
Chapter 36
Bioengineered Cell-Derived Vesicles as Drug Delivery Carriers
Vipul Gujrati and Sangyong Jon
36.1 Introduction
Following a quarter century of rapid advances, the field of nanotechnology has witnessed rapid growth in the area of nanocarrier-based medicines for treating cancers [1–3]. We foresee cancer research developing into a rational science, reflecting the development of nanomedicines based on an understanding of the complexities of the disease as revealed in laboratory and clinical studies. Although synthetic drug carriers are being proactively studied, attention has been drawn toward natural nanocarriers developed from a variety of cells, including prokaryotes and higher eukaryotes [1–8].
This review focuses on recent progress in the development of versatile drug delivery vehicles developed from natural sources, such as cellular compartments, including membranes and cellular vesicles. Both the cellular membrane and vesicles enclose diverse substances that are effectively protected from the exterior environment and associated stresses. In addition, cellular vesicles play a central role in communicating biological information, both inter- and intracellularly. Such natural barrier- and transporter-like properties of cellular components are being explored in diverse ways. Various unmodified and modified cellular particles have been tested for possible application in passive or active targeting. Several attributes make natural carriers a better choice, including the fact that these carriers show much better biocompatibility and reduced toxicity compared with synthetic carriers. The process of production and purification is simple, well controlled, and cost-effective. Moreover, it is possible to package diverse therapeutic agents using molecular engineering and physical approaches. Although these biological carriers are in the early developmental stage, studies discussed in the current review emphasize the potential of exploiting biological carriers for therapeutic drug delivery applications. Here we summarize the biological carriers derived from diverse cellular sources, ranging from bacteria and yeast to higher eukaryotic cells. In addition, we also discuss examples of recently reported cell membrane-camouflaged nanoparticles (NPs).
36.2 Prokaryotic Cell-Derived Nanocarriers
36.2.1 Bacterial Minicells as Drug Carrier
MacDiarmid and colleagues described a promising technology that effectively encapsulates diverse anticancer agents into bacterial minicells, which are then selectively targeted to cancer cells using bispecific antibodies [9–11]. Minicells are anucleate NPs that have a uniform diameter of approximately 400 nm, generated by inactivating genes that control normal cell division in bacteria [9, 12]. Interestingly, they can be produced in high yields from both gram-positive and gram-negative bacteria. However, obtaining sterile minicell preparations requires extensive purification to remove contaminants, such as parental bacteria, membrane blebs, cellular debris, nucleic acids, and free endotoxin [9]. For cancer targeting, a bispecific antibody is linked to minicells via the O-polysaccharide component of lipopolysaccharide (LPS) exposed on the surface, allowing the other free end to target the cell surface receptor. A range of anticancer agents with diverse physicochemical properties (structure, charge, solubility), such as chemotherapeutic drugs and small inhibitory RNA (siRNA), have been packaged into minicells by co-incubation [9, 10]. In vitro studies have revealed that these minicells actively target cancer cells, undergo endocytosis, and are degraded in endosomes or lysosomes to release their payload, which escapes into the cytoplasm or nucleus to exert toxic effects. Interestingly, in vivo xenograft studies have shown that potent antitumor effects can be achieved with amounts of drugs more than 1000-fold lower than that required for the respective free drugs.
36.2.2 Bioengineered Bacterial Outer Membrane Vesicles for Cancer Targeting and Drug Delivery
In recent years, strategies have been developed using bacterial blebs or outer membrane vesicles (OMVs) to deliver vaccines and siRNA [13, 14]. OMVs are nanosized proteoliposomes with diameters in the range of 50–200 nm. They are composed of outer membrane proteins, LPS, outer membrane lipids, periplasmic proteins, DNA, and RNA and are constitutively released from the outer membrane of bacteria during normal growth [15–18]. Gujrati and colleagues developed bioengineered bacterial OMVs for siRNA delivery to treat HER2-overexpressing cancers (Figure 36.1) [14]. They utilized a genetically modified K12-derived W3110 Escherichia coli strain, which harbors a mutation in the msbB gene that leads to the production of the very low immunogenicity, penta-acylated LPS [19, 20]. In addition, for cancer-specific targeting, they engineered the E. coli strain to express an HER2-specific affibody on its surface and vesicles derived from it. The cell-specific binding and uptake was also evaluated following in vitro cell binding and uptake assay. Additionally, ELISA was done to analyze HER2 specificity of the modified OMVs. Next, siRNA loading into OMVs was achieved by electroporation. The sub-nanometer size and HER2-selective accumulation of AffiHER2OMVsiRNA in tumor tissue after intravenous (i.v.) administration ensured potent anticancer effects.

Figure 36.1 Bacterial OMVs as drug delivery carriers. (a) The bioengineering strategy for OMVs is shown. The anti-HER affibody was expressed on the outer membrane of msbB mutant W3110 derived K12 strain. The purified AffiHER2OMV's were loaded with siRNA (AffiHER2OMVsiRNA) and kinetics of AffiHER2OMVsiRNA was evaluated in vitro and in vivo. (b) In vivo therapeutic efficacy of AffiHER2OMVsiRNA was evaluated against different controls. HER2 targeted AffiHER2OMVsiRNA showed significant tumor regression effect compared with controls. (Gujrati et al. 2014 [14]. Reproduced with permission of American Chemical Society.)
36.3 Eukaryotic Cell-Derived Nanocarriers
36.3.1 Bioengineered Yeast for Development of Nanocarriers
Preclinical studies using bacterially derived nanovectors have clearly demonstrated the benefits of genetic engineering in the development of drug delivery carriers from bacterial strains; however, such pathogen-derived vehicles are always accompanied by immunogenicity concerns [9, 14]. Addressing the issues that directly affect the clinical feasibility of nanocarriers, Gujrati et al. developed bioengineered nanocarriers from a cellular compartment (vacuole) of budding yeast, a nonhazardous and nonpathogenic strain of yeast [21]. In this application, yeast were genetically engineered by deleting yeast protein transport 7 (YPT7), a Rab GTPase that plays a crucial role in membrane fusion processes [22–25], and expressing a cancer-targeting ligand (anti-HER2 affibody) as a fusion partner with the vacuole membrane alkaline phosphatase, PHO8. Deletion of YPT7 and expression of an anti-HER2 affibody resulted in a yeast strain that generated a large number of fragmented vacuoles expressing the cancer-targeting anti-HER2 affibody on their surface. The combination of anti-HER2 affibody expression on vacuole membranes and similarities in the lipid composition of yeast vacuoles and mammalian cell membranes achieved enhanced cancer targeting and tissue penetration [21]. Moreover, anti-HER2 affibody-expressing vacuoles (AffiHER2VacuoleDox) incorporating the anticancer agent doxorubicin (Dox) showed potent antitumor effects, mainly owing to active targeting and enhanced tumor-specific penetration and distribution. Importantly, vacuoles are considered safe because of their endomembrane composition and are capable of being produced on a large scale by increasing the yeast fermentation capacity.
36.3.2 Bioengineered Extracellular Vesicles for the Development of a Drug Delivery Platform
Almost all cell types release vesicles into their surrounding environment. These vesicles, referred to as extracellular vesicles (EVs), are derived from endosomal or plasma membranes and are crucial for intercellular communication [26–28]. EVs are classified based on their origin, function, and physicochemical properties. As part of regular cellular communication systems, EVs can transport proteins, lipids, and nucleic acids and have thus recently been explored for their potential as drug delivery carriers [26–28]. In the following section, we discuss strategies developed using exosomes as nanomedicines.
Exosomes are nanosized vesicles with diameters in the range of 30–120 nm; each has a unique lipid and protein composition that often varies based on its source of origin. Exosomes are secreted by cultured cells and are also found in body fluids, including blood, urine, saliva, and breast milk. Because exosomes are relatively stable, they remain intact and maintain their morphology for a long time in the circulation and thus can be used as drug delivery carriers [29, 30].
Erviti et al. reported the utility of engineered exosomes for brain-specific targeting and siRNA delivery [31]. They isolated exosomes secreted from immature dendritic cells (DCs) that were engineered to generate exosomes with active cancer-targeting properties. Immature DCs express a low level of immunostimulatory components on their surface and, because of their unique endomembrane composition, are considered safe for in vivo use. To achieve active targeting, these researchers genetically fused the exosomal membrane protein Lamp2b with a peptide derived from the nicotinic acetylcholine receptor-binding rabies virus glycoprotein (RVG), which has shown its ability to deliver NPs across the blood–brain barrier (BBB). These authors also reported an electroporation protocol for loading siRNA into nanovehicles. After i.v. injection, the siRNA-packaged exosomes were effectively delivered to neurons in the brain and exhibited significant gene-silencing effects, decreasing target gene expression by approximately 60%. Thus, an engineered exosome-based approach is a promising technology for targeted gene therapy.
In another study, Ohno et al. developed engineered exosomes for delivery of microRNA (miRNA) [32]. To achieve epidermal growth factor receptor (EGFR)-specific targeting, these researchers transfected exosome donor human embryonic kidney 293 (HEK293) cells with pDisplay encoding a GE11 or EGF construct. The pDisplay vector uses the transmembrane domain of the platelet-derived growth factor receptor (PDGFR) to ensure the expression of recombinant proteins on plasma membranes. Exosomes expressing GE11 or EGF peptides were purified using a well-established ultracentrifugation method. The modified exosomes were loaded with the tumor suppressor, miRNA-Let-7a, introduced into exosomes by using lipofection, and drug loading was determined by quantitative real-time reverse transcription–polymerase chain reaction (qRT–PCR). Upon i.v. injection, these engineered exosomes successfully delivered the miRNA to the xenograft cancer tissue, markedly suppressing tumor growth. This report thus represents a successful strategy for engineering exosomes for EGFR-specific targeting and packaging of nucleic acid drugs for cancer therapy. In a similar vein, Tian et al. developed engineered exosomes for delivery of the chemotherapeutic drug, Dox [33]. These authors demonstrated improvement in the therapeutic index owing to the cancer-targeting ability of exosomes compared with the same dose of free drug. In this application, immature DCs were transfected with a vector expressing iRGD-Lamp2b (pEGFP-C1-iRGD-Lamp2b). The resulting bioengineered iRGD-Lamp2b exosomes, isolated and purified from DC cultures using multiple centrifugation and ultracentrifugation protocols, showed high specificity for target αν integrin-positive cancer cells. Dox was loaded into bioengineered iRGD-Lamp2b exosomes via electroporation, and its delivery by iRGD exosomes and inhibitory effects on cancer cell proliferation were analyzed in vitro in various cell types. A further evaluation of biodistribution and antitumor effects in vivo showed that the modified, Dox-packaged exosomes specifically targeted the tumor and exerted powerful tumor growth inhibition effects, suggesting that iRGD exosomes effectively accumulate Dox in tumor sites and inhibit tumor growth by virtue of their cancer-targeting ability.
In a study, Jang et al. reported exosome mimetic nanovesicles (NVs) for targeted delivery of chemotherapeutic agents [34]. Although exosomes can be loaded with diverse agents and utilized for targeted drug delivery purposes, low production yields and tedious purification methods hamper their clinical translation. Jang et al. developed NVs from different cell types that were pre-incubated with chemotherapeutic drug and subjected to serial extrusions through filters of decreasing pore size. Using a step gradient and ultracentrifugation protocol, these authors obtained drug-loaded NVs with an overall yield that was manifold higher than of exosomes obtained from the same cell type. In order to achieve cell-specific delivery, they proposed generation of NVs from cells harboring cognate receptors of ligands expressed on target cells. To this end, they prepared NVs from macrophages and monocytes expressing receptors for endothelial cell adhesion molecules (CAMs). Rapidly proliferating, abnormal endothelial cells within tumors express CAMs, which are targeted by macrophages and monocytes through cognate receptors such as LFA-1 (lymphocyte function-associated antigen 1). In this report, the authors successfully demonstrated the high-yield production of Dox-loaded NVs that were capable of inducing significant cytotoxic effects by actively targeting human umbilical vein endothelial cells (HUVECs), treated with tumor necrosis factor (TNF)-α to promote CAM expression. They also demonstrated the possibility of incorporating diverse agents into these NVs, such as 5-fluoruricil (5-FU), gemcitabine, and carboplatin.
An obstacle in the development of biological NV-based drug delivery systems is the difficulty of loading hydrophobic compounds into preformed NVs. Using liposomes with membrane fusogenic potential, Lee et al. developed a platform technology in which exogenous hydrophobic and hydrophilic agents were packaged into the plasma membrane and cytoplasm of donor cells, respectively, and subsequently into their derived membrane vesicles (MVs) (Figure 36.2) [35]. The membrane fusogenic liposomes (MFLs) that are loaded with hydrophobic and hydrophilic compounds were treated on cells, which were delivered into the plasma membrane and cytosol, respectively. It was found that the exosomes and microvesicles generated from MFL-treated cells could carry both components in its membrane and cytosol based on respective physicochemical property. Next, the packaged agents were delivered to neighboring cells via secreted MVs that could be isolated from cultures using standard ultracentrifugation protocols developed for exosome isolation. This liposome-mediated engineered MV system was further tested for therapeutic applications in vitro and in vivo.

Figure 36.2 (a) Schematic showing MV generation and loading of agents with different physicochemical properties. Cancer-specific distribution of MVs in vitro (a) and in vivo (b). (Lee et al. 2015 [35]. Reproduced with permission of American Chemical Society.)
Various studies have also shown that exosomes secreted by different cells are able to target specific tissues or cells. An exploratory study of an exosomal drug delivery system was reported by Sun et al. [36] in which the hydrophobic drug curcumin was loaded into exosomes derived from the EL-4 (mouse lymphoma) cell line using a direct mixing method. Such exosomal delivery systems enhance the anti-inflammatory activity of curcumin by increasing its solubility, stability, and bioavailability. In addition, the delivery of curcumin to activated monocyte-derived myeloid cells was substantially enhanced. In the LPS-induced septic shock model, it was demonstrated that curcumin-loaded exosomes, injected intraperitoneally (i.p.), were capable of targeting CD11b+Gr-1+ cells and promoting apoptosis, resulting in anti-inflammatory activity. Such anti-inflammatory effects may be beneficial in treating various inflammatory diseases, including cancers. These authors further exploited this exosomal drug delivery system to deliver anti-inflammatory drugs (curcumin or JSI124) to the brain via the intranasal route [37].
Microparticles (MPs), shed by almost all cell types in response to various stimuli, fall in diverse sizes ranging from 100 to 1000 nm. Tang et al. demonstrated that MPs derived from apoptotic cancer cells can package chemotherapeutic agents and deliver them to cancer cells [38]. For this purpose, mouse hepatocarcinoma cells were treated with methotrexate (MTX) and irradiated with UV light for 1 h to induce apoptosis. After 12 h, MPs were isolated form cultures using a multistep centrifugation protocol. Apoptotic tumor cells (5 × 106) were found to release drug-packaged MPs with high yield (3 × 105). They also confirmed the packaging of other drugs into MPs by cancer cells of diverse origin; examples include MPs loaded with cisplatin or DOx produced by H22 and A2780 cells. This study showed that MPs derived from apoptotic cancer cells can act as carriers to deliver diverse chemotherapeutic drugs and are relatively safe, highlighting their potential for clinical applications. Strategies are also reported for preparing EVs encapsulating diverse NPs regardless of their physicochemical properties [39]. Such NP-loaded EVs show potential for use as theranostics.
36.4 Cell Membrane-Camouflaged Nanoparticles
36.4.1 Erythrocyte Membrane-Coated Nanocarriers
In order to achieve sustained systemic delivery and prolonged circulation, researchers have developed different approaches that generally involve altering the physicochemical properties of NPs. The use of polyethylene glycol (PEG) for the development of long-circulating nanocarriers has led to the development and clinical translation of various synthetic NPs [40]. However, the immune response triggered by PEG has motivated research into other appropriate biocompatible options [41]. Accordingly, erythrocytes have been explored as drug carriers owing to their long circulation potential and natural origin. In pioneering work, Hu and colleagues developed a red blood cell (RBC) membrane-coated-based platform technology for preparing camouflaged NPs [42]. In this application, polymeric NPs were coated with a bilayered RBC membrane consisting of RBC lipids and proteins. The preparation process is divided in two parts: first, isolation of MVs from RBCs and second, fusion with NPs. The RBC membrane was ruptured using a hypotonic medium, and hemoglobin was removed by centrifugation. The resulting RBC ghosts were sonicated and serially extruded using 400 and 100 nm polycarbonate porous membranes. Finally, RBC NVs were coextruded with poly(lactic-co-glycolic acid) (PLGA) NPs. The resulting RBC membrane-coated PLGA NPs were characterized with respect to size, shape, surface charge, and membrane composition. This analysis showed that the NPs were effectively coated with RBC membranes. These RBC membrane-coated PLGA NPs showed greater stability in serum than bare PLGA NPs, which immediately aggregated in serum. Furthermore, RBC membrane-coated NPs exhibited a prolonged circulation time in vivo, indicating that these NPs are less susceptible to clearance because of the RBC membrane components, and therefore can be utilized in various biomedical applications. A similar extrusion strategy was applied to coat inorganic Au nanoparticles (AuNPs) with RBC membranes (Figure 36.3) [43]. As shown (Figure 36.3), mouse RBCs were given hypotonic shock to remove intracellular components and ghost cells were repeatedly extruded to generate nanosized RBC vesicles. Next, citrate-stabilized AuNPs were mixed with RBC vesicles and extruded to facilitate membrane coating due to fusion of membrane on AuNPs and formation of RBC-AuNPs. Such RBC membrane-camouflaged NPs can be utilized for a range of biomedical applications. However, in cancer therapy, cell-specific targeting is preferred to minimize nonspecific toxic effects of the drug. In this context, Zhang and colleagues reported functionalizing biological membrane-camouflaged NPs with targeting ligands using a lipid-insertion approach [44]. In this method, targeting moieties of diverse size, including folate (MW 441 Da) or AS1411 aptamer (MW 9000 Da), were incorporated in a controlled manner into RBC membranes with the help of tethered lipid molecules, and the resulting ligand-functionalized membranes were coated over a polymeric core using an extrusion method. Diverse biomedical applications have been reported using RBC membrane-coated NPs, such as detoxification treatments and preparation of vaccines [45, 46], indicating the potential of this approach for development as a new class of nanotherapeutics and for bridging the gap between synthetic and biological materials.
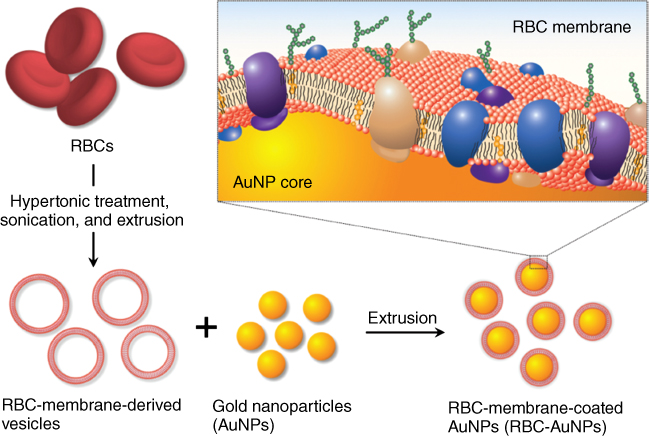
Figure 36.3 Functionalization of AuNPs with RBC membranes. RBC vesicles were developed by removing intracellular contents of RBCs and repeatedly extruding the RBC ghosts. Next, citrate-stabilized AuNPs were extruded with RBC vesicles resulting in membrane coating of AuNPs and formation of RBC-AuNPs. (Reproduced with permission [43]. Copyright 2013, John Wiley & Sons.)
36.4.2 Leukocyte Membrane-Camouflaged Nanoparticles
RBCs have a simple structure compared with other eukaryotic cells, but only a few reports on eukaryotic membrane-coated NPs are available. In one such study, Parodi et al. reported leukocyte membrane-coated porous silicon particles, referred to as leuko-like vectors (LLVs) [47]. The leukocyte membrane coating helps NPs overcome challenges associated with the immune system, such as opsonization and uptake by phagocytes, allowing them to pass through various biological barriers. The electrostatic interaction between the negatively charged proteolipid membrane and positively charged NP ensures complete surface coating. Camouflaged NPs exhibited a dramatic reduction in the adsorption of serum albumin and marked inhibition of phagocytic uptake. Delayed in vivo liver clearance and enhanced tumor-specific uptake were also observed.
36.4.3 Platelet Membrane-Camouflaged Nanoparticles
Zhang and colleagues also introduced platelet membrane-enclosed PLGA nanoparticles (PNPs) carrying diverse therapeutic agents [48]. Similar to LLVs, PNPs acquired platelet-mimicking functionality owing to their enrichment in membrane proteins. These PNPs exhibited increased immune compatibility of NPs, targeting of diseased vasculature, and specific binding to platelet-adhering pathogens, demonstrating the potential utility of this PNP platform technology.
36.4.4 Cancer Cell Membrane-Camouflaged Nanoparticles
Several reports have described the utility of cancer antigen-mimicking NPs for achieving therapeutic benefits [49, 50]. Among the several bio-inspired nanocarriers discussed to date are those engineered to target cancer cells; however, in an interesting application, Fang and colleagues utilized this approach to prepare cancer cell membrane-coated nanoparticles (CCNPs) [51] using PLGA as the polymeric core. The membrane-associated cancer antigen provided efficient delivery of such modified NPs to antigen-presenting cells and evoked potent immune responses. In addition, it was found that CCNPs carry CAMs, which enabled CCNPs to deliver a packaged fluorescent dye specifically to cancer cells owing to their ability to bind homotypically to source cancer cells. Importantly, the polymeric core can be utilized to load a range of agents.
36.5 Conclusions
Natural cell-based carriers offer various advantages, mainly in terms of bioengineering, safety, and toxicity. The limitations associated with clinical translation of synthetic carriers can be addressed using bio-inspired carriers. However, several hurdles must be overcome for the effective clinical translation of such biological carriers. Generating biological carriers of uniform quality requires the establishment of good manufacturing practice (GMP) parameters to ensure reproducibility and minimize batch-to-batch variations. Importantly, conditions for large-scale drug loading, purification, and storage should be defined. Biological vesicles have produced promising results on a laboratory scale; however, careful preclinical safety and toxicity assessments will be needed for translation to clinical practice.
Acknowledgments
The authors acknowledge financial support from the Intelligent Synthetic Biology Center of the Global Frontier Project (No. 2013M3A6A8073557) funded by the Ministry of Science, ICT & Future Planning.
References
- 1. Davis, M.E., Chen, Z., and Shin, D.M. (2008) Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug. Discov., 7, 771–782.
- 2. Chauhan, V.P. and Jain, R.K. (2013) Strategies for advancing cancer nanomedicine. Nat. Mater., 12, 958–962.
- 3. Chow, E.K.H. and Ho, D. (2013) Cancer nanomedicine: from drug delivery to imaging. Sci. Transl. Med., 5, 216rv4.
- 4. Shi, J.J., Votruba, A.R., Farokhzad, O.C., and Langer, R. (2010) Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett., 10, 3223–3230.
- 5. Adair, J.H., Parette, M.P., Altinoglu, E.I., and Kester, M. (2010) Nanoparticulate alternatives for drug delivery. ACS Nano, 4, 4967–4970.
- 6. Petros, R.A. and DeSimone, J.M. (2010) Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov., 9, 615–627.
- 7. Desai, N. (2012) Challenges in development of nanoparticle-based therapeutics. AAPS J., 14, 282–295.
- 8. Yoo, J.W., Irvine, D.J., Discher, D.E., and Mitragotri, S. (2011) Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat. Rev. Drug Discov., 10, 521–535.
- 9. MacDiarmid, J.A., Mugridge, N.B., Weiss, J.C., Phillips, L. et al. (2007) Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell, 11, 431–445.
- 10. MacDiarmid, J.A., Amaro-Mugridge, N.B., Madrid-Weiss, J., Sedliarou, I. et al. (2009) Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug. Nat. Biotechnol., 27, 643–651.
- 11. Karagiannis, E.D. and Anderson, D.G. (2009) Minicells overcome tumor drug-resistance. Nat. Biotechnol., 27, 620–621.
- 12. de Boer, P.A., Crossley, R.E., and Rothfield, L.I. (1989) A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell, 56, 641–649.
- 13. Kim, J.Y., Doody, A.M., Chen, D.J., Cremona, G.H. et al. (2008) Engineered bacterial outer membrane vesicles with enhanced functionality. J. Mol. Biol., 380, 51–66.
- 14. Gujrati, V., Kim, S., Kim, S.H., Min, J.J. et al. (2014) Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano, 8, 1525–1537.
- 15. Lee, E.Y., Choi, D.S., Kim, K.P., and Gho, Y.S. (2008) Proteomics in gram-negative bacterial outer membrane vesicles. Mass Spectrom. Rev., 27, 535–555.
- 16. Kuehn, M.J. and Kesty, N.C. (2005) Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev., 19, 2645–2655.
- 17. Schertzer, J.W. and Whiteley, M. (2013) Bacterial outer membrane vesicles in trafficking, communication and the host-pathogen interaction. J. Mol. Microbiol. Biotechnol., 23, 118–130.
- 18. Chen, D.J., Osterrieder, N., Metzger, S.M., Buckles, E. et al. (2010) Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc. Natl. Acad. Sci. U. S. A., 107, 3099–3104.
- 19. Kim, S.H., Kim, K.S., Lee, S.R., Kim, E. et al. (2009) Structural modifications of outer membrane vesicles to refine them as vaccine delivery vehicles. Biochim. Biophys. Acta, 1788, 2150–2159.
- 20. Hajjar, A.M., Ernst, R.K., Tsai, J.H., Wilson, C.B. et al. (2002) Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol., 3, 354–359.
- 21. Gujrati, V., Lee, M., Ko, Y.J., Lee, S. et al. (2016) Bioengineered yeast-derived vacuoles with enhanced tissue-penetrating ability for targeted cancer therapy. Proc. Natl. Acad. Sci. U. S. A., 113, 710–715.
- 22. Wickner, W. (2002) Yeast vacuoles and membrane fusion pathways. EMBO J., 21, 1241–1247.
- 23. Wickner, W. (2010) Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu. Rev. Cell Dev. Biol., 26, 115–136.
- 24. Haas, A., Scheglmann, D., Lazar, T., Gallwitz, D. et al. (1995) The Gtpase Ypt7p of saccharomyces-cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J., 14, 5258–5270.
- 25. Jun, Y. and Wickner, W. (2007) Assays of vacuole fusion resolve the stages of docking, lipid mixing, and content mixing. Proc. Natl. Acad. Sci. U.S.A., 104, 13010–13015.
- 26. Raposo, G. and Stoorvogel, W. (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol., 200, 373–383.
- 27. Johnsen, K.B., Gudbergsson, J.M., Skov, M.N., Pilgaard, L. et al. (2014) A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta Rev. Cancer, 1846, 75–87.
- 28. Tan, S., Wu, T., Zhang, D., and Zhang, Z. (2015) Cell or cell membrane-based drug delivery systems. Theranostics, 5, 863–881.
- 29. El Andaloussi, S., Maeger, I., Breakefield, X.O., and Wood, M.J.A. (2013) Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov., 12, 348–358.
- 30. Vlassov, A.V., Magdaleno, S., Setterquist, R., and Conrad, R. (2012) Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta Gen. Subjects, 1820, 940–948.
- 31. Alvarez-Erviti, L., Seow, Y.Q., Yin, H.F., Betts, C. et al. (2011) Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol., 29, 341–U179.
- 32. Ohno, S., Takanashi, M., Sudo, K., Ueda, S. et al. (2013) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther., 21, 185–191.
- 33. Tian, Y., Li, S., Song, J., Ji, T. et al. (2014) A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials, 35, 2383–2390.
- 34. Jang, S.C., Kim, O.Y., Yoon, C.M., Choi, D.S. et al. (2013) Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano, 7, 7698–7710.
- 35. Lee, J., Kim, J., Jeong, M., Lee, H. et al. (2015) Liposome-based engineering of cells to package hydrophobic compounds in membrane vesicles for tumor penetration. Nano Lett., 15, 2938–2944.
- 36. Sun, D., Zhuang, X., Xiang, X., Liu, Y. et al. (2010) A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther., 18, 1606–1614.
- 37. Zhuang, X., Xiang, X., Grizzle, W., Sun, D. et al. (2011) Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther., 19, 1769–1779.
- 38. Tang, K., Zhang, Y., Zhang, H.F., Xu, P.W. et al. (2012) Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun., 3, 1282.
- 39. Silva, A.K.A., Corato, R.D., Pellegrino, T., Chat, S. et al. (2013) Cell-derived vesicles as a bioplatform for the encapsulation of theranostic nanomaterials. Nanoscale, 5, 11374–11384.
- 40. Hu, C.M., Fang, R.H., Luk, B.T., and Zhang, L. (2014) Polymeric nanotherapeutics: clinical development and advances in stealth functionalization strategies. Nanoscale, 6, 65–75.
- 41. Knop, K., Hoogenboom, R., Fischer, D., and Schubert, U.S. (2010) Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl., 49, 6288–6308.
- 42. Hu, C.M., Zhang, L., Aryal, S., Cheung, C. et al. (2011) Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U. S. A., 108, 10980–10985.
- 43. Gao, W., Hu, C.M., Fang, R.H., Luk, B.T. et al. (2013) Surface functionalization of gold nanoparticles with red blood cell membranes. Adv. Mater., 25, 3549–3553.
- 44. Fang, R.H., Hu, C.M., Chen, K.N., Luk, B.T. et al. (2013) Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale, 5, 8884–8888.
- 45. Hu, C.M., Fang, R.H., Copp, J., Luk, B.T. et al. (2013) A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol., 8, 336–340.
- 46. Hu, C.M., Fang, R.H., Luk, B.T., and Zhang, L. (2013) Nanoparticle-detained toxins for safe and effective vaccination. Nat. Nanotechnol., 8, 933–938.
- 47. Parodi, A., Quattrocchi, N., van de Ven, A.L., Chiappini, C. et al. (2013) Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol., 8, 61–68.
- 48. Hu, C.M., Fang, R.H., Wang, K.C., Luk, B.T. et al. (2015) Nanoparticle biointerfacing by platelet membrane cloaking. Nature, 526, 118–121.
- 49. Oldenborg, P.A., Zheleznyak, A., Fang, Y.F., Lagenaur, C.F. et al. (2000) Role of CD47 as a marker of self on red blood cells. Science, 288, 2051–2054.
- 50. Hamdy, S., Molavi, O., Ma, Z., Haddadi, A. et al. (2008) Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine, 26, 5046–5057.
- 51. Fang, R.H., Hu, C.M., Luk, B.T., Gao, W. et al. (2014) Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett., 14, 2181–2188.