
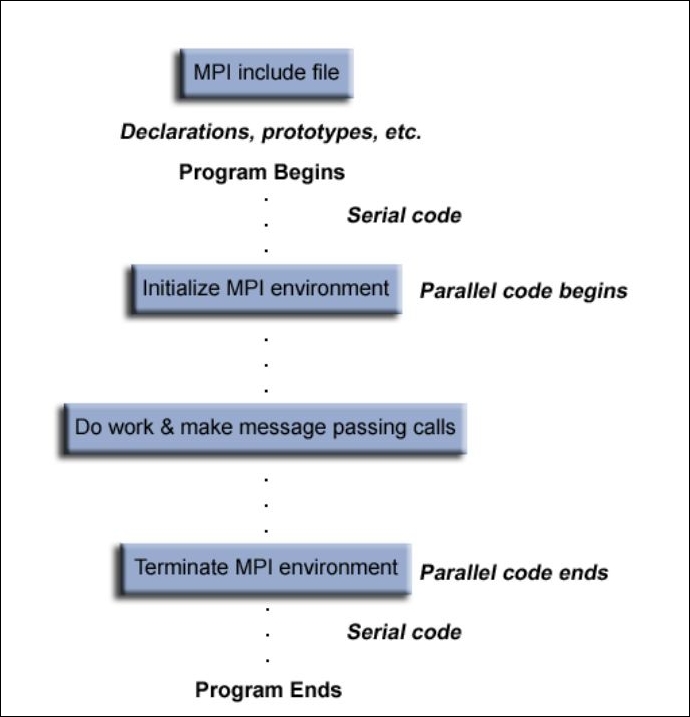
The general structure of MPI programs is depicted here (please visit the links provided in the introduction for a more in-depth discussion on the topic). Your MPI codes will have elements of this structure:
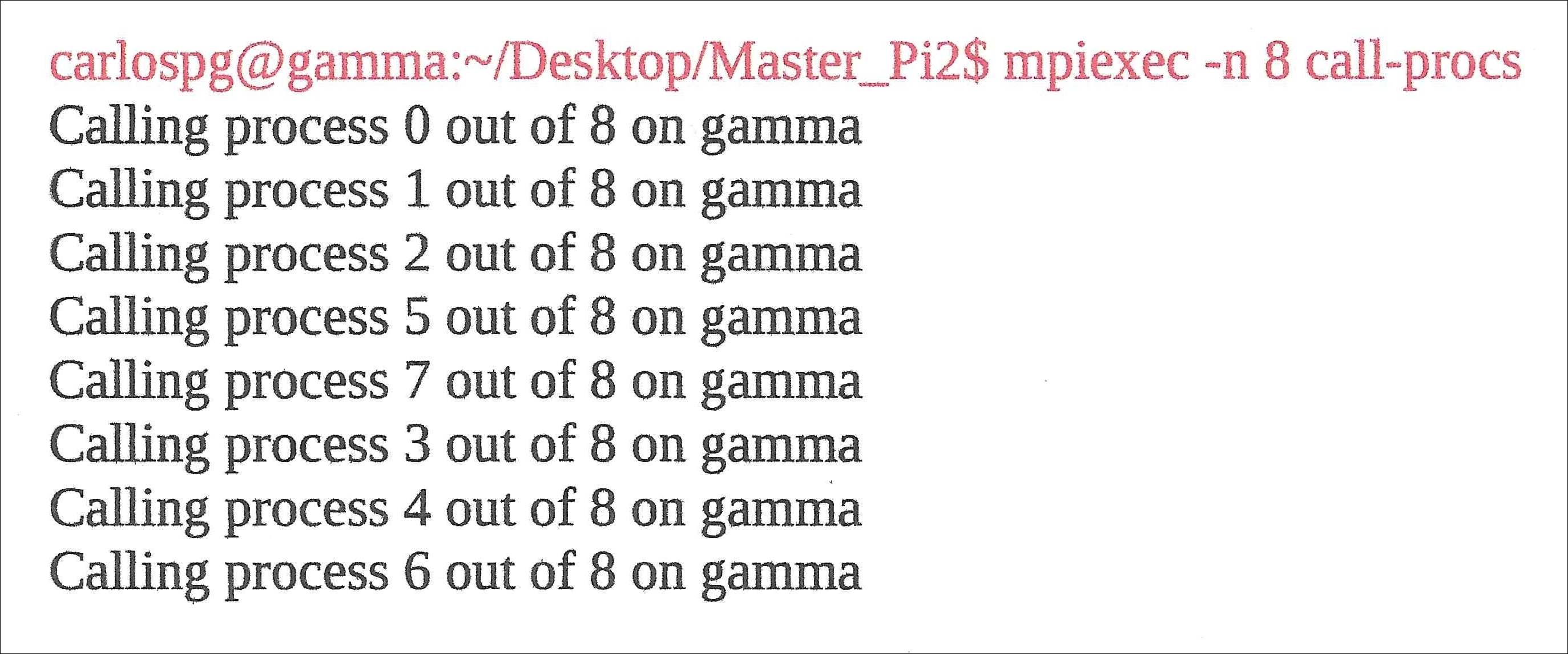
A relatively simple MPI code is shown here. You will notice how the code mirrors the general structure of MPI programs:
call-procs.c: #include <math.h> // math library #include <stdio.h>// Standard Input/Output library int main(int argc, char** argv) { /* MPI Variables */ int num_processes; int curr_rank; int proc_name_len; char proc_name[MPI_MAX_PROCESSOR_NAME]; /* Initialize MPI */ MPI_Init (&argc, &argv); /* acquire number of processes */ MPI_Comm_size(MPI_COMM_WORLD, &num_processes); /* acquire rank of the current process */ MPI_Comm_rank(MPI_COMM_WORLD, &curr_rank); /* acquire processor name for the current thread */ MPI_Get_processor_name(proc_name, &proc_name_len); /* output rank, no of processes, and process name */ printf("Calling process %d out of %d on %srn", curr_rank, num_processes, proc_name); /* clean up, done with MPI */ MPI_Finalize(); return 0; }
Your typical MPI code must contain, in large part, as the statements highlighted. There are however, many more statements/functions available for use, depending on the task you want to accomplish. We will not concern ourselves with those other statements, except for the following statements/functions:
"MPI_Bcast(&n, 1, MPI_INT, 0, MPI_COMM_WORLD);" and "MPI_Reduce(&rank_integral,&pi, 1, MPI_DOUBLE, MPI_SUM, 0, MPI_COMM_WORLD);",
You will add these to the π version of the code. The basic MPI code depicted previously was given the filename call-procs.c. Compile the code using the mpicc call-procs.c -o call-procs command. Run it using the mpiexec -n 8 call-procs command; the -n designates n threads or processes, followed by the number 8 − the number of threads or processes you want to use in the calculation. The result is shown here. Here, the author is using all eight threads in the four cores in his PC processor. The code randomly calls on the various processes, out of a total of 8, on the host computer gamma on which the code is running:
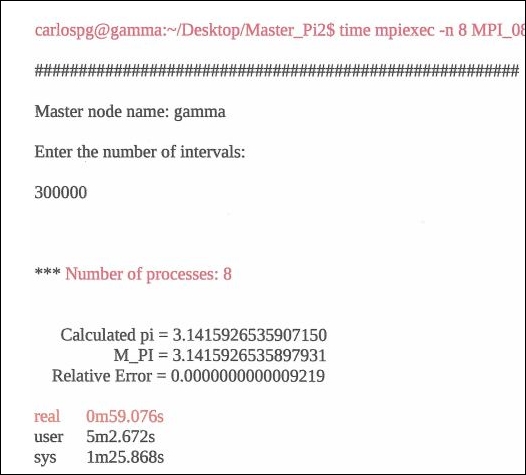
The MPI π code (filename MPI_08_b.c), with its run, is depicted on the following pages:
MPI_08_b.c:
#include <mpi.h> // (Open)MPI library
#include <math.h> // math library
#include <stdio.h>// Standard Input/Output library
int main(int argc, char*argv[])
{
int total_iter;
int n, rank, length, numprocs, i;
double pi, width, sum, x, rank_integral;
char hostname[MPI_MAX_PROCESSOR_NAME];
MPI_Init(&argc, &argv);// initiates MPI
MPI_Comm_size(MPI_COMM_WORLD, &numprocs);// acquire
number of processes
MPI_Comm_rank(MPI_COMM_WORLD, &rank);// acquire current
process id
MPI_Get_processor_name(hostname, &length);// acquire
hostname
if(rank == 0)
{
printf("n");
printf("#######################################################");
printf("nn");
printf("Master node name: %sn", hostname);
printf("n");
printf("Enter the number of intervals:n");
printf("n");
scanf("%d",&n);
printf("n");
}
// broadcast to all processes, the number of segments you
want
MPI_Bcast(&n, 1, MPI_INT, 0, MPI_COMM_WORLD);
// this loop increments the maximum number of iterations, thus providing
// additional work for testing computational speed of the processors
for(total_iter = 1; total_iter < n; total_iter++)
{
sum=0.0;
width = 1.0 / (double)total_iter; // width of a segment
// width = 1.0 / (double)n; // width of a segment
for(i = rank + 1; i <= total_iter; i += numprocs)
// for(i = rank + 1; i <= n; i += numprocs)
{
x = width * ((double)i - 0.5); // x: distance to
center of i(th) segment
sum += 4.0/(1.0 + x*x);// sum of individual segment
height for a given
// rank
}
// approximate area of segment (Pi value) for a given rank
rank_integral = width * sum;
// collect and add the partial area (Pi) values from all processes
MPI_Reduce(&rank_integral, &pi, 1, MPI_DOUBLE, MPI_SUM,
0, MPI_COMM_WORLD);
}// End of for(total_iter = 1; total_iter < n; total_iter++)
if(rank == 0)
{
printf("nn");
printf("*** Number of processes: %dn",numprocs);
printf("nn");
printf(" Calculated pi = %.30fn", pi);
printf(" M_PI = %.30fn", M_PI);
printf(" Relative Error = %.30fn", fabs(pi-M_PI));
// printf("Process %d on %s has the partial result of
%.16f n",rank,hostname,
rank_integral);
}
// clean up, done with MPI
MPI_Finalize();
return 0;
}// End of int main(int argc, char*argv[])
Compile and run this MPI C code. The result is depicted following. Notice the time parameter used for displaying the execution time beneath the program result. You will notice the gradual reduction in execution time as successively more processes/threads are brought online:

We will now discuss the most critical structure of the MPI version of the serial π code - the for (i = rank + 1; i <= total_iter; i += numprocs) statement. This construct assigns the data among the various processes. The variable numprocs has the value 8 - the author chose all eight threads (two per core) to do his digital bidding. So, for the eight processes/ranks, that is, 0, 1, 2, 3, 4, 5, 6, 7, and the ![]() segment (total 300,000) of the area under the
segment (total 300,000) of the area under the ![]() function curve, is assigned as follows:
function curve, is assigned as follows:
|
Rank |
Loop 1 |
Loop 2 |
Loop 3 |
Loop 4 |
Loop 5 |
Loop 6 |
Loop 7 |
Loop 8 |
Loop 9 |
|
0 |
1 |
9 |
17 |
25 |
33 |
41 |
49 |
57 |
65 |
|
1 |
2 |
10 |
18 |
26 |
34 |
42 |
50 |
58 |
66 |
|
2 |
3 |
11 |
19 |
27 |
35 |
43 |
51 |
59 |
67 |
|
3 |
4 |
12 |
20 |
28 |
36 |
44 |
52 |
60 |
68 |
|
4 |
5 |
13 |
21 |
29 |
37 |
45 |
53 |
61 |
69 |
|
5 |
6 |
14 |
22 |
30 |
38 |
46 |
54 |
62 |
70 |
|
6 |
7 |
15 |
23 |
31 |
39 |
47 |
55 |
63 |
71 |
|
7 |
8 |
16 |
24 |
32 |
40 |
48 |
56 |
64 |
72 |
Rank 0 is assigned the 1st, 9th, 17th, 25th, 33rd, 41st, 49th, 57th, 65th, and so on segments; rank 1 is assigned the 2nd, 10th, 18th, 26th, 34th, 42nd, 50th, 58th, 66th, and so on segments; and so on and so forth. This table extends to the 300,000th iteration/loop. So, for each rank, the distance x to the center of the ![]() segment as follows: width of a segment (width)
segment as follows: width of a segment (width) ![]() . This
. This x value is now used to calculate the height of the ![]() segment within its rank or processor core. The calculated heights for a given rank are continuously summed (sum += 4.0 / (1.0+x*x)) with each iteration/loop, and stored in that process temporary memory. Note that even though the
segment within its rank or processor core. The calculated heights for a given rank are continuously summed (sum += 4.0 / (1.0+x*x)) with each iteration/loop, and stored in that process temporary memory. Note that even though the sum variable is used by each process, it is a unique memory location for that process, so the data from one process will not contaminate the data in another process within a core, even though the processes use the same variable name. The author appreciates how perplexing this may seem to you; however, as they say in street parlance, that's how MPI rolls. After the 300,000th iteration/loop, the partial sum from each process/core is then combined/summed by the MPI_Reduce function to give the overall value for ![]() . Note that we ask the 0 rank or process to print/display the result. This is standard operating procedure.
. Note that we ask the 0 rank or process to print/display the result. This is standard operating procedure.
With regards to the outer for loop, it is used to extend the computational time for the simple pi calculation - the astute reader would have noticed that the computational time for one core, using the MPI protocol, is slower than the serial code using the same core; that is, 2m37.693s, as opposed to 0m0.039s. This occurs because the outer for loop incrementally increases the value of the upper limit total_iter - the maximum number of iterations - as the code executes. This intentional increase in processing time is to aid in making any improvement in the computational time compression perceptible, as you successively add more cores. Otherwise, for this simple π calculation, computation would be over in a very short time when using the maximum number of cores - not much fun if you are a novice in supercomputing - you want to observe your eventual newly minted Pi2 or Pi3 supercomputer producing digital sweat by working hard. You can always remove this artificial limitation (restrained mode) when you desire maximum computing power while performing legitimate work. Feel free to experiment by commenting out the outer for statement and replacing the inner for statement with for (i = rank + 1; i <= n; i += numprocs), where the value of n is the maximum number of iterations you entered at the initial run of the code (don't forget to also comment out width = 1.0/(double)total_itar, while uncommenting the statement immediately following). You should now notice a substantial drop in computational time for π.
Note how the run time shrinks with the activation of additional threads, processes, and cores. We now have undisputed evidence of the advantages of parallel processing. Be audacious, and improve your coding skills by modifying your codes, or writing new codes with other MPI functions listed on the websites provided in the introduction.
Now that you've had a taste of running MPI, lets now write and run three more exotic π equations that were discovered by the famous mathematicians Euler, Leibniz, and Nilakantha. Let's tackle first the Euler (Swiss mathematician, physicist, astronomer, logician, and engineer) equation:
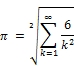
Copy or create the file using the; vim Euler.c command. Compile the file using mpicc Euler.c -o Euler -lm. The -lm links the math library header, <math.h>. Run the program using; time mpiexec -n x Euler, where x is replaced by the number of threads/cores you want to employ.
Here is the code for Euler:
Euler.c:
#include <mpi.h> // (Open)MPI library
#include <math.h> // math library
#include <stdio.h>// Standard Input/Output library
int main(int argc, char*argv[])
{
int total_iter;
int n,rank,length,numprocs,i;
double sum,sum0,rank_integral,A;
char hostname[MPI_MAX_PROCESSOR_NAME];
MPI_Init(&argc, &argv);// initiates MPI
MPI_Comm_size(MPI_COMM_WORLD, &numprocs); // acquire
number of processes
MPI_Comm_rank(MPI_COMM_WORLD, &rank); // acquire
current process id
MPI_Get_processor_name(hostname, &length);// acquire
hostname
if(rank == 0)
{
printf("n");
printf("#######################################################");
printf("nn");
printf("Master node name: %sn", hostname);
printf("n");
printf("Enter the number of intervals:n");
printf("n");
scanf("%d",&n);
printf("n");
}
// broadcast the number of iterations "n" in the
infinite series
MPI_Bcast(&n, 1, MPI_INT, 0, MPI_COMM_WORLD);
// this loop increments the maximum number of
iterations, thus providing
// additional work for testing computational speed of
the processors
for(total_iter = 1; total_iter < n; total_iter++)
{
sum0 = 0.0;
for(i = rank + 1; i <= total_iter; i += numprocs)
{
A = 1.0/(double)pow(i,2);
sum0 += A;
}
rank_integral = sum0;// Partial sum for a given rank
// collect and add the partial sum0 values from all
processes
MPI_Reduce(&rank_integral, &sum, 1, MPI_DOUBLE,
MPI_SUM, 0,
MPI_COMM_WORLD);
} // End of for(total_iter = 1; total_iter < n;
total_iter++)
if(rank == 0)
{
printf("nn");
printf("*** Number of processes: %dn",numprocs);
printf("nn");
printf(" Calculated pi = %.30fn", sqrt(6*sum));
printf(" M_PI = %.30fn", M_PI);
printf(" Relative Error = %.30fn",
fabs(sqrt(6*sum)-M_PI));
}
// clean up, done with MPI
MPI_Finalize();
return 0;
}// End of int main(int argc, char*argv[])
Let's check out the code run for Euler now:
carlospg@gamma:~/Desktop$ time mpiexec -n 1 Euler
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 1
Calculated pi = 3.141589470484041246578499340103
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003183105751869419464128441
Real 3m51.168s
user 3m45.792s
sys 0m0.012s
carlospg@gamma:~/Desktop$ time mpiexec -n 2 Euler
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 2
Calculated pi = 3.141589470484066115574250943610
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003183105727000423712524935
real 2m0.190s
user 3m55.128s
sys 0m0.020s
carlospg@gamma:~/Desktop$ time mpiexec -n 3 Euler
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 3
Calculated pi = 3.141589470484025703456154587911
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003183105767412541808880633
real 1m32.108s
user 4m27.736s
sys 0m0.016s
carlospg@gamma:~/Desktop$ time mpiexec -n 4 Euler
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 4
Calculated pi = 3.141589470484047019738227390917
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003183105746096259736077627
real 1m14.463s
user 4m51.544s
sys 0m0.040s
Let's write and run Leibniz (German polymath, inventor of differential and integral calculus along with Sir Isaac Newton, and philosopher):
Copy or create the file using the command; vim Leibniz.c. Compile the file using; mpicc Leibniz.c -o Leibniz -lm. The -lm links the math library header <math.h>. Run the program using the command; time mpiexec -n x Leibniz, where x is replaced by the number of threads/cores you want to employ.
The Gregory-Leibniz code is follows:
Leibniz.c:
#include <mpi.h> // (Open)MPI library
#include <math.h> // math library
#include <stdio.h>// Standard Input/Output library
int main(int argc, char*argv[])
{
int total_iter;
int n, rank,length,numprocs;
double rank_sum,pi,sum,A;
unsignedlong i,k;
char hostname[MPI_MAX_PROCESSOR_NAME];
MPI_Init(&argc, &argv); // initiates MPI
MPI_Comm_size(MPI_COMM_WORLD, &numprocs); // acquire number of processes
MPI_Comm_rank(MPI_COMM_WORLD, &rank); // acquire current process id
MPI_Get_processor_name(hostname, &length);// acquire hostname
if (rank == 0)
{
printf("n");
printf("#######################################################");
printf("nn");
printf("Master node name: %sn", hostname);
printf("n");
printf("Enter the number of intervals:n");
printf("n");
scanf("%d",&n);
printf("n");
}
// broadcast the number of iterations "n" in the infinite series
MPI_Bcast(&n, 1, MPI_INT, 0, MPI_COMM_WORLD);
// this loop increments the maximum number of iterations, thus providing
// additional work for testing computational speed of the processors
for(total_iter = 1; total_iter < n; total_iter++)
{
sum = 0.0;
for(i = rank + 1; i <= total_iter; i += numprocs)
{
k = i-1;
A = (double)pow(-1,k)* 4.0/(double)(2*k+1);
sum += A;
}
rank_sum = sum;// Partial sum for a given rank
// collect and add the partial sum values from all
processes
MPI_Reduce(&rank_sum, &pi, 1, MPI_DOUBLE, MPI_SUM, 0,
MPI_COMM_WORLD);
} // End of for(total_iter = 1; total_iter < n;
total_iter++)
if(rank == 0)
{
printf("nn");
printf("*** Number of processes: %dn",numprocs);
printf("nn");
printf(" Calculated pi = %.30fn", pi);
printf(" M_PI = %.30fn", M_PI);
printf(" Relative Error = %.30fn", fabs(pi-M_PI));
}
// clean up, done with MPI
MPI_Finalize();
return 0;
}
Now, let's go through the code run for Gregory-Leibniz:
carlospg@gamma:~/Desktop$ time mpiexec -n 1 Gregory_Leibniz
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 1
Calculated pi = 3.141595986934242024091190614854
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003333344448908093227146310
real 25m13.914s
user 25m8.500s
sys 0m0.012s
carlospg@gamma:~/Desktop$ time mpiexec -n 2 Gregory_Leibniz
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 2
Calculated pi = 3.141595986933658934958657482639
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003333343865818960694014095
real 13m5.893s
user 26m5.500s
sys 0m0.052s
carlospg@gamma:~/Desktop$ time mpiexec -n 3 Gregory_Leibniz
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 3
Calculated pi = 3.141595986934202056062304109219
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003333344408940064340640674
real 8m57.444s
user 26m46.880s
sys 0m0.040s
carlospg@gamma:~/Desktop$ time mpiexec -n 4 Gregory_Leibniz
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 4
Calculated pi = 3.141595986934321516059753776062
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000003333344528400061790307518
real 7m14.118s
user 28m31.732s
sys 0m0.116s
Let's now write and run Nilakantha (brilliant Indian mathematician and astronomer):
Copy or create the file using; vim Nilakantha.c command. Compile the file using mpicc Nilakantha.c -o Nilakantha -lm. The -lm links the math library header, math.h. Run the program using; time mpiexec -n x Nilakantha, where x is replaced by the number of threads/cores you want to employ.
The code for Nilakantha is given as follows:
Nilakantha.c:
#include <mpi.h> // (Open)MPI library
#include <math.h> // math library
#include <stdio.h>// Standard Input/Output library
int main(int argc, char*argv[])
{
int total_iter;
int n,rank,length,numprocs,i,k;
double sum,sum0,rank_integral,A,B,C;
char hostname[MPI_MAX_PROCESSOR_NAME];
MPI_Init(&argc, &argv);// initiates MPI
MPI_Comm_size(MPI_COMM_WORLD, &numprocs); // acquire number of processes
MPI_Comm_rank(MPI_COMM_WORLD, &rank); // acquire current process id
MPI_Get_processor_name(hostname, &length);// acquire hostname
if(rank == 0)
{
printf("n");
printf("#######################################################");
printf("nn");
printf("Master node name: %sn", hostname);
printf("n");
printf("Enter the number of intervals:n");
printf("n");
scanf("%d",&n);
printf("n");
}
// broadcast to all processes, the number of segments you want
MPI_Bcast(&n, 1, MPI_INT, 0, MPI_COMM_WORLD);
// this loop increments the maximum number of iterations, thus providing
// additional work for testing computational speed of the processors
for(total_iter = 1; total_iter < n; total_iter++)
{
sum0 = 0.0;
for(i = rank + 1; i <= total_iter; i += numprocs)
{
k = i-1;
A = (double)pow(-1,k+2);
B = 4.0/(double)((k+1)*(k+2)*(k+3));
C = A * B;
sum0 += C;
}
rank_integral = sum0;// Partial sum for a given rank
// collect and add the partial sum0 values from all processes
MPI_Reduce(&rank_integral, &sum, 1, MPI_DOUBLE, MPI_SUM, 0,
MPI_COMM_WORLD);
} // End of for(total_iter = 1; total_iter < n; total_iter++)
if(rank == 0)
{
printf("nn");
printf("*** Number of processes: %dn",numprocs);
printf("nn");
printf(" Calculated pi = %.30fn", (3+sum));
printf(" M_PI = %.30fn", M_PI);
printf(" Relative Error = %.30fn", fabs((3+sum)-M_PI));
}
// clean up, done with MPI
MPI_Finalize();
return 0;
}// End of int main(int argc, char*argv[])
Let's have a look at the code run for Nilakantha;
carlospg@gamma:~/Desktop$ time mpiexec -n 1 Nilakantha
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 1
Calculated pi = 3.141448792732286499074234598083
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000143860857506616923728870461
real 24m37.130s
user 24m31.772s
sys 0m0.008s
carlospg@gamma:~/Desktop$ time mpiexec -n 2 Nilakantha
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 2
Calculated pi = 3.141448792732290939966333098710
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000143860857502176031630369835
real 12m36.831s
user 25m6.088s
sys 0m0.008s
carlospg@gamma:~/Desktop$ time mpiexec -n 3 Nilakantha
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 3
Calculated pi = 3.141448792732290051787913398584
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000143860857503064210050069960
real 9m0.872s
user 26m30.512s
sys 0m0.052s
carlospg@gamma:~/Desktop$ time mpiexec -n 4 Nilakantha
#######################################################
Master node name: gamma
Enter the number of intervals:
300000
*** Number of processes: 4
Calculated pi = 3.141448792732288275431073998334
M_PI = 3.141592653589793115997963468544
Relative Error = 0.000143860857504840566889470210
real 6m56.071s
user 27m10.824s
sys 0m0.052s
Now that you have sampled the extreme power that awaits you at your beck and call, let's now proceed to build that much anticipated Pi2 or Pi3 supercomputer.