
1
Graphene Chemical Derivatives Synthesis and Applications: State‐of‐the‐Art and Perspectives
Maxim K. Rabchinskii1, Maksim V. Gudkov2, and Dina Yu. Stolyarova3
1Ioffe Institute, Centre of Nanoheterostructure Physics, 26 Politekhnicheskaya, 194021 Saint Petersburg, Russia
2Russian Academy of Sciences, N.N. Semenov Federal Research Center for Chemical Physics, Department of Polymer and Composite Materials, 4 Kosygina Street, Building 1, 119991, Moscow, Russia
3NRC “Kurchatov Institute”, Complex of NBICS Technologies, 1 Akademika Kurchatova pl., 123182, Moscow, Russia
1.1 Introduction
After its rediscovery in 2004 by Geim and Novoselov s[1], graphene has provided a stunning scientific and technological excitement for researchers in various disciplines, becoming the first two‐dimensional (2D) crystal broadly studied [2, 3]. However, after the first decade of extensive research, an explosion of interest has been followed by the fade of enthusiasm. Such a shift is due to several reasons. Firstly, a large set of new 2D materials, such as hexagonal boron nitride (h‐BN), molybdenum disulfide (MoS2), silicene, etc., has been isolated and widely investigated, demonstrating to have new unprecedented properties, differing from those in graphene [4, 5]. Secondly, despite a large number of techniques developed for the graphene synthesis, it has appeared that transition from obtaining a single ideal laboratory sample to large‐scale production of high‐quality graphene still exhibiting its unique properties is a major challenge, still not resolved [6]. This limits the realization of the proposed revolutionary applications of graphene. Finally, thanks to great attention attracted at the start, during almost two decades of graphene history the key features of its physics has been thoroughly studied, leaving, as seemed, only details and questions of its further integration into practical applications.
Given these facts, the main focus within the field of graphene research has progressively shifted to another key feature of this material – versatile chemistry and its influence on the properties of graphene. Apart from being a 2D crystal derived from the graphite structure, graphene at the same time represents a quasi‐infinite π‐conjugated polyaromatic hydrocarbon macromolecule [7]. This means graphene can undergo a large set of chemical reactions from organic synthesis and chemistry of aromatic compounds, even though several limitations due to the difference in the overall reactivity have to be considered. Using these reactions, various organic groups, containing halogens (Br, Cl, F, I), chalcogens (O, S), pnictogens (N, P, Sb, Bi), or silicon as well as more complex functionalities, such as alkyl and aryl hydrocarbons, can be covalently attached to edges or basal plane of graphene [8, 9]. Furthermore, substitutional modification of graphene, during which carbon atoms are replaced by atoms like nitrogen, phosphorous, sulfur, or boron, can be performed as well (doping process) [10].
In turn, the introduction of functional groups or doping provides the reconfiguration of the π‐conjugated electron cloud due to the partial conversion of sp2‐hybridized carbon atoms into sp3‐hybridized ones, as well as to the electron‐withdrawing or electron‐donating effect of the modifying moieties [11]. Accordingly, an intriguing opportunity to tailor the electronic structure of the graphene layer and, thus, its physical and chemical properties, arises. Graphene transport characteristics and band structure, optical absorption and luminescence, field emission, magnetic, and thermal properties can be tuned in a desirable manner via modification of graphene chemistry [8, 9, 12]. In other words, chemical derivatization allows to render the physics of graphene significantly more variable, altering the intrinsic properties of graphene and giving rise to new inspiring features, not observable in pristine graphene. The introduction of organic groups to graphene edges or basal plane also provides an opportunity to controllably modify the reactivity of material, its chemiresistive, and wetting properties [13]. This extends the performance of graphene in various applications due to the adjustment of its characteristics for certain conditions.
Hence, it is not a surprise that a large family of graphene chemical derivatives, chemically modified graphenes (CMGs) or functionalized graphenes (FGs), has appeared during the last decades, displacing pristine graphene from the position of a central research subject within the field of nanocarbon science. Stoichiometric CMGs (C1X1, where X – modifying functionality), such as fluorographene and graphane, have been synthesized as well as a large number of non‐stoichiometric (C1X<1) graphene derivatives has been synthesized. Speaking about graphene functionalization, one cannot circumvent graphene oxide (GO) – graphene layer, which basal plane and edges are covalently functionalized with a set of oxygen‐containing groups [14]. Starting as just a precursor for the graphene synthesis, GO promptly appeared to be a facile starting platform for the CMG synthesis. This feature arises from the ease of large‐scale production of GO via liquid‐phase exfoliation of graphite and the presence of oxygenic groups [15], which can be substituted with other organic groups via simple methods from organic chemistry. This is in contrast to the functionalization of pristine graphene, which requires special reactive species that can form covalent adducts with the sp2 carbon structures in graphene [16]. As a result, the modification of GO has given rise to numerous CMGs and up to date is a key approach for the synthesis of the functionalized graphenes [8].
Despite the progress made within the field of graphene derivatization, the research on CMGs synthesis and characterization is still far from over. The detailed understanding on the effects of modifying functionalities on the physical properties of CMGs still not achieved [17]. Particularly, the origin of optical absorbance or fluorescence in GO or fluorographene is still under discussion with many controversial models suggested to explain the particular role of functionalization parameters in this question [18, 19]. New methods for the synthesis of new stoichiometric CMGs apart from fluorographene and graphane as well as for the precise introduction of a single certain type of functional group in the desired concentration are still under development. This section summarizes and compares the recent results within the field of graphene chemical derivatives, starting from features of GO synthesis and concluding by some remarks on the perspectives of CMGs applications.
1.2 Graphene Oxide: Synthesis Methods and Chemistry Alteration
The history of GO began long before the term “graphene” was proposed. It started with graphite oxide, the synthesis of which was proposed back in 1859 by Brodie [20]. The product was named graphitic acid or graphite oxide and was moderately studied for over 150 years [21]. The boom around graphene among other things has revived the interest of the scientific community to graphite oxide. It was discovered that upon an increase in pH from acidic to neutral graphite oxide exfoliates to monolayer GO sheets due to the Coulomb repulsion of similarly charged dissociating functional groups.
Here it must be emphasized that GO and graphite oxide are distinct materials, although these names are often used as synonyms. The former one is a single graphene layer covalently modified with oxygen moieties, whereas the latter one is a stack of non‐exfoliated GO sheets. Generally speaking, the nomenclature for two‐dimensional carbons and CMGs is still not aligned well, with improper and arbitrary naming of graphene derivatives. This topic is finely discussed by the Carbon editorial board in the recent article by Bianco et al. [22].
GO is a non‐stoichiometric graphene derivative with a general formula of the type CxHyOz with the amount of hydrogen on average estimated as y = 0.8, whereas the carbon‐to‐oxygen ratio (so‐called oxidation degree, C/O) can vary from 1.5 to 2.5 [23]. GO carries a large set of oxygen‐containing functional groups, such as hydroxyls, epoxides, carboxyls, carbonyls, ester and lactol structures, peroxides, etc. Numerous models, ranging from the Hofmann and Ruess models to Lerf–Klinowski and Dekany ones [24, 25], have been being progressively developed to describe the chemistry of GO, reflecting the evolution of the synthesis protocols and methods applied for the studying of GO. This process has pointed out that the oxidation degree, defectiveness of graphene layer, and the specific composition of oxygenic groups can be controlled by the choice of a certain method of synthesis and parameters within it.
Particularly, the Brodie method based on the heating of graphite at 60 °C for few days (three to four days) in the mixture of KClO3 and HNO3 results in GO with a high C/O ratio of approximately 2.5–2.7 with an introduction of N‐containing functionalities, particularly nitrates. Application of the modifications of the Brodie method by Staudenmaier (addition of concentrated H2SO4 and portioned introduction of KClO3) and Hofmann (the use of non‐fuming nitric acid) results in an almost equal C/O ratio and functionalization predominantly by basal‐plane epoxides with low content of edge‐located carboxyls and carbonyls (less than 1 at.%) [26, 27]. At the same time, the elemental analysis of the GO synthesized by these methods has indicated the absence of sulfur and nitrogen species in the material.
Higher oxidation degree and introduction of a substantial amount of carboxyl and carbonyls is achieved by the GO synthesis via the method proposed by Hummers and Offerman with the replacement of KClO3 and fuming HNO3 by KMnO4 and KNO3 in concentrated H2SO4 [28]. This has allowed to reduce the C/O ratio of the synthesized GO to two and less with simultaneous increase in the content of carbonyls/carboxyls by almost three times to approximately 3–4 at.% [29]. However, the Hummers method also results in the appearance of the traces of sulfur and nitrogen in GO (up to 6 at.%) [30], likely to be due to the covalently bonded sulfates and nitrates or adsorbed sulfuric and nitric acids [31]. This effect, despite intuitive being a disadvantage for the GO synthesis, can be applied for the doping and chemical modification of GO, which will be discussed further.
Noteworthy, in the case of large graphite flakes >50 μm, the product of the Hummers method inevitably contains under‐oxidized particles with a graphite core and an oxidized shell, carrying oxygenic groups. Moreover, the materials produced by Brodie, Staudenmaier, or Hummers methods have shown a defective structure of the graphene layer with the presence of holes, rips, and wrinkles due to the oxidation reaction [32]. A solution for these problems was for the first time proposed by Kovtyukhova based on the pre‐expansion of graphite in a concentrated H2SO4, K2S2O8, and P2O5 for several hours at 80 °С [33]. This increased the yield of the fully oxidized graphite flakes. Another approach was proposed by Marcano et al. [15], which has become the most popular among other variations of the Hummers method and is known as a Tour method. The authors proposed using a mixture of H2SO4 and H3PO4 (9 : 1) instead of concentrated sulfuric acid as well as six equivalents of KMnO4 instead of three according to the standard Hummers procedure and excluding NaNO3 from the reaction. This has resulted in the further reduction of the C/O ratio to 1.7–1.9, an increase in the concentration of the edge‐located oxygen moieties (carbonyls/carboxyls) [29], and a more regular structure with fewer defects in the basal plane compared to the other methods. However, the volume of acids required for the procedure according to the proposed method is incredibly high (400 ml per 3 g of graphite vs. 69 ml in the standard Hummers method), which is a significant drawback that hinders to apply this process for the mass production of GO.
Substantial alteration in the relative content of basal‐plane and edge‐located oxygenic groups can be achieved by the graphite oxidation using K2Cr2O7 in the H2SO4 solution in the presence of NaNO3 first proposed in 2010 [34]. It turned out that the GO obtained by this method has a low oxidation degree [35, 36], C/O = 2.8–3.1, due to the minor number of the introduced basal‐plane hydroxyls and epoxides. At the same time, the overall concentrations of carboxyls and carbonyls appear to be higher than those in the GO produced by the conventional Hummers or Tour methods, reaching up to 7 at.% as was demonstrated by X‐ray photoelectron and X‐ray absorption spectra (Figure 1.1). Nevertheless, the material is fairly stable in aqueous solutions, and GO platelets have a lateral size of 10–100 μm. As a result, this method provides an intriguing opportunity to obtain partially oxidized GO with a high number of chemically reactive carboxyl groups without the need for additional steps of GO treatment. Furthermore, it can be suggested that oxidation of graphite with the mix of K2Cr2O7 and KMnO4 introduced in different relative concentrations can be performed, allowing one to manage the oxidation degree of GO and, more importantly, the relative content of carboxyls, carbonyls, and hydroxyls/epoxides.
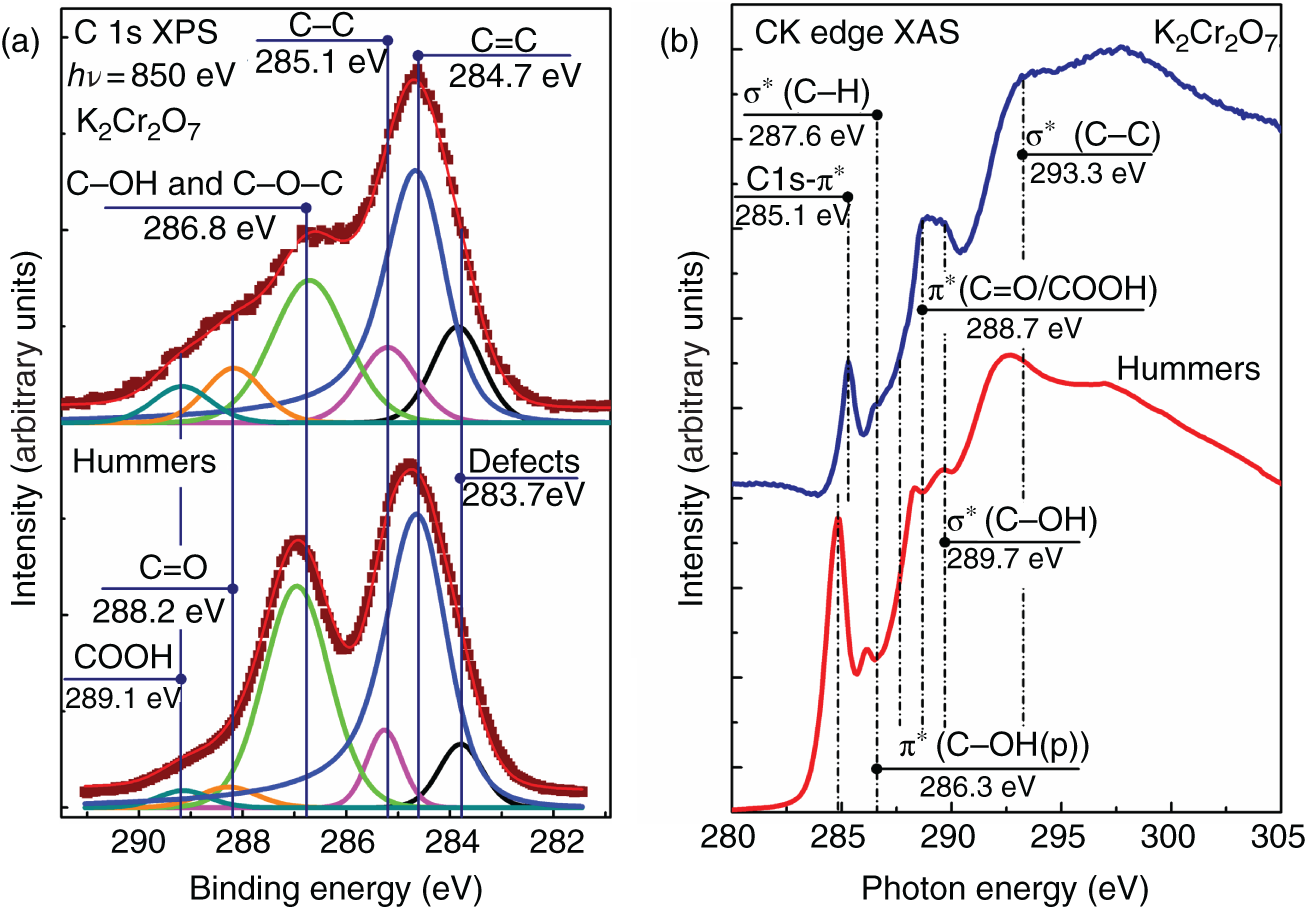
Figure 1.1 (a) C 1s X‐ray photoelectron and (b) CK edge X‐ray absorption spectra of GO samples, synthesized via Hummers and K2Cr2O7 oxidation methods.
Besides the choice of the specific method of graphite oxidation, the chemistry of GO can be tuned also by the adjustment of the water amount, temperature and duration of the reaction, type of the graphite used, and exclusion of a certain reagent. Chen et al. have shown that a careful control of the water added during the oxidation allows one to modulate the content of hydroxyl and epoxide groups on GO sheets without sacrificing their structural integrity [37]. At the same time, the increase of the temperature up to 95 °C in the lack of water results in the selective formation of carboxyl groups. Recently, we have shown that the increase in the intercalation time in the modified Hummers method from several hours to about a week with the use of NaNO3 leads to first nitration and then doping of GO with introduction of up to 5 at.% of nitrogen, predominantly in the graphitic form [38]. At the same time, covalent attachment of sulfuric acid esters during the Hummers oxidation with their retention upon the washing in organic solvents was demonstrated by Dimiev et al. [39] and further discussed by Groveman et al. [40]. This demonstrates that apart from oxygenic groups, other functional groups can be introduced to GO during its synthesis for various practical purposes even though at first sight they can be regarded as contaminating impurities.
Nevertheless, despite these achievements, GO synthesis via various methods of liquid‐phase exfoliation gives only limited opportunities in the adjustment of the chemistry of GO and its derivatives. The exact principles and mechanisms lying behind the graphite oxidation are still not well understood and are under discussion, restricting the development of new strategies for the GO synthesis with a required set of oxygenic or other organic groups [41]. Given this, electrochemical synthesis of GO has begun to be regarded as a facile strategy for this purpose. Electrochemically synthesized graphene oxide (EGO) having a C/O ratio of 1.7, lower than that in GO obtained via liquid‐phase exfoliation, can be obtained in high yield with few defects [42, 43]. However, the power of the electrochemical synthesis methods is not only the production of highly oxidized GO but the synthesis of various functionalized graphene layers. Owing to the possibility of using various electrolytes, such as ammonium, chloride, hydroxide, phosphate, nitrate, bromic and chloric acid, etc., one can obtain graphene layers functionalized by nitrogen, sulfur, or halogen (Cl, Br, I) containing organic groups. Moreover, once obtained EGO can be again subjected to the electrochemical oxidation or reduction using another electrolyte, allowing complex functionalization of the graphene layer with various types of organic moieties. Thanks to this and simplicity of the method, the possibility of re‐using of the electrolytes, and absence of necessity of excessive washing, the electrochemical methods are regarded as a next step in the large‐scale synthesis of GO and CMGs with a controllable chemistry [44].
1.3 Graphene Oxide Reduction and Functionalization
The very first strategy proposed for the further GO processing was its conversion into pristine graphene by the so‐called reduction process – elimination of all oxygenic groups to restore the π‐conjugated graphene network. Expectations were that such an approach would be a facile route to large‐scale and low‐cost production of graphene with dozens of various reduction methods proposed [23, 45]. However, the obtained material, reduced graphene oxide (rGO), appeared to be far from both the theoretically investigated ideal graphene and pristine graphene synthesized via mechanical cleavage of graphite or chemical vapor deposition (CVD) method [46]. This is due to the incomplete removal of the oxygenic groups and the introduction of structural defects during the reduction process whichever method is applied. The contamination of GO with the residuals of the reducing agent appeared to be another significant problem. The typical values of the C/O ratio reached upon the GO reduction are about 12–40 [23], typically one order of magnitude lower compared to CVD graphene. The only method for GO reduction demonstrated up to date to provide the C/O > 75 and a minor number of structural defects is high‐temperature annealing at 1000–1100 °C in the inert atmosphere – a process comparable by its complexity and yield with CVD growth of graphene with the still worse structural quality of the material. Apparently, the GO reduction has entrenched in the graphene community as a second rate method to manufacture graphene for several applications where the quality of graphene is not the determining factor. Despite a large number of new eco‐friendly, facile, and high‐yield methods for GO reduction are still being developed up to date, this strategy for the synthesis of pristine graphene, demonstrating such desired physical properties, becomes regarded as a dead end.
However, all changes if we proceed from considering GO reduction as just a route to obtain pristine graphene to regarding it as a versatile method for the synthesis of graphene layer functionalized with various organic groups. Within this concept, the initial presence of oxygenic groups in GO, their tendency to retain or being substituted by other moieties, and incorporation of other elements into the graphene network upon reduction drastically transform from a problem into an advantageous feature. The goal of the GO processing shifts to the alteration of the type, number, and spatial distribution of modifying moieties, taking the control on the π‐conjugation rate and local electronic structure of graphene layers along with their chemical reactivity (Figure 1.2) [8, 9, 11]. The introduction of defects also can be translated into a beneficial feature within this concept, owing to their influence on the electronic structure and properties of graphene.
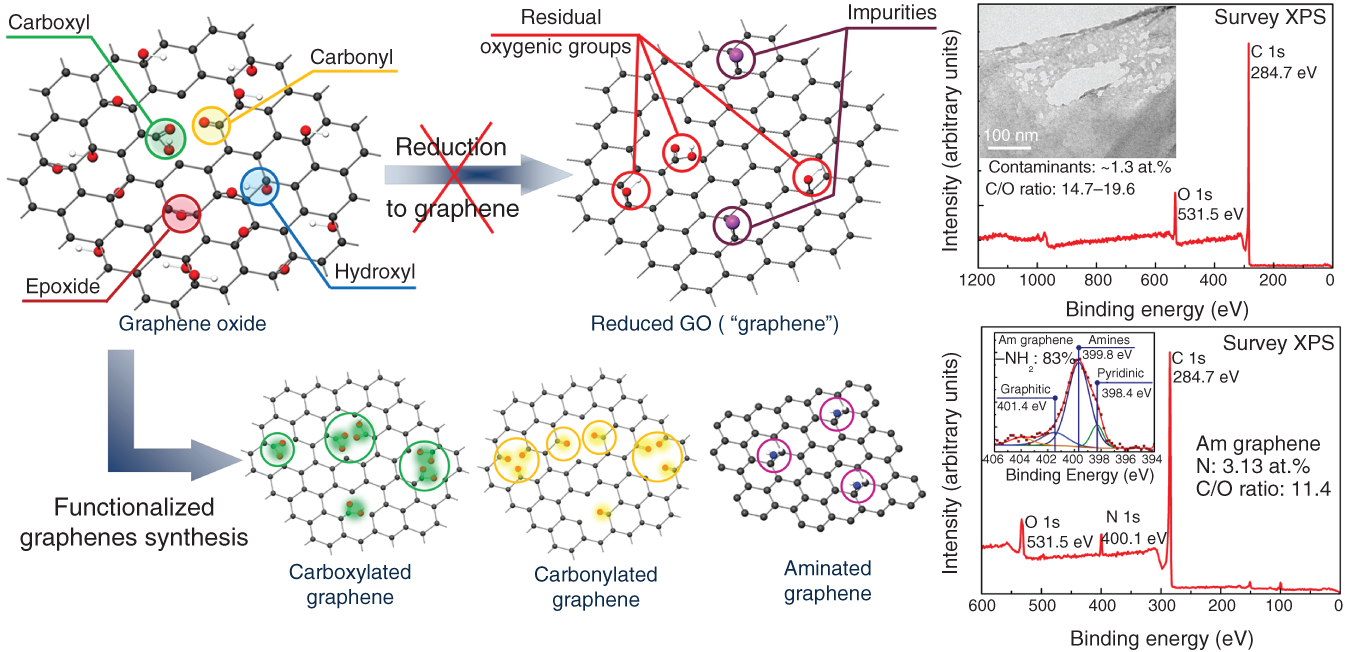
Figure 1.2 Two strategies for the GO processing: reduction and functionalization. In the former case, the desired pristine graphene cannot be obtained due to the retained oxygenic moieties, contamination of graphene layer, and formations of defects. Oppositely, these features become advantageous in the case of graphene functionalization to synthesize graphene chemical derivatives.
Source: Adapted from Refs. [8, 9, 11]
.
Note, GO inherits this versatility in further modification from its initial chemistry, whereas pristine graphene is chemically inert, with chemical modification requiring either complicated radiative methods or radical chemistry [16, 47].
Two general lines can be distinguished in the conversion of GO into CMG with the covalently attached functional groups: (i) selective reduction or oxidation of GO with the retention or increase in concentration of a certain oxygenic group; (ii) substitutional modification of GO with the removal and partial replacement of the oxygen‐containing functionalities by other organic groups. Synthesis of carboxyl‐rich CMGs is widely disseminated within the selective GO reduction. Such an interest arises from the well‐established chemistry of carboxylic acid derivatives in organic synthesis and efficiency of carboxyls as anchoring points for the further covalent modification with biomolecules, polymers, etc. Typically, selective reduction of GO with the preservation of carboxyls can be performed by low‐temperature annealing at 150–200 °C, which is lower than the temperature of their dissociation [48] or, chemically, by using NaBH4 [49], thiourea dioxide, or hydrazine [50, 51]. However, the concentration of carboxyls in the initial GO is low, varying from 2 to 4 at.% with the only exception of GO synthesized via aforementioned oxidation with potassium dichromate, which contains up to 6 at.% of carboxyls [29, 35]. Thus, reduction methods that additionally increase the number of carboxyls are actively developed nowadays, although only few were presented. Pumera et al. have synthesized carboxyl‐enriched graphene from GO using the Kolbe–Schmitt reaction [52], resulting in up to 11 at.% of carboxyl groups with a negligible amount of other oxygenic groups. Another approach was suggested by Jankovsky et al. by repetitive oxidation of GO with potassium permanganate in an acidic environment [53]. The applied method allows to reach the content of carboxyls of up to 11–15 at.%, although a large number of hydroxyls are also present in the material. A careful treatment with a combination of hydrogen peroxide and ammonia or another base can be applied to obtain carboxylated graphene, although this method also provides cutting of graphene layer to nanoflakes [54]. Apart from the liquid chemistry methods, the photochemical synthesis of the carboxylated graphene can be performed. Particularly, we have shown that concentration of carboxyls can be engineered within the range of 3–11 at.% with simultaneous reduction of basal‐plane groups via the UV irradiation of GO films in the argon atmosphere (Figure 1.3) [55, 57]. The proposed method is compatible with the lithography techniques, but its yield is sufficiently lower compared to liquid‐phase carboxylation protocols.

Figure 1.3 (a) C 1s, (b) O 1s X‐ray photoelectron, and (c) CK edge X‐ray absorption spectra of the initial GO, carbonylated graphene (C‐ny), carboxylated graphene (C‐xy) synthesized according to the methods described in Refs. [55, 56].
Less attention is gained to the carbonylated graphene derivatives, although carbonyl groups can act as centers for the non‐covalent coordination bonding and contribute to the reversible pseudocapacitance or bonding of metal cations [58–60]. Carbonyls are more stable than carboxyls, and basal‐plane functionalities [48] easily retain in the GO upon its reduction via soft reducing agents or low‐temperature annealing. The problem is, analogously to the case of carboxyls, the low initial content of carbonyls – of about 2–3 at.%. To overcome this, GO reduction methods, inducing the elimination of hydroxyls according to the E1cB mechanism with the progressive formation of carbonyls, can be applied. Recently, we have shown that such a process proceeds in the case of GO reduction using sodium silicate [61], resulting in the carbonylated graphene with up to 9 at.% of carbonyls (Figure 1.3). Almost at the same time, Yu et al. have presented an alternative way to synthesize carbonylated graphene covered by up to 6 at.% via the GO treatment with the dilute H3PO4 and NaH2PO4 solutions [62]. In both the synthesized CMGs, carbonyl is the predominant oxygenic group, with negligible content of other functionalities.
Carboxylated and carbonylated CMGs are relatively stable graphene derivatives regarding low‐temperature heating, and other external effects may be applied during their application. However, the drawback is either perforation of the graphene layer or disruption of graphene flakes to smaller one during the synthesis since carboxyls and carbonyls locate at the edges of the graphene network (Figure 1.4), requiring the increase of its extent to appear in large numbers [54, 55, 57]. Carboxyls can be attached to the GO basal plane as well [63], but in a lower number and being less stable. Thus, structural modification of the graphene layer upon carboxylation and carbonylation and corresponding alteration of the graphene charge transport must be considered. On the other hand, performed in a controllable way, this feature expands the application of CMGs within the field of desalination, considering the perforated structure of carboxylated/carbonylated graphene with chemically active edges of holes.

Figure 1.4 Transmission electron microscopy images of the (a) carbonylated graphene and (b) carboxylated graphene platelets obtained according to the method discussed in Refs. [55, 56].
Apart from these two oxygenic CMGs, preferential functionalization by hydroxyls or epoxides is demonstrated to be performed. One such type of reaction is hydroboration which can give GO with an almost stoichiometric composition of (C1(OH)1)n [64]. Hydroxyl‐enriched graphene layers were also synthesized by lengthy immersion of GO in a NaOH solution with the conversion of most of the epoxy groups into hydroxyls [65]. Oppositely, mild oxidation of preliminary reduced GO using treatment with nitric acid is recently demonstrated to give an opportunity to control the surface concentration of epoxide groups in the range of 10–15 at.% [66]. However, the fundamental problem of graphene functionalization with hydroxyls and epoxides is their intrinsic instability. Hydroxyls and epoxides easily convert into each other upon the changes in the pH value of the suspension [67], migrate through the basal plane of the graphene layer to undesirably form clusters [18], and eliminate event at room temperature and under light radiation [68]. These facts make the engineering of the composition and properties of epoxide and hydroxyl‐derived CMGs a rough task and limiting the conditions of their further application. The problem of the reproducibility of the chemistry of such CMGs during their large‐scale synthesis is also a question.
Besides the engineering of the number of four main oxygenic groups of GO, their substitution with functional groups containing halogens (F, Cl, Br, I), chalcogens (O, S), pnictogens (N, P), silicon, and other elements is another strategy, as one can consider – the main one – within the field of the synthesis of CMGs. To date, halogenated graphenes are one of the hugest families of CMGs with fluorinated graphene (fluorographene) as the main derivative. This originates from a long history of fluorographene and fluorographite by itself, analogously to graphite oxide. Graphite fluoride, a well‐known graphite derivative (fluorine‐intercalated compound) with covalent C—F bonds, was known and successfully applied as a lubricant for more than 100 years. However, only in 2010 first isolation of fluorographene, a monolayer of graphite fluoride, was achieved with subsequent synthesis using graphene plasma treatment with XeF2 [69]. This, among other things, points out that fluorinated graphene can be prepared via various methods apart from GO treatment giving CMGs with a stoichiometric composition (C1F1) [70]. Nevertheless, GO and rGO are still facile precursors for the synthesis of fluorographene via using various reagents including hydrofluoric acid, diethylaminosulfurtrifluoride (DAST), fluorides of sulfur‐based reagents, N2/F2, etc. [71, 72]. Controlling the degree of oxidation of the treated GO and the type of the reagent used, selectively reacting with a certain oxygenic group, one can obtain fluorographenes with the desired content of fluorine atoms. Note that such CMGs will contain CF, CF2, and CF3 groups, with two latter ones located on the edges of the graphene layer [73]. The approaches based on GO/rGO fluorination are easily scalable and simple in comparison to conventionally used plasma treatment of CVD graphene [74]. Moreover, fluorine atoms retain the hydrophilic nature of the GO after its derivatization, providing its dispersibility in aqueous solutions, additionally facilitating this approach for the fluorographene synthesis. The drawback of such a method is difficulties in obtaining fluorographene with stoichiometric composition [70].
The main purpose of the synthesis of fluorographene from the GO even though modifying groups in both materials alter the electronic structure of graphene is higher stability of the former one and the ability to obtain its stoichiometric variation [75]. The latter fact originates from the steric issues which arise for the oxygenic groups and absent in the case of fluorine. The same reason limits the synthesis of stoichiometric forms of other halogen‐derived CMGs. Besides chlorinated graphene, having not well‐expressed chemical reactivity, bromine‐derived CMGs are of interest nowadays. The introduction of bromines allows one to subsequently perform a large variety of chemical modifications of graphene by substitutional reactions with no need in using strong chemicals [70]. Brominated graphenes can be easily obtained via GO treatment with bromine solutions or hydrobromic acid, resulting in a simultaneous reduction and functionalization by bromine with ∼5–10 at.% bromine concentration (Figure 1.5) [56, 76]. The use of the Hunsdiecker reaction for the introduction of bromine on the edges of the GO flakes was also reported by Jankovsky et al., leading to the introduction of up to 6.4 at.% [77]. Despite the level of functionalization by bromine does not exceed 10 at.%, it is sufficient enough to substantially alter the chemical reactivity of the graphene layer and modify its electrophysical properties. This fact along with the steric limitations of bromine atoms and the discovered influence of covalently bonded bromine atoms on the morphology of the graphene layer suggests the absence of strong necessity in the further increase of the bromination rate via new methods [57].
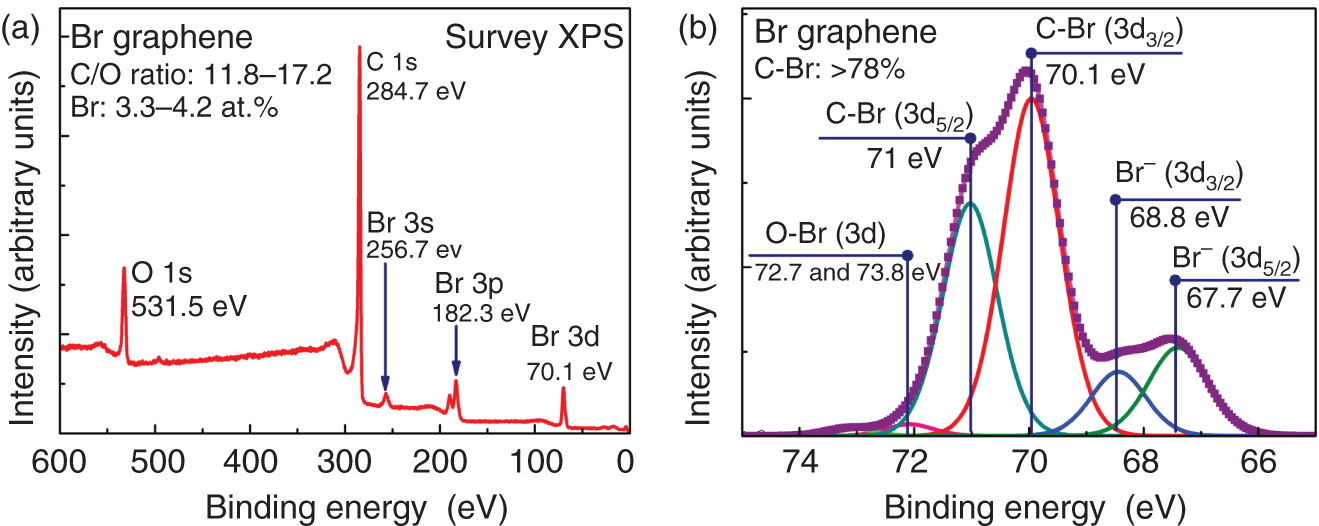
Figure 1.5 (a) Survey and (b) high‐resolution Br 3d X‐ray photoelectron spectra of the brominated graphene synthesized according to the method described in Ref. [56].
Source: Based on Rabchinskii et al. [56].
Regarding the iodine functionalization of graphene, several reports on the iodination of GO can be found. Particularly, Simek et al. have shown the covalent attachment of iodine atoms by iodine or hydroiodic acid under reflux or in an autoclave at elevated temperatures (240 °C) and pressures (over 100 bar) [78]. The C/O ratio of the obtained CMGs was estimated to be about 20–24, but the concentration of iodine is less than 3.3 at.%. Such a low concentration reached arises from the large atomic radius of an iodine atom and the weakness of carbon–iodide bond. Due to this fact iodine more preferentially just physically adsorbs on the surface of the graphene layer as I− and I2 with no covalent bonding. As a result, GO/rGO treatment via hydroiodic acid by itself or in the mixture with the acetic or trifluoroacetic acids has proved itself as just a facile method for the elimination of oxygenic groups [79, 80].
Amines represent another group of attractive functionalities that enable an easy CMGs' grafting through amide coupling or so‐called “click” reactions and provide tunable modification of its electronic and optical properties. Various aliphatic and aromatic amines can be introduced to GO with its simultaneous reduction by the nucleophilic reaction between amine‐containing moieties and epoxides [81]. A straightforward method for the introduction of up to 3–5 at.% of amines is the hydrothermal treatment of GO with aqueous ammonia [82], leading to the substitution of epoxides by amines [83]. Another approach was suggested by Zhang et al. with the formation of amino‐functionalized graphene via Hoffman rearrangement in GO [84], reaching an amine concentration of around 4 at.%. Furthermore, substitutional amination using halogen‐derived CMGs can be carried out [85]. In this case, primary aliphatic amines are predominantly covalently attached to the graphene layer [57]. Recently, one‐pot GO amination and reduction via reductive amination of carbonyl groups using Leuckart reaction was reported by Aguilar‐Bolados et al. [86]. Considering carbonyls are located exclusively at the edges of the graphene network, this approach allows one to introduce aromatic amines to the graphene layers. Additionally, combining this method with the introduction of amines by epoxide‐ring opening one can obtain amine‐derived CMGs with the tunable rate of aliphatic and aromatic primary amines, differently modifying the electronic structure of the graphene layer [57]. It is noteworthy, that nowadays a large number of GO and graphene derivatives with amine group attached through an oligomeric linker can be found, even as a commercial product, although strictly speaking they do not refer to amine‐derived CMGs.
Graphene functionalization with organic groups containing another pnictogen, phosphorous, is reported much scarcer. Some et al. have prepared graphene phosphonic acid by the treatment of GO with a polyphosphoric acid/phosphoric acid mixture [87]. Graphene oxide diphenylphosphinite was reported to be prepared via reaction of GO with Ph2PCl [88]. Despite these results, the synthesis of phosphorous‐containing CMGs still retains a challenging task. The introduction of sulfur‐containing groups appeared to be a less complicated problem. In the previous section, we have mentioned the possible preservation of sulfonic acid groups covalently attached to the GO layers after the synthesis [39]. However, the concentration of these functionalities lies within the range of 1–3 at.%. A prospective approach to add sulfonic acid groups was suggested by Yeo et al. with the introduction of these functionalities to the edges and basal plane of graphene via a one‐step reduction and functionalization of GO using p‐hydrazinobenzene sulfonic acid hemihydrate (HBS) as a reducing agent [89]. Another approach was demonstrated by Garg with coauthors who have synthesized sulfonated graphene (Gr‐HSO3) using fuming H2SO4 acid at 120 °C for three days [90]. Along with sulfonic acid groups, thiol groups can be covalently attached to the graphene layer through the conversion of epoxy and hydroxyl via the reaction with thiourea and HBr, followed by treatment with NaOH [91]. Furthermore, halogenated CMGs, such as brominated graphene, can serve as a platform for the thiolation of graphene by subjecting them simple substitution reactions leading to thiofluorographene [92].
Numerous other CMGs, less common within the field of graphene derivatives, have been reported up to date as well [6], such as graphane (hydrogenated graphene, boron or silicon functionalized graphenes, etc.) [8]. The main efforts within the field of CMGs synthesis are now devoted to the increase of the functionalization rate, which is still low for the most part of modifying organic groups. Besides steric effects, difficulties within intense graphene derivatization are related to the dynamic modification of the local areas of the graphene layer due to the reconfiguration of its morphology and electronic structure upon the introduction of a certain organic group. This feature requires adjustment of the conventional methods taken from organic synthesis to functionalize GO/rGO or graphene and suggests more preferable use of a multistep modification protocols. The interplay between the modifying moieties and oxygenic groups retained in GO/rGO upon the modification are to be considered as well, which commonly leads to the degradation of the synthesized CMGs.
Apart from the improvements of the functionalization rate, the developments in graphene derivatization are seemed to be concerned on the complex functionalization of graphene layer with combinations of several type of different types of organic groups and precise control of their spatial distribution. Introduction of two and more different organic groups besides those already presented in GO/rGO is still a challenging task due to both their interaction and undesired effects on the reaction mixture on the already introduced moieties. The use of protecting groups can be suggested to overcome these difficulties. The control of the relative rate of the edge and basal‐plane functionalization by several organic groups has been achieved due to the distinct reactivity of these sites in graphene layer. However, patterning the desired geometry of the distribution of functional groups to precisely tune the electronic structure of the graphene layer is still an ambitious task for the future of graphene derivatization.
1.4 Applications of CMGs
The efficient and diverse functionalization of graphene has substantially enhanced its performance in conventional practical applications intrinsic to nanocarbon materials. CMGs with an isolated oxygenic group, particularly carboxyls and carbonyls, have been demonstrated to be efficient materials for the sorption and selective detection of various metal ions and salts, such as uranyl, cadmium, plumbum, etc. [93]. This is due to the high affinity of carboxyls and carbonyls toward these compounds along with the large specific surface area of graphene, especially in the form of aerogels [94]. These features combined with reasonable high conductivity make CMGs on the base of GO selective reduction to be prospective materials to be applied as electrodes with the reversible pseudocapacitance for the energy storage applications [9, 62]. Flexible and transparent electrodes for photovoltaic devices are another field of CMGs application, particularly sulfonated and aminated ones. Owing to such a functionalization graphene layer demonstrate better interaction with the active layer, whereas its band gap and charge mobility can be tuned in the desired way to fit the certain configuration of the device [95]. CMGs containing phosphonic acid groups on the edges of the graphene sheet exhibited flame‐retardant properties and were used as material for the improvement of fire safety in cotton fabrics [96].
Gas sensing is probably one of the most promising and native fields of CMGs applications. GO and rGO have already been demonstrated to be a perspective gas sensing layer, exhibiting an excellent sensing performance toward numerous gases and volatile organic compounds (VOCs), including NO2, NH3, CO, ethanol, H2O, trimethylamine, hydrogen cyanide (HCN), and dimethyl methylphosphonate [97]. Employing CMGs, which carry a certain type of organic group, having definite affinity to gas molecules, and being in favor to rather specifically detect them, improves the selectivity of the gas sensors. Moreover, selectivity can be further enhanced if CMG layers will be utilized in multisensor on‐chip arrays by a simple segmentation of the graphene by electrode stripes granting an analyte‐specific vector response to a large number of gases [98]. In this case, the intrinsic spatial inhomogeneity of CMGs becomes an advantageous feature due to the induced variation of both the adsorption of gas molecules and the electronic structure of the CMG in different segments of the chip.
Besides the conventional graphene applications, graphene functionalization and derivatization also advance the appearance of new applications, especially in the field of smart materials [99]. Both graphene and GO have a negative coefficient of thermal expansion (CTE), but in the latter one, this effect is more pronounced due to the thermally induced removal or insertion of water molecules between GO layers [100]. This phenomenon can be applied for the application of GO and its derivatives as thermoresponsive actuators. At the same time, the alteration of the GO chemistry can allow one to tune the CTE value as well as the compatibility of the CMG layer with the polymer matrix [101]. Furthermore, thanks to the chemical reactivity, high and tunable photothermal conversion efficiency, and high mechanical strength of CMGs [102], graphene derivatives functionalized with oxygenic groups, sulfur, or amines are seemed to be perfect fillers for the self‐healing hydrogels for medical applications and light‐triggered mechanical actuators [103]. Ferromagnetic and paramagnetic properties of the sulfur‐modified CMGs and fluorographenes endow such materials' magnetic responsiveness, suggesting their application in magneto‐responsive actuators [104].
The development of electrorheological fluids (ERFs) is another practical application where the advantageous features intrinsic to CMGs reveal. ERFs are suspensions composed of the dielectric or semiconducting particles dispersed in electrically insulating oils, which change their rheological properties under applying the external electric field – demonstrate the electrorheological (ER) effect [105, 106]. The efficiency of the ERFs is generally governed by the dispersibility of the filler particles in the used oils, their low conductivity to avoid electrical short circuits between the electrodes, and high aspect ratio and polarizability to demonstrate high ER response [106]. CMGs perfectly fits these criteria due to its insulating behavior, two‐dimensional structure, amphiphilic nature, and tunable chemistry. GO has demonstrated facile performance as a filler for the ERFs with low filler concentrations [107, 108]. However, GO suffers from the instability of its chemistry upon external effects, leading to the undesired increase of its conductivity [18]. Furthermore, the ER response of GO is still not so high compared to other applied materials [105]. To overcome these drawbacks, the use of more stable CMGs, such as fluorographene, as well as extensive functionalization of GO with more polarizable sulfonic acid and nitrate groups to enhance the ER response, can be performed. The grafting of GO and other CMGs with various oligomers and polymers, such as polyaniline (PANI), glycidyl methacrylate, polydimethyl siloxane, etc. is also widely studied for this purpose [108].
Regarding further developments within the field of CMGs applications, we can assert that one of the tendencies would be formation of optoelectronic or sensing devices within a single CMG flake. Given the size of 10–100 μm combined with a controllably patterned structure and chemical composition, CMG flakes would operate as individual devices. Such an approach allows to overcome the fundamental problem of the degradation of the properties of CMGs and graphene due to its polycrystallinity and approach further miniaturization of the graphene‐based devices with their combination in complex systems. However, significant efforts are still to be made within the field of CMGs synthesis, deposition, and patterning to achieve this goal.
1.5 Concluding Remarks
In the past decade, the family of CMGs has extensively grown and emerged as an important part of nanocarbon science. The interest in the synthesis and applications of CMGs is evident from the significant increase in the number of related publications within the last six years. The continuing main driving force for this area arises from the versatility of the CMGs, providing large opportunities for both carrying out fundamental studies on the physics and chemistry of nanocarbon materials and the practical use of graphenes in various practical technologies.
The further progress within the field of the CMGs' synthesis, characterization, and application lies in the development of new more complex graphene derivatives, achieving higher rates of functionalization, the introduction of sets of various organic groups with the controlled relative concentrations, and techniques for the spatial patterning of the modifying moieties in CMGs. In practice, the precise control over reaction kinetics, regioselectivity, and stability of CMGs during and after the synthesis is still a challenging task. On the other hand, the detailed understanding of the effects of the introduced organic groups on the physical and chemical properties of CMGs is also still to be discovered. As a net result, much of the recent work has just laid the groundwork for further development in the field of graphene functionalization to offer the potential basis for truly revolutionary applications beyond the silicon age.
Acknowledgments
M.K. Rabchinskii and D.Yu. Stolyarova thank the Russian Foundation for Basic Research (Grant No. 20‐04‐60458) for the support. D.Yu. Stolyarova thanks for support NRC “Kurchatov Institute.” M.V. Gudkov acknowledge the Russian Foundation for Basic Research (Grant No. 18‐29‐19159).
References
- 1 Novoselov, K.S., Geim, A.K., Morozov, S.V. et al. (2004). Science 306: 666–669.
- 2 Geim, A.K. and Novoselov, K.S. (2007). Nat. Mater. 6: 183–191.
- 3 Ruoff, R. (2008). Nat. Nanotechnol. 3: 10–11.
- 4 Kim, S.M., Hsu, A., Araujo, P.T. et al. (2013). Nano Lett. 13: 933–941.
- 5 Song, I., Park, C., and Choi, H.C. (2015). RSC Adv. 5: 7495–7514.
- 6 Backes, C., Abdelkader, A.M., Alonso, C. et al. (2020). 2D Materials 7: 022001.
- 7 Fitzer, E., Kochling, K.‐H., Boehm, H.P., and Marsh, H. (1995). Pure Appl. Chem. 67: 473–506.
- 8 Sturala, J., Luxa, J., Pumera, M., and Sofer, Z. (2018). Chem. Eur. J. 24: 5992–6006.
- 9 Dasari, B.L., Nouri, J.M., Brabazon, D., and Naher, S. (2017). Energy 140: 766–778.
- 10 Xu, Y. and Shi, G. (2011). J. Mater. Chem. 21: 3311–3323.
- 11 Fan, M., Feng, Z.‐Q., Zhu, C. et al. (2016). J. Mater. Sci. 51: 10323–10349.
- 12 Xu, H., Ma, L., and Jin, Z. (2018). J. Energy Chem. 27: 146–160.
- 13 Kulia, T., Bose, S., Mishra, A.K. et al. (2012). Prog. Mater. Sci. 57: 1061–1105.
- 14 Eigler, S. and Hirsch, A. (2014). Angew. Chem. Int. Ed. 53: 7720–7738.
- 15 Marcano, D.C., Kosynkin, D.V., Berlin, J.M. et al. (2010). ACS Nano 4: 4806–4814.
- 16 Park, J. and Yan, M. (2013). Acc. Chem. Res. 46: 181–189.
- 17 Yu, W., Sisi, L., Haiyan, Y., and Jie, L. (2020). RSC Adv. 10: 15328–15345.
- 18 Kumar, P.V., Bardhan, N.M., Tongay, S. et al. (2014). Nat. Chem. 6: 151–158.
- 19 Mei, Q., Liu, B., Han, G. et al. (2019). Adv. Sci. 6: 1900855.
- 20 Brodie, B.C. (1859). On the atomic weight of graphite. Philos. Trans. R. Soc. London, Ser. B, Great Britain 149: 249–259.
- 21 Brisebois, P.P. and Siaj, M. (2020). J. Mater. Chem. C 8: 1517–1547.
- 22 Bianco, A., Cheng, H.‐M., Enoki, T. et al. (2013). Carbon 65: 1–6.
- 23 Chua, C.K. and Pumera, M. (2014). Chem. Soc. Rev. 43: 291–312.
- 24 Lerf, A., He, H., Forster, M., and Klinowski, J. (1998). J. Phys. Chem. B 102: 4477–4482.
- 25 Szabó, T., Berkesi, O., Forgo, P. et al. (2006). Chem. Mater. 18: 2740–2749.
- 26 Staudenmaier, L. (1898). Ber. Dtsch. Chem. Ges. 31: 1481–1487.
- 27 Hofmann, V.L. and Konig, E. (1937). Z. Anorg. Allg. Chem. 31: 311–336.
- 28 Hummers, W.S. and Offeman, R.E. (1958). J. Am. Chem. Soc. 80: 1339.
- 29 Chua, C.K., Sofer, Z., and Pumera, M. (2012). Chem. Eur. J. 18: 13453–13459.
- 30 Eigler, S., Dotzer, C., Hof, F. et al. (2013). Chem. Eur. J. 19: 9490–9496.
- 31 Poh, H.L., Sanek, F., Ambrosi, A. et al. (2012). Nanoscale 4: 3515–3522.
- 32 Dreyer, D.R., Park, S., Bielawski, C.W., and Ruoff, R.S. (2010). Chem. Soc. Rev. 39: 228–240.
- 33 Kovtyukhova, N.I., Ollivier, P.J., Martin, B.R. et al. (1999). Chem. Mater. 11: 771–778.
- 34 Chandra, S., Sahu, S., and Pramanik, P. (2010). Mater. Sci. Eng., B 167: 133–136.
- 35 Sitko, R., Turek, E., Zawisza, B. et al. (2013). Dalton Trans. 42: 5682–5689.
- 36 Muzyka, R., Kwoka, M., Smedowski, L. et al. (2017). New Carbon Mater. 32: 15–20.
- 37 Chen, J., Zhang, Y., Zhang, M. et al. (2016). Chem. Sci. 7: 1874–1881.
- 38 Rabchinskii, M.K., Ryzhkov, S.A., Gudkov, M.V. et al. (2020). 2D Mater. 7: 045001.
- 39 Dimiev, A., Kosynkin, D.V., Alemany, L.B. et al. (2012). J. Am. Chem. Soc. 134: 2815–2822.
- 40 Groveman, S., Peng, J., Itin, B. et al. (2017). Carbon 122: 411–421.
- 41 Dimiev, A.M. and Tour, J.M. (2014). ACS Nano 8 (3): 3060–3068.
- 42 Pei, S., Wei, Q., Huang, K. et al. (2018). Nat. Commun. 9: 145.
- 43 Ambrosi, A. and Pumera, M. (2016). Chem. Eur. J. 22: 153–159.
- 44 Nishina, Y. and Eigler, S. (2020). Nanoscale 12: 12731–12740.
- 45 Compton, O.C. and Nguyen, S.T. (2010). Small 6: 711–723.
- 46 Deokar, G., Avila, J., Razado‐Colambo, I. et al. (2015). Carbon 89: 82–92.
- 47 Willke, P., Amani, J.A., Sinterhauf, A. et al. (2015). Nano Lett. 15: 5110–5115.
- 48 Acik, M., Lee, G., Mattevi, C. et al. (2011). J. Phys. Chem. C 115: 19761–19781.
- 49 Jankovsky, O., Simek, P., Luxa, J. et al. (2015). Chem. Plus Chem. 80: 1399–1407.
- 50 Xu, C., Yuan, R.‐S., and Wang, X. (2014). Carbon 71: 345.
- 51 Pan, N., Guan, D., Yang, Y. et al. (2014). Chem. Eng. J. 236: 471–479.
- 52 Eng, A.Y.S., Sofer, Z., Sedmidubský, D., and Pumera, M. (2017). ACS Nano 11: 1789–1797.
- 53 Jankovsky, O., Novacek, M., Luxa, J. et al. (2016). Chem. Eur. J. 22: 17416–17424.
- 54 Jiang, F., Chen, D., Li, R. et al. (2013). Nanoscale 5: 1137–1142.
- 55 Rabchinskii, M.K., Shnitov, V., Dideikin, A.T. et al. (2016). J. Phys. Chem. C 120: 28261–28269.
- 56 Rabchinskii, M.K., Ryzhkov, S.A., Kirilenko, D.A. et al. (2020). Sci. Rep. 10: 6902.
- 57 Rabchinskii, M.K., Shnitov, V.V., Stolyarova, D.Y. et al. (2020). Fullerenes Nanotubes Carbon Nanostruct. 28: 221–225.
- 58 Fang, Y., Luo, B., Jia, Y. et al. (2012). Adv. Mater. 24: 6348–6355.
- 59 Divyaa, V. and Reddy, M.L.P. (2013). J. Mater. Chem. C 1: 160–170.
- 60 Yan, R., Qiu, S., Tong, L., and Qian, Y. (2016). Chem. Speciation Bioavailability 28: 72–77.
- 61 Rabchinskii, M.K., Dideikin, A.T., Kirilenko, D.A. et al. (2018). Sci. Rep. 8: 14154.
- 62 Yu, M., Zhang, S., Chen, Y. et al. (2018). Carbon 133: 101–108.
- 63 Bonanni, A., Chua, C.K., and Pumera, M. (2014). Chem. Eur. J. 20: 217–222.
- 64 Poh, H.L., Sofer, Z., Simek, P. et al. (2015). Chem. Eur. J. 21: 8130–8136.
- 65 Lee, D. and Seo, J. (2014). Sci. Rep. 4: 7419.
- 66 Jin, Y., Zheng, Y., Podkolzin, S.G., and Lee, W. (2020). J. Mater. Chem. C 8: 4885–4894.
- 67 Taniguchi, T., Kurihara, S., Tateishi, H. et al. (2015). Carbon 84: 560–566.
- 68 Kim, S., Zhou, S., Hu, Y. et al. (2012). Nat. Mater. 11: 544–549.
- 69 Nair, R.R., Ren, W., Jalil, R. et al. (2010). Small 6: 2773.
- 70 Karlicky, F., Datta, K.K.R., Otyepka, M., and Zboril, R. (2013). 15021ACS Nano 7: 6434–6464.
- 71 Jankovsky, O., Simek, P., Sedmidubsky, D. et al. (2014). RSC Adv. 4: 1378–1387.
- 72 Poh, H.L., Sofer, Z., Klimova, K., and Pumera, M. (2014). J. Mater. Chem. C 2: 5198–5207.
- 73 Mazanek, V., Jankovsky, O., Luxa, J. et al. (2015). Nanoscale 7: 13646–13655.
- 74 Robinson, J.T., Burgess, J.S., Junkermeier, C.E. et al. (2010). Nano Lett. 10: 3001–3005.
- 75 Shen, B.S., Chen, J.T., Yan, X.B., and Xue, Q.J. (2012). RSC Adv. 2: 6761–6764.
- 76 Li, Y., Chen, H., Voo, L.Y. et al. (2012). J. Mater. Chem. 2012, 22: 15021–15024.
- 77 Jankovsky, O., Lojka, M., Luxa, J. et al. (2017). Chem. Eur. J. 23: 10473–10479.
- 78 Simek, P., Klímova, K., Sedmidubsky, D. et al. (2015). Nanoscale 7: 261–270.
- 79 Moon, I.K., Lee, J., Ruoff, R.S., and Lee, H. (2010). Nat. Commun. 1: 73.
- 80 Cui, P., Lee, J., Hwang, E., and Lee, H. (2011). Chem. Commun. 47: 12370–12372.
- 81 Bourlinos, A.B., Gournis, D., Petridis, D. et al. (2003). Langmuir 19: 6050–6055.
- 82 Megawati, M., Chua, C.K., Sofer, Z. et al. (2017). Phys. Chem. Chem. Phys. 19: 15914–15923.
- 83 Vacchi, I.A., Spinato, C., Raya, J. et al. (2016). Nanoscale 8: 13714–13721.
- 84 Zhang, W., Ma, J., Gao, D. et al. (2016). Prog. Org. Coat. 94: 9–17.
- 85 Ye, X., Ma, L., Yang, Z. et al. (2016). ACS Appl. Mater. Interfaces 8: 7483–7488.
- 86 Aguilar‐Bolados, H., Vargas‐Astudillo, D., Yazdani‐Pedram, M. et al. (2017). Chem. Mater. 29: 6698–6705.
- 87 Some, S., Shackery, I., Kim, S.J., and Jun, S.C. (2015). Chem. Eur. J. 21: 15480–15485.
- 88 Fareghi‐Alamdari, R., Haqiqi, M.G., and Zekri, N. (2016). New J. Chem. 40: 1287–1296.
- 89 Yeo, J.‐S., Yun, J.‐M., Jung, Y.‐S. et al. (2014). J. Mater. Chem. A 2: 292.
- 90 Garg, B., Bisht, T., and Ling, Y.‐C. (2014). RSC Adv. 4: 57297.
- 91 Chua, C.K. and Pumera, M. (2015). ACS Nano 9: 4193–4199.
- 92 Urbanov, V., Hol, K., Bourlinos, A.B. et al. (2015). Adv. Mater. 27: 2305–2310.
- 93 Klimova, K., Pumera, M., Luxa, J. et al. (2016). J. Phys. Chem. C 120: 24203–24212.
- 94 Gorgolis, G. and Galiotis, C. (2017). 2D Mater. 4: 032001.
- 95 Wan, X., Huang, Y., and Chen, Y. (2012). Acc. Chem. Res. 45: 598–607.
- 96 Wang, W., Wang, X., Pan, Y. et al. (2017). Ind. Eng. Chem. Res. 56: 6664–6670.
- 97 Yuan, W.J., Liu, A.R., Huang, L. et al. (2013). Adv. Mater. 25: 766–771.
- 98 Lipatov, A., Varezhnikov, A., Wilson, P. et al. (2013). Nanoscale 5: 5426–5434.
- 99 Yu, X., Cheng, H., Zhang, M. et al. (2017). Natl. Rev. 2: 17046.
- 100 Zhu, J., Andres, C.M., Xu, J. et al. (2012). ACS Nano 6: 8357–8365.
- 101 Saravanan, N., Rajasekar, R., Mahalakshmi, S. et al. (2014). J. Reinf. Plast. Compos. 33: 1158–1170.
- 102 Liang, J., Xu, Y., Huang, Y. et al. (2009). J. Phys. Chem. C 113: 9921–9927.
- 103 Huang, L., Yi, N., Wu, Y. et al. (2013). Adv. Mater. 25: 2224–2228.
- 104 Lee, S.‐H., Jung, J.‐H., and Oh, I.‐K. (2014). Small 10: 3880–3886.
- 105 Wen, W., Huang, X., and Sheng, P. (2008). Soft Matter 4: 200–210.
- 106 Sheng, P. and Wen, W. (2012). Annu. Rev. Fluid Mech. 44: 143–174.
- 107 Zhang, W.L., Liu, Y.D., Choi, H.J., and Kim, S.G. (2012). ACS Appl. Mater. Interfaces 4: 2267–2272.
- 108 Zhang, W.L., Liu, J., and Choi, H.J. (2015). J. Nanomater. 2015: 574637.