules to be implemented on experimentally obtained heat of formation values for individual groups of atoms to calculate the entire heat of formation of a molecule under investigation. The group additivity relation is generally obeyed within 0.6 kCal/mol for estimation of
as the separation between R and S becomes large compared to their dimensions.” The basis for the law is analogous to the fact that in infinitely dilute multicomponent systems, molecular properties can be reasonably and accurately estimated by adding contribution from individual atoms toward these properties. This is possible because of the short-range intermolecular forces in the absence of substantial Coulombic interaction, such as in the case of hydrocarbons. However, with the presence of long-range Coulombic forces, such as for polar molecules, such an approach will lead to erroneous molecular property estimation. While CHETAH's principal aim is to predict deflagration/detonation potential from molecular structure, it is as well a useful tool to assess chemical reactivity hazard owing to its ability to estimate heat of reaction and thermodynamic properties of individual substances through the same principles. It identifies and estimates the greatest energy release scenario of reactants decomposing into different product species. Benson group increment theory (Benson and Buss, 1958; Benson et al., 1969), developed additivity rules to be implemented on experimentally obtained heat of formation values for individual groups of atoms to calculate the entire heat of formation of a molecule under investigation. The group additivity relation is generally obeyed within 0.6 kCal/mol for estimation of 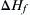 (Benson and Buss, 1958).
(Benson and Buss, 1958).
CHETAH can calculate three major impact sensitivity indicators (Harrison, 2005) to assess chemical reactivity hazard as illustrated below:
1. Maximum enthalpy of decomposition with the assumption that in the worst-case scenario, the chemical decomposes into a set of products releasing maximum amount of energy
2. Modified version of calculating maximum enthalpy of decomposition, taking into account, stoichiometry, number of peroxide bonds, etc.
3. Net plosive density, defined as a group contribution index weighting stabilizing and destabilizing molecular fragments
Accuracy of CHETAH, or put differently, applicability of Benson's group activity method, in assessing chemical reactivity hazard has been tested against experimental data and implementation of all three approaches resulted in more than 80% of correct predictions as illustrated in Figure 3.13 (Harrison, 2005).
A subset of data presented in (Wei et al., 2004b) is presented in Table 3.10 to illustrate effectiveness of CHETAH in estimating heat of formation of various chemical compounds.
While implementation of Benson's group activity method through CHETAH has been very successful, it can fail short of expectation where the values of Benson group properties are missing or erroneous and do not take into account sensitivities of molecules to the different initiation modes or variation in process conditions. To illustrate, let us recreate the example from Saraf et al. (2003) in Figure 3.14. The calculated maximum heat of decomposition (energy released and denoted by the negative sign) for hydrogen peroxide yields a value of −0.743 kcal/g or −3.109 kJ/g, which is higher than the required −2.929 kJ/g criteria used in CHETAH to define high reactivity hazard. Note that, such an approach does not address the effect of change in process conditions or estimation of heat of decomposition under various process conditions to guide toward safer process conditions while working with any potential reactivity hazard such as hydrogen peroxide at certain conditions.

Figure 3.13 Assessment of CHETAH accuracy against experimental data for predicting the presence of reactive chemical (Harrison, 2005).
Table 3.10
Comparison of Heat of Formation Values from CHETAH with Experimental Data (Wei et al., 2004b)
| Chemical | Structure |
|
|
| CHETAH | Experimental | ||
| Hydrogen cyanide |

|
130.54 | 135.14 |
| Formaldehyde |

|
−125.31 | −115.9 |
| Methanol |
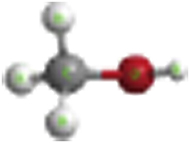
|
−200.83 | −201.17 |
| Butadiene |

|
110.16 | 108.78 |
| Nitric acid |
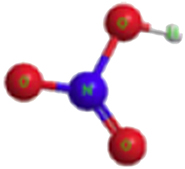
|
−134.31 | −134.31 |

In addition, in scenarios where significant long-range molecular interaction is expected, such an approach is not recommended to use as the basic assumption is violated.
Heat of decomposition, or the heat released during the decomposition process, is the energy that is released on the decomposition of the material and is often chosen as a basis for estimating stability of the chemical in question. QSPR-based approaches have been implemented in assessing chemical or material stability (or the lack of) through development of predictive models for estimating heat of decomposition. As discussed before, the basis for QSPR-based approaches is their molecular descriptors. The estimation of these molecular descriptors can be performed in various ways including first principle calculations and employing experimental data sets. In Fayet et al. (2010a), a QSPR-based approach targeted toward nitroaromatic molecules was developed to assess their thermal stability and electric spark sensitivity. The authors used topological, geometrical, electronic, and quantum chemical molecular descriptors to associate adequate molecular descriptors with the experimental properties of the molecules.

Figure 3.14 Hazard evaluation for hydrogen peroxide using CHETAH 7.2 (Saraf et al., 2003).
While numerous molecular descriptors have been utilized until date (Karelson, 2000), the work, as shown in Figure 3.15 attempted to build a QSPR model based on a subset of such molecular descriptors. In this particular study, the authors employed the use of Quantum chemical calculations to optimize the molecular structures of the molecules of interest prior to carrying out regression analyses for mapping a set of molecular descriptor values of a specific property of interest. The form of the multiple linear regression as implemented in this study was

![]()
where Y is predicted property, Xi is ith molecular descriptor, ai is regression constant for ith molecular descriptor, and a0 is universal regression constant.
Based on data obtained from differential scanning calorimetry results, in a separate work (Fayet et al., 2009b), the researchers established the following correlation for enthalpy of decomposition:
![]()
The coefficient of determination, or R squared (R2), was calculated to be 0.91 in this case. The descriptors used are defined as follows:
![]()
![]()
It must be noted that, while developing a multiple linear regression model for QSPR model development, one may even omit the presence of a universal regression constant, that is a constant, which is not associated with any particular molecular descriptors, and formulate it as
![]()
As an example, in Saraf et al. (2003), a QSPR model for energy of reaction to nitro compounds was developed as
![]()
The two major sources of estimation of these molecular descriptors typically are:
• Experimental thermokinetic data
• Quantum mechanical calculations
Another very important factor, while developing QSPR models, is to determine which and how many molecular descriptors should contribute toward the development of a QSPR model to the satisfactory confidence level. Descriptors for reactivity hazard assessment can be classified in two different types:
• Local
• Global
Local descriptors are specific to a type of compounds and hence may be undefined for a different kind of compounds. An example of local descriptor could be the number of nitro groups in a compound. Global descriptors, as the name suggests, is applicable to any compound. One such example of global descriptor could be the molecular weight of a compound. Another one, a conventional empirical descriptor used in screening technique for hazard assessment of energetic materials is the oxygen balance as defined in the work by Lothrop and Handrick (1949). Using stoichiometry, it is defined as
![]()
3.6.2.2. Choosing descriptors
It is recommended that the act of choosing molecular descriptors should be done keeping the following in mind:
• Proven physics-based strong correlation between property and descriptor
• Minimized number of descriptors
• Uncorrelated and unique descriptors
• Invariant with respect to atom indexing, molecular translation, and rotation
Several methods are availed by researchers to help them choose appropriate descriptors such as correlation, F-score, genetic algorithm, and random forest. Note the presence of “physics-based” wording. A good correlation between a descriptor and a property should be explainable physically.
Let us examine the applicability of OB descriptor, which has been defined before. Since in here, fundamentally combustion is taking place, a measure of oxygen balance and attempt to correlate that with explosive properties and intuitive. However, note that there are several factors, which results into a clarion call for in corporation of other molecular descriptors. OB descriptor value does not distinguish between oxygen that is loosely bound and easily available for combustion versus one that is already bound to carbon or hydrogen in a very stable manner. Nor does that addresses reactivity hazard associated oxygen free hazardous compounds such as acetylene, acetylides, azides, and explosive nitrides (Shanley and Melhem, 1995). Sensitivity to reaction initiation can also be not addressed appropriately with oxygen balance approach alone. Thermochemical and kinetic relations based descriptors need to be considered for a better model development.
In addition to choosing molecular descriptors, another important factor to consider is when to stop adding descriptors. A simple technique, called “breaking rule,” as employed in several studies (Katritzky et al., 2007; Fayet et al., 2010c), looks into the improvement in statistical quality of the model. It compares the change in validation correlation coefficient R2 (or cross-validation correlation coefficient 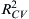 ), with addition of new descriptors.
), with addition of new descriptors.
Based on the findings as illustrated in Figure 3.16, the authors of the work developed a four parameter–based model for estimating heats of decomposition of nitroaromatic compounds:
![]()
where 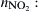 number of nitro groups,
number of nitro groups,  average one-electron reactivity index for a carbon atom,
average one-electron reactivity index for a carbon atom,  mean polarizability, and
mean polarizability, and  : average information content, a topological index.
: average information content, a topological index.
3.6.2.3. First principle–based approaches
We have illustrated the importance of accurate estimation of heat of reaction in assessing a chemical reactivity hazard. One of the major challenges in this area lies in the form of unavailable data. If prior data are available, model development as demonstrated before is possible through training and validating a model. In the absence of available data, quantum mechanical calculation based approaches can accurately estimate the heat of reaction, formation or combustion as required.

Figure 3.16 Change in R2 with increment in descriptors for QSPR model development of Heats of decomposition (Fayet et al., 2010c).
Let us consider the study done by Miranda et al. (2012). Noting the importance of 2,1,3-benzothiadiazole as organic light emitting diodes, organic photovoltaic and biosensor, the authors pointed out the lack of information on enthalpy of formation for compounds with sulfur–nitrogen bonds, which are not sulfonamides. To address, the authors employed quantum mechanical approach, validated against experiments, in efforts to find out the heat of combustion. In this benzothiadiazole problem, as mentioned above, the authors followed the following steps in order to estimate the gas phase enthalpy formation:
• The structure of 2,1,3-benzothiadiazole molecule is fully optimized with DFT at the B3LYP/6-31G (d) level. The obtained coordinates of the stationary points are characterized as minima through frequency calculation at the same level. It cannot be stressed enough, that obtaining an accurate structure of a molecule is a critical part in this approach.
• Estimate single point energy of the molecule using higher levels of theory such as QCISD (T)/6-31G (d) and MP2/GTMP2Large
• Correct electronic energies through introduction of vibrational, rotational, translational, and the pV terms computed at the B3LYP/6-31G (d) level to obtain final enthalpies at 298.15 K
• Use appropriate reactions demonstrating the formation of 2,1,3-benzothiadiazole and implementing information obtained from above-mentioned steps, estimate standard molar enthalpy of formation at 298.15 K
![]()
Implementation of the above-mentioned steps for a reaction process would result in estimation of heat of reaction, formation or combustion. However, it is of paramount importance to note that, while quantum mechanical calculations has the potential to provide very accurate information, appropriate selection of model and basis sets are fundamental toward an efficient and accurate set of results. Table 3.11 illustrates how various quantum mechanics models at the semiempirical level compare with respect to their accuracy in energy calculations and their scaling with number of atoms. One can observe the positive correlation between increase in scaling with model accuracy in estimating molecular energetics. Please note that the table is not comprehensive in nature and presented to provide an understanding in cost versus accuracy tradeoff in quantum mechanical calculations.
Let us also revisit the work on evaluation of thermal decomposition hazard associated with hydroxylamine nitrate as done by employing quantum mechanics as a screening tool prior to conducting more expensive experiments (Wei et al., 2006). The authors performed reactivity screening using semiempirical quantum mechanical calculation package, MOPAC (Molecular Orbital Package), and ASTM Chemical Engineering Thermodynamics and Hazard Evaluation (CHETAH). We have already discussed on the usage of CHETAH in the previous section. For the quantum mechanical calculations, the levels of theory used in MOPAC were AM1 and PM3. Similar to the steps described above, the approach applied was as provided in a Gaussian white paper (Ochterski, 2000). Put differently, the methodology consisted of structure optimization and energy minimization, energy estimation, incorporation of energy correction factor through introduction of translational, electronic, rotational, and vibrational motion partition function, and, finally, estimation enthalpy of reaction at the desired temperature. A detailed description with a slight variation of the steps is also provided in Foresman and Frisch (1996).
Table 3.11
Comparison of Various Semiempirical Quantum Mechanical Models
| Model | Name | Description | Error | Scaling | Cost |
| Equil Geom, kcal/mol | ∼Basis function | Relative, 1–10 | |||
| AM1 | Austin Model 1 | Semiempirical | 10–30 | N2 | 1 |
| HF | Hartree Fock | Averaged electron–electron | 10–20 | N4 | 2 |
| B3LYP | Becke, Lee, Yang, Parr | Density functional | 5 | N4 | 3 |
| MP2 | Moller–Plesset | Pertubation, correlated, 2nd order | 10 | N5 | 3 |
| MP3 | Moller–Plesset | Pertubation, correlated, 3rd order | 5 | N6 | 5 |
| CISD | CI singles, doubles | Configuration interaction, correlated | 5.8 | N6 | 5 |
| CCD | CC doubles | Coupled cluster, correlated | 2.4 | N6 | 5 |
| CCSD | CC singles, doubles | Coupled cluster, correlated | 1.9 | N6 | 6 |
| QCISD | QC singles, doubles | Quadratic configuration interaction | 1.7 | N6 | 6 |
| MP4SDQ | Moller–Plesset | Perturb, corr, 4th order, ignore triples | 2.7 | N7 | 6 |
| MP4 | Moller–Plesset | Pertubation, correlated, 4th order | 1.3 | N7 | 7 |
| G2 | G2, multilevel corrections | Composite method | 1.2 | N7 | 6 |
| G3+ | G3, multilevel corrections | Composite method, Truhlar, et al. | 1.0 | N7 | 7 |
| QCISD (T) | QC singles, doubles, (triple) | Quadratic configuration interaction | 0.3 | N7 | 7 |
| MP5 | Moller–Plesset | Pertubation, correlated, 5th order | 0.8 | N8 | 8 |
| MP6 | Moller–Plesset | Pertubation, correlated, 6th order | 0.3 | N9 | 9 |
| CCSD (T) | CC singles, doubles (triple) | Coupled cluster, singles, doubles, triple | 0.3 | N8 | 8 |
| CCSDT | CC singles, doubles triples | Coupled cluster, singles–triple | 0.2 | N8 | 9 |
| MP7 | Moller-Plesset | Pertubation, correlated, 7th order | N10 | 10 | |
| CCSDTQ | CC singles, doubles triples, quadruples | Coupled cluster, singles–quadruples | 0.01 | N10 | 10 |

Source: Argonne National Laboratory, US Department of Energy, http://www.cse.anl.gov/
In addition to assisting estimation of heat of formation of a chemical, first principle calculations can also be put to work in places where considerable disagreement between experimental values exists. Consider the case for formaldehyde and acetaldehyde (da Silva et al., 2006). As demonstrated by Silva et al., the disagreement among experimentally estimated values for heat of formation of formaldehyde and acetaldehyde exceeds the allowed standard error margins. The widely reported values for standard enthalpy of formation of formaldehyde are −26.0 and −27.7 kcal/mol at 298 K with acceptable error limits are less than the difference in these values, that is, 1.7 kcal/mol. This illustrates that even with the availability of experimental data, care must be taken in ensuring utilizing accurate information. To address these discrepancies, in da Silva et al. (2006), the authors implemented first principle calculations such as DFT and ab initio–level semiempirical theories to carefully examine the accurate heat of formation for formaldehyde and acetaldehyde. The approach taken by the authors can be broken down in the following set of steps:
1. Identify reactions with known enthalpies from literature involving molecule of interest, that is, formaldehyde in da Silva et al. (2006)
2. Calculate enthalpies of reactions using various levels of quantum mechanical calculations. Interested readers can find the details in the article
3. Compare computationally estimated reaction enthalpies
a. Among various computational calculations
b. Among experimentally estimated and computationally estimated values
4. Identify a subset of reactions where reasonable agreement is established among all computational approaches and experimental results
5. Identify a subset of reactions where enthalpies of formation are available for all species participating in the reaction
6. Calculate enthalpy of formation of the molecule of interest using enthalpy of formation of formation of other species from experiments for each reaction
7. Average estimated heat of formation for the molecule of interest over all the reaction as calculated in the previous step.
The work by Wei et al. (2004a) demonstrated the use of semiempirical quantum chemical methods such as AM1 (Dewar and Storch, 1985) and PM3 (Stewart, 1989) to screen reactive chemicals successfully. The low-level quantum mechanical approach was used in this particular study, as the primary objective was to screen the chemicals. The authors also observed better quality prediction of heat of formation with PM1 and AM3 as opposed to MNDO (Dewar and Thiel, 1977) and MINDO/3 (Bingham et al., 1975a,b) methods. This is where understanding the applicability of various methods becomes important.
3.6.3. Practical Considerations
Depending on the approach, calculation of heat of reaction should take into consideration of its associated caveats. At the macro-scale modeling level, reasonably accurate values of parameters such as compressibility factor, acentric factor, and critical constants are required as an input to Gibbs free energy minimization calculations, which results in accurate multiphase calculations of reactive systems. Other concerns relate to choice of appropriate minimization technique with minimal dependence on quality of initial guess on final convergence. While several books exist discussing on minimization and optimization techniques (Bertsekas, 1999; Luenberger and Ye, 2008), for our practical purposes, it is important to be conversant with available computer programs to calculate heat of reaction from thermophysical data.
..................Content has been hidden....................
You can't read the all page of ebook, please click here login for view all page.