
Chapter 10
Single-Cell Analysis with the BioPen*
Irep Gözen1, Gavin Jeffries2, Tatsiana Lobovkina2, Emanuele Celauro3, Mehrnaz Shaali2, Baharan Ali2 Doosti and Aldo Jesorka3
1University of Oslo, Department of Biosciences and Department of Chemistry, Gaustadalléen 21 Forskningsparken 0349, Oslo, Norway
2Chalmers University of Technology, Biophysical Technology Laboratory, Department of Chemistry and Chemical Engineering, Kemivägen 10, 41296 Göteborg, Sweden
3Chalmers University of Technology, Department of Biology and Biological Engineering, SE-412 96 Göteborg, Sweden
* This chapter is dedicated to our friend and colleague Prof. Silviu T. Balaban, †17 November 2016.
10.1 Introduction
In this chapter, an overview of recently published application examples of the multifunctional pipette [1, 2] (BioPen), whose conceptual and technical details have been reviewed in Chapter 9, is presented. The BioPen fills a gap in single-cell research technology, as it addresses specific problems encountered in research on adherent single cells and tissue samples. While a large variety of single-cell techniques exist for cell suspension, ranging from flow cytometry to advanced high-throughput robotic systems, which can reliably address, process, and characterize large numbers of individual cells in rapid succession, the access to cells adhered to surfaces in typical culture environments remains challenging. Major reasons for this discrepancy are on one hand the availability of well-developed commercial fluidic technologies, coupled to established and optimized optical end electric field-based analytical and manipulation instrumentation, and on the other hand the problematic exclusive access to individual cells in confluent cultures, where each single entity is usually surrounded by many others. Open-volume microfluidics has in the past decade opened new possibilities in this area, and the BioPen has been developed in particular with adherent cells in common culture environments and microscopy-based investigations in mind. It combines some benefits of glass needle technologies, such as the pointed shape of the device and the coupling to micromanipulator setups, and of microfluidics, namely, the chip format, and inherent versatility of microflow circuits, and the fabrication options provided by modern clean room equipment and processes.
Typical applications of interest in adherent single-cell studies are selective superfusion, harvesting of cell membrane samples and contents extraction, studies of intracellular processes, single-cell electroporation (SCE), and transport phenomena between cells. Often, multistep processes are desired, where changes in the chemical environment, for example, for dose–response studies, and in temperature need to be applied in sequence. In this context, viability testing of affected cells is also of importance. Figure 10.1 shows at a glance the application space that has been covered by our experimental work with the BioPen. Each icon represents one or more concluded studies, which demonstrates the versatility of the tool in the context of single-cell studies. A separate subchapter is dedicated to each of the applications. A brief outlook projects the use of the BioPen into foreseeable future application areas, which include integration of sensing devices, construction of biointerfaces, and embedded cell content analysis.
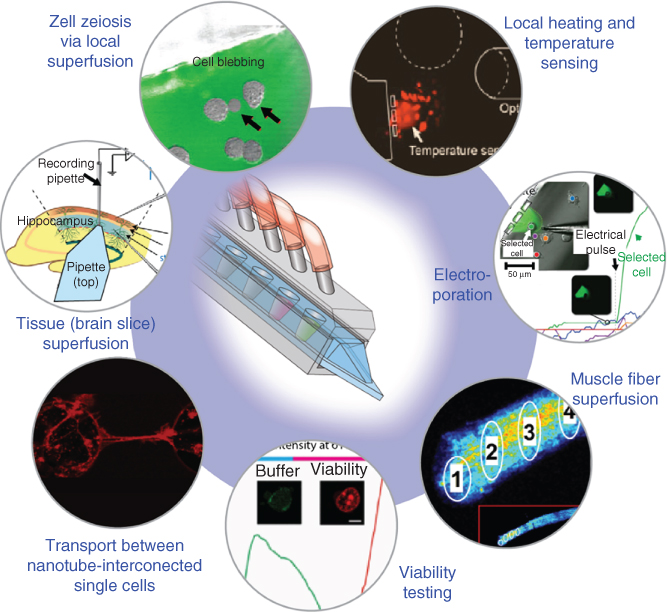
Figure 10.1 Overview over the application of the multifunctional pipette (BioPen) in adherent single-cell and tissue research. Each icon represents an individual application area, in which specific features and benefits of open-volume microfluidic technology can be utilized with significant benefits.
To this date, the focus of our studies resided mainly on analytical applications that rely on superfusion, that is, the exchange of the chemical environment around a single cell or group of cells in their native culture or tissue. Such applications include the study of ion channel and organelle function as well as quantification of intercellular enzymatic processes. In one example, transport processes between nanotube-interconnected cells have been initiated and characterized. Work on tissue samples, specifically brain tissue, served the purpose of evaluating the capabilities of the BioPen, in direct comparison with conventional superfusion chambers and closed channel chip devices, also in the context of improved efficiency of tissue use, which is relevant considering the desirable reduction of animal sacrifices for such studies.
Since its first reported use in 2010 [1], the BioPen has reached a mature state of development and is currently in use by multiple laboratories worldwide. The applications presented in this chapter not only are intended to review the state of the art but should also provide inspiration for researchers interested in adherent single-cell experiments and lead the way to the expansion of the current application space into thus far uncharted territory, as well as to encourage further technological advancement of open-volume devices in order to address the multitude of challenges in single-cell research.
10.2 The Single-Cell Challenge
The cell is the fundamental structured biological unit. Under the influence of various external or internal factors or processes, cells develop into different types and undergo changes between different states. Proliferation and differentiation, originating from a solitary cell, lead to the numerous individual cell types in multicellular organisms, which interact in complex ways. Malfunctions in these regulated development processes can alter both individual cellular function and intercellular interactions, leading to diseases such as cancer. For the understanding of the origins of diseases, and the processes involved in their progression, analysis of single-cell properties and functions are of fundamental importance. Cellular heterogeneity in pathological tissue has also been linked to disease progression. Cancer stem cells, for example, which give rise to the different cell types present in cancer tissue, may survive attempts of chemotherapy and become the cause of tumor relapse. Identifying such unique cells, and developing specific therapeutic approaches to their elimination, has become a strong research focus in the life sciences, which is one of the main driving forces for the development of single-cell analytical methods and technologies.
10.2.1 Single-Cell Analysis
The classical approach in biology is to study a process in a controlled system that mimics, on a smaller scale and in a reproducible way, the local physiology of a real tissue or the pathological phases of a disease. For ethical and practical reasons, this is generally performed using a simple model organism or a population of cultured cells (either on a 2D surface or in a 3D matrix), which are assumed to be mostly homogeneous, in terms of morphology and expression profiles for that specific experimental setup. Nevertheless, even if all the cells share the same genome, actual tissues are composed of many distinct types of cells, with specific transcriptome modulations [3], interacting with each other in complex signaling networks and on a tiny timeframe [4], thus leaving the scientist with a narrow analysis window and making him prone to introduce unwanted biases or oversimplifications. An obvious solution would be to bring cells out of such a noisy background, in a manner that does not affect their individual physiology, but still allows for extensive manipulations to be performed. In conventional sequencing studies, which analyze DNA or RNA samples extracted from large cell populations, the individual character of the cells is no longer represented. Advances in single-cell manipulation, often referred to as next-generation sequencing techniques, have enabled rapid high-resolution genome analysis, which unraveled complexity and revealed details on cellular heterogeneity and its function. These approaches typically involve suspensions of single cells, isolated by disaggregation of primary tissue. Currently, measurements of single-cell suspensions are state of the art and have led to significant insights into transcriptional genomic and epigenomic variation at the level of individual cells. However, a similar state of development is still lacking for adherent cells and intact tissues, which are of particular interest in the context of intercellular interactions, where removing single cells from their environment leads to the loss of much, if not all contextual information.
10.2.2 Technology Overview
During the last decade, rapid progress has been made in the field of suspended single-cell analysis, like the scaling up of the “omics” sequencing throughput that can now deliver impressive amounts of data using few cells as a starting sample [5] or the technical innovations occurred in the most used ways to isolate single cells for further analysis: micromanipulation, fluorescence-activated cell sorting (FACS), and microfluidics.
Micromanipulation is the simplest, but slowest, method of choice to manually pick individual cells or fractions of them. This is accomplished using a standard setup composed of an inverted microscope and a microstepped three-axis system to handle thin glass capillaries, which are connected to a liquid aspirating/dispensing device. This method is very flexible and useful for expanding clonal colonies from a specific cell, to inject picoliters of reagents into the cell itself or to analyze cellular compartments (e.g., patch clamp technique).
To date, FACS is the most common way to sort cells [6] since it allows the automation of the entire process, to reproducibly get well-separated subpopulations of cells from an original mix. This system uses laser excitation to sort a stream of cells passing through a pressure-driven flow cell; by hydrodynamic focusing the cells are lined up and subjected to optical excitation by the laser beam, providing a signal that is used to choose whether to discard that specific cell or to collect it. This is achieved by forcing the aligned cells through small channels and then a nozzle, to create small droplets of carrier solution, which will be then sorted by deviating wanted/unwanted ones by electrically charged plates to collection tubes or plates. Recent advances include some commercially available solutions, like the Fluidigm system that couples a lysis chip after the single-cell isolation processing line. Microfluidics is nowadays the most prolific field of innovation, with many different devices already available. As reviewed in [6], there is a wide choice of techniques, and the most promising to separate individual cells is “droplet in oil-based” isolation, “pneumatic membrane valving,” and “hydrodynamic cell traps.” Anyway, these methods require a consistent tailoring for every specific cell line or the experimental conditions design; this means that a “lab-on-a-chip” needs to be redesigned and reprinted if a carrier solution is not compatible with the device's material or if the cells have a diameter that does not fit the micro-traps. The overall aim of these different approaches is clearly to bring the analysis window toward the single cell, yet they still do not allow to keep the connection between individual contribute of millions of single cells and the resulting complexity of a tissue. Besides, these approaches still exclusively rely on physically dislodging cells from their original growth position, involving an enzymatic or mechanical dissociation step that affects viability, breaking any ongoing cell–cell interaction, and triggering big changes in the cellular homeostasis [7].
It's also important to note that in the in vivo state, cells are constantly maintained in a stable environment by a modulated supply of nutrients and a coupled removal of waste products.
10.2.3 Adherent Cells
In comparison with a three-dimensionally organized living tissue, adherent cells are accepted as a good compromise between loss of physiological architecture and the convenience of a scalable system, amplifiable at will and that can be handled in standardized plastic hardware. Historically this method always consisted in seeding cells on a rigid plastic surface, which could be also coated with specific compounds to help cells to attach, and incubating them in an appropriate atmosphere. However, it has been widely proven [8, 9] that such a hard and flat support does not offer the proper stimuli for the cells to grow and show their native physiology, and in most cases additional controls need to be introduced. Therefore many efforts have been put to introduce a third dimension in cell culture, with the addition of natural or synthetic jelly/soft matrices, thus recreating the complex mix of extracellular molecules and textures that cells are living on and interacting with. These improvements in creating an artificial model organism with only a bunch of cultured cells unfortunately also introduce new problems for the aforementioned techniques. This is mainly because of the dissociation steps required to process the individual cells and technical constrains in arranging an automated pipeline that does not introduce too many manipulations on cells and slows down the whole analysis.
Given this, there is an evident need of a tool that allows investigating processes in a specific cell during a specific treatment, which cannot be done by standard methods that always involve temporal separation between experiment and analysis. The BioPen offers the opportunity to work by proximity and in real-time on single cells, even in more complex scenarios like a multilayered matrix with interspersed cells while providing an enclosed dynamic environment to keep the cell alive and properly responding. This approach is clearly useful to improve research on small model organism, as for a whole mouse embryo, which is extremely sensitive to changes in its local microenvironment, in terms of nutrients availability and removal of toxic metabolic compounds.
10.3 Superfusion Techniques
Superfusion is a solution exchange technique widely used in studies of adherent cells and tissues. It involves running a flow of liquid around or across the surface of a biological sample. This permits exposing the object to active compounds, for example, ligand-gated ion channels in a cell to the ligands required to actuate them. Rapidly exchanging the chemical environment surrounding, a cell creates access to the functions of the ion channel and allows determining binding profiles, effective concentrations, and channel kinetics. Superfusion systems are also essential in order to control the physiological environment in tissue culture experiments, where they maintain slice viability by providing nutrients and oxygen and removing waste. Most commonly used are flow-through perfusion chambers, where fluid is continuously supplied to the chamber and leaves simultaneously. The solutions today are typically stored in racks, some above the superfusion chamber, where the flow of the selected solution is driven by gravity. Cells or tissue in the connected chamber receive the active compounds together with oxygen and nutrients from the passing stream. There is a variety of commercially available devices, such as the Quick StageTM chamber by AutoMate Scientific, others for more specific applications, for example, the field stimulation chamber by Warner Instruments. Recently, new, more versatile perfusion systems became available, which make use of microfluidic technology. One example is the Dynaflow platform, which allows for solution exchange on the millisecond timescale around single cells suspended at the tip of a patch pipette. Other examples include platforms specifically developed for tissue slices. The improvements in terms of reduced exchange time and reagent amounts required for a sample are dramatic.
10.3.1 Hydrodynamic Confinement
However, the use of a flow cell or chamber is indeed associated with a considerable number of limitations. The flow chamber cannot be of arbitrary size and geometry. Its use requires either the culturing of cells inside its boundaries prior to the experiments (e.g., in the Dynaflow device) or the transfer of biological sample from their growth environment. The problem of greatest magnitude is the nonlocalized nature of the superfusion. The entire chamber interior is simultaneously affected. A more gentle way of exchanging the chemical environment would be local superfusion only in the vicinity of a selected cell and ideally in its culture dish. This is, for example, possible by using backfilled, pressurized microinjection capillaries (puffer needles), which are employed to dispense (puff) a very small amount of active solution across the adherent cell. This concept is well applicable to open volumes, allows for approaching individual adherent cells of interest, and permits the simultaneous use of additional probes, such as microelectrodes and patch clamp capillaries, on the same cell. The fragility of glass needles poses severe problems, in particular due to the operation close to the surface. Broken capillary releases its contents instantly, flooding the reservoir and exposing all cells confined in it. Furthermore, while preparation of needles by means of automated pullers is an established process, it remains cumbersome, and the quality of the needles needs to be constantly monitored. The limitation of one unique solution per needle could only be addressed by constructing multibarrel systems, which makes the fabrication, filling, and interfacing much more complicated, or, alternatively, by switching junctions in the supply line, which makes use of the laminarity of microflows but increases the dead volume. The risk of damage due to surface contact remains. Although puffer needles supply only tiny amounts of chemical compounds, the chemicals accumulate over time, where they cause desensitization of ion channels or pH changes. In 2004, this particular aspect was addressed through a coaxial double barrel system with differential pressures [10]. This provided the technical framework for a hydrodynamically confined volume at the tips of the inner needle. It can be viewed as an on-demand, or virtual, flow cell, since its boundaries are not physical, but defined by the borders of coexisting laminar flows. Its properties and conditions for formation have been described in detail in previous chapters. The latest developments in this area, which comprises the transition from glass capillaries to microfluidic devices, fabricated in technology-specific material, such as silicone elastomers, started in 2005 with the publication of the microfluidic probe (MFP) [11], also reviewed in an earlier chapter. Specific distinguishing feature of this, and so far all subsequently presented microfabricated devices, for example, the chemistrode [12], is the more or less rectangular channel profiles, and the side-by-side, rather than coaxial, arrangements of channels. This geometry is largely due to constrains imposed by the 2D fabrication process involved. With this transition from needle-based superfusion to microfluidic devices suitable for adherent cultures, new pathways have been opened for user-friendly, robust devices that life scientists outside the microfluidic laboratory would find acceptable.
10.4 The BioPen
As introduced in the previous chapter, the multifunctional pipette [2], or BioPen (TM), took advantage of the concept of hydrodynamic confinement in fluid delivery. As detailed earlier, the idea of localized fluid delivery by means of hydrodynamically confined flow was carried further, so that the benefits of the glass needles and microfluidic circuitry were emphasized, and many of the disadvantages of earlier superfusion devices could be eradicated or reduced. An elastomeric engineering material makes the tip insensitive to deformation, microfluidic circuitry allows for switching of active solution, and the low flow rate allows the fluids to be driven and adjusted by static pressure. With integrated on-chip wells, which make external solution reservoirs and connecting tubes with large dead volumes obsolete, the BioPen is a consumable device designed for single use that not only avoids cross-contamination but also requires only very small amounts of reagents to operate. The following sections provide examples of how the locally created and confined fluid spheroid is advantageously used to selectively treat individual cells, allowing for variable superfusion sequences, typically in the presence of additional probes or manipulation modes.
10.5 Application Areas
10.5.1 Cell Zeiosis and Ion Channel Activation
In this section, we describe the earliest application of the BioPen. In the experiments, the device was used for the first time to stimulate biological cells, with a special focus on zeiosis (“cell blebbing,” i.e., the disconnection of the plasma membrane from the cytoskeleton) and for demonstrating the control of ion channel activity in selected cells. Cells form bulges or protrusions outward, casually referred to as “blebs,” typically during apoptosis (programmed cell death), cell locomotion, division, or exposure to chemical stress [13, 14]. The blebs originate from the plasma membrane and can be perceived as cellular spherical pockets of bulky morphology, encapsulating the intracellular components. These blebs later may de-attach from the cell and get degraded via phagocytosis, where the cytoplasmic content can be recycled for other cellular processes. The cell blebs preserve the membrane proteins and lipids in their native form and orientation and therefore are widely used in studies associated with the cell membrane and its components. Some protocols focus on collecting the blebs, which are released from the cells as vesicles, for proteomic analysis.
Zeiosis can be artificially induced upon incubation with chemicals such as dichlorodiphenyltrichloroethane (DTT), formaldehyde, tumor necrosis factor (TNF), or staurosporine [13, 15]. Conventional experimental approaches to induce blebbing require time-consuming steps, and chemical treatments often lead to altered yield and functionality of the membrane-associated components. In such experiments, the entire cell culture is affected by the treatment, and it is typically difficult to control the extent of blebbing, and the viability of the cells is therefore strongly affected [14–16].
The BioPen can be used to induce blebs in human embryonic kidney (HEK) cells in a more controlled and rapid way, and in a less-invasive manner with respect to the rest of the cell culture (Figure 10.2a–c). Initially the microfluidic pipette supplies a bleb-inducing mixture of 20 mM DTT and 25 mM formaldehyde, in the arrangement depicted in Figure 10.2a,b. These agents cause the reduction of protein disulfide bonds and block free thiol groups. As a result, the plasma membrane of the cells decouples from the cytoskeleton, leading to zeiosis, over the course of several minutes (Figure 10.2c).
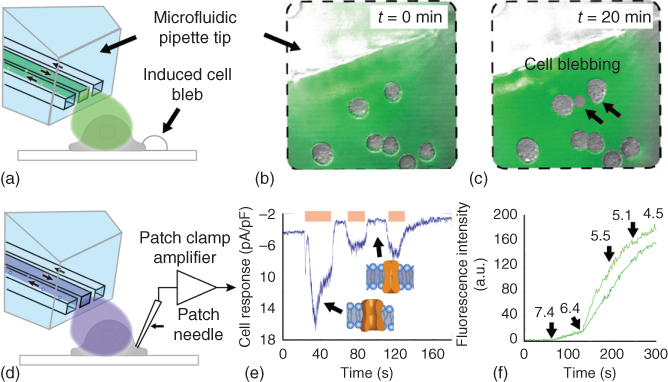
Figure 10.2 Cell zeiosis, single-cell patch clamp, and cell group fluorescence uptake assays supported by the BioPen. Schematically shown are the respective experimental configurations (a,d), the formation of cell blebs after prolonged exposure to selected cells in the hydrodynamically confined volume at the tip of the BioPen (b, preexposure; c, postexposure), the current response from a patch clamp experiment (e), where the states of the ion channel at different time points are depicted in insets, and the fluorescence response due to YO-PRO-1 uptake upon gradual pH changes (f) in the extracellular local environment in a group of CHO cells. The arrows in (c) point to formed cell blebs, in (e) to the current response associated with the ion channel state, and in (f) to the time points at which pH changes were applied.
The second part of the 2010 report focused on activation of ion channels in selected cell groups by local superfusion. Ion channels are pore-forming proteins, located across the entire cell membrane. They mediate the transportation of ions through the hydrophobic cell membrane, which is otherwise impermeable to charged species. Mostly being selective to one or two types of ions, the ion channels can be categorized as “resting” or “gated,” depending on the pore-opening mechanism. While resting channels are always open, for example, K+ channels, the gated channels only open upon stimuli, such as mechanical input, binding of ligands, change in temperature, or in membrane potential. The cation channel transient receptor potential vanilloid 1 (TRPV1), also known as the capsaicin receptor, is an example that is associated with the pain sensation (nociception). TRPV1 can be activated by moderate heat, low pH, capsaicin, or similar compounds [17, 18]. Ion channels play a prominent role in nerve impulse, muscle contraction, T-cell activation, and pancreatic beta-cell insulin release, which make them ideal drug targets, putting them in focus of several research studies [19, 20]. The currently most common instrumental technique to measure real-time electrical currents passing through an ion channel at the surface of a single cell has been “patch clamp.” Introduced in 1976 by Neher and Bert Sakmann [21] in this method, a glass micropipette, filled with an electrolyte, is applied on a cell membrane to make a gigaohm seal. The electrical properties of the cell are then measured by a patch clamp amplifier that is connected to two electrodes, one inserted in micropipette and the other placed in the surrounding solution of the cell [22]. Therefore, the ion channel activity can be recorded when the channel is triggered by a stimulus. Effectively, a combination of this type of electrophysiological measurement and rapid superfusion is required for the determination of ion channel response to a changing extracellular environment. Over the previous decades, several microfluidic approaches have been introduced in conjunction with the conventional patch clamp amplifier to provide a more rapid superfusion [23] (Dynachip Cellectricon, Sweden) [24]. Such techniques can provide fast exposure to solutions (<100 ms, [24]) and have managed to decrease the exposure of ligands to non-clamped cells during recordings, but have not eliminated it completely.
The BioPen has been suggested as a solution to this specific problem (Figure 10.2d–f). As mentioned earlier, TRPV1 can be activated by noxious heat (>43 °C), 0.1 µM capsaicin (EC50) or reduced pH (5.4). Figure 10.2e shows the trace of the patch clamp recorded current from a single CHO cell over time, as a result of repeated activation of TRPV1 by means of the BioPen. The activation has been performed by exposing the cell to a pH 5.4 HCl solution contained in the recirculation zone of the pipette. The durations for which the channels were activated are displayed as bands on top of the current over time graph. The current response, on the order of 10 pA/pF, and its decrease in magnitude upon repeated stimulation are typical for the cell type expressing TRPV1. The insets depict the activated (open) and deactivated (closed) form of channels. In Figure 10.2f, the dependency of intracellular fluorescence intensity of two independent CHO cells as a response to pH stimuli has been plotted. For the experiment, influx of the DNA-binding stain YO-PRO-1 through the pH-activated ion channels and subsequent binding to intracellular DNA were used for quantification of ion channel activity. The time points at which the pH was altered with the BioPen are indicated with arrows. The noticeable decrease in fluorescence intensity at pH 4.5, corresponding to ∼300 s, are results of saturation of the cell's nuclei with the applied stain.
10.5.2 Single Cell Enzymology
As indicated earlier in the patch clamp experiments, the small footprint and the free-standing nature of the BioPen allows it, similar to glass needles, be used in conjunction with an assortment of additional sample interrogation probes. One such probe is a pulled optical fiber, which can be used to direct infrared light to a localized region of interest (ROI), capable of selectively heating a small volume close to its tip [25]. We implemented this combination to generate a novel method to directly probe the influence of temperature on enzymes within individual living mammalian cells, by simultaneously controlling both the thermal and chemical environment. We used the BioPen to generate a hydrodynamically confined zone of a specified reagent, and to switch the solution about the cells within 100 ms. A flat-ended (non-focusing) optical fiber, connected to an IR-B diode laser, was directed toward the targeted cells, to enable control of the thermal environment close to its tip, while minimizing any influence away from the exposed cells. Five temperature point enzyme activity curves were generated for alkaline phosphatase (AP) in both single intact NG108-15 and HEK 293 cells, using the pore-forming agent α-hemolysin, to deliver a substrate into the target cells. Enzyme activity was monitored through the introduction of a pre-fluorescent substrate, fluorescein diphosphate (FDP), into the cell under investigation. AP hydrolyzes FDP, resulting in the fluorescent product fluorescein.
This single cell enzyme activity platform is outlined in Figure 10.3, where panel (a) highlights the probe arrangement. Briefly, the BioPen was positioned directly in front of each chosen cell, forming a patterned flow zone. The optical fiber was then positioned adjacent to the cell of interest, to direct the IR-B heating to influence a region of a few 100 µm. Each targeted cell experienced a well-defined chemical and thermal environment, through modulation of solution and exposure times, in combination with precise calibration of the thermal responses to the IR-B irradiation.
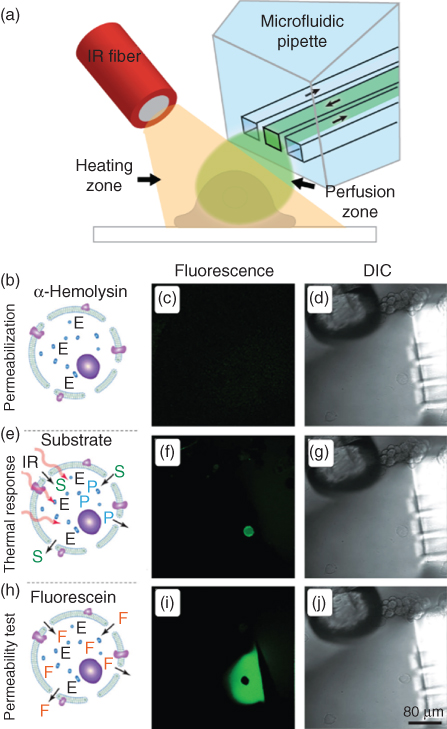
Figure 10.3 Single cell enzymology platform, wherein an adherent cell is held in an isolated chemical and thermal environment. (a) Schematic perspective view of the platform setup consisting of two crucial components: the localized superfusion device (Biopen), and an optical fiber delivering IR-B light, generating a localized heating area. The experimental scheme for measuring single-cell enzymatic activity is shown in panels (b–j). Schematic illustrations (b,e,h) represent the three distinct stages in the experimental procedure. Fluorescent images (c,f,i) correspond to the DIC images of (d,g,j). (b) A cell is mildly permeabilized by exposure to α-hemolysin (200 mg/mL); (e) this permeabilized cell is exposed to the enzyme substrate (S) FDP (150 µM), while simultaneously being heated, through IR exposure to five different temperatures in sequence (22°, 27°, 32°, 37°, and 42°). The enzyme (E) kinetics was extracted by following the formation of the fluorescent product (P). (h) The permeability of the cell was probed through exposure to a 10 µM fluorescein solution. The scale bar in image (j) represents 80 µm and is applicable to all the fluorescent and DIC images.
The targeted cells were exposed to sequences of α-hemolysin (pore-forming agent), extracellular buffer (ECB) buffer (wash solution), FDP (substrate), and fluorescein (permeabilization test, upon conclusion of experiment). The various stages are shown in Figure 10.3b,e,h, where fluorescent images (c,f,i) and differential interference contrast (DIC) images (d,g,j) are displayed for each stage. To probe the enzyme activity, it was first measured at a resting state (room temperature, 22 °C). The temperature was then increased to 27°, 32°, 37°, and 42° in sequence. The dwell time at each temperature was approximately 40 s, and the activity of the enzyme was extracted from the slopes of the fluorescence response curve, as the intracellular product concentration is directly proportional to the rate of product formation. The enzyme activity was calculated from a fluorescence ROI analysis, using a mean value the cell cytosol. The measured enzyme kinetics directly correlates to the cell permeability, placing an emphasis on the need to have a well-controlled and timed exposure to the poration agent and the pre-fluorescent substrate.
In addition to the absolute rate determination, we were able to interrogate the effects of the cellular environment on the thermal response of AP, through utilization of two different cell lines, NG108-15 and HEK 293. The enzymes were found to have a similar response within the temperature range 22–42 °C. However, the efficiency of AP in HEK cells was notably higher at room temperature than in NG cells. The ability to investigate each cell response independently allowed for the construction of single-cell efficiency distribution plots, which highlighted the differences between the two cell lines. NG cells at 22 °C had a tight clustering of efficiencies around 40% of maximum, whereby in HEK cells, this distribution is broader and centers closer to 60%. At 42 °C, the NG cells exhibited a more normal distribution response; however in HEK cells, the distribution changes drastically, centering at close to the same value as NG cells (about 70%), but with a much broader cellular response. The shape of the distributions for both enzymes has a wide range of efficiencies, which would be expected for biological samples, having various states of the cell cycle; however a change of the distribution at 42 °C for HEK cells, and not for NG cells, may indicate that the NG are better able to stabilize the structure of the enzyme at elevated temperatures, shifting its efficiency peak, but not significantly skewing the distribution.
This application example highlights the ability to use multiple positionable probes about an adherent cell sample, in particular for the measurement of single cell enzyme activity, by locally varying the chemical and thermal environment. This approach was validated in situ by probing AP, within individual NG and HEK 293 cells, using α-hemolysin to deliver substrate. The cells required no preincubation with dyes, and the membrane structure is undisturbed until the moment of permeabilization to allow for substrate delivery. Moreover, the enzymes remain in their intracellular environment, and the cells are not detached nor moved during the measurement. This unique combinatorial approach has great potential as a simple analytical tool for investigating the role of thermal modulation on cell signaling, receptor response, and signal pathway regulation at the microscale. It enables the construction of varying condition data sets from a single-cell population, aiding heterogenic analysis, while simultaneously allowing a greater number of single cell.
10.5.3 Local Temperature Adjustment and Measurement in a Single-Cell Environment
The example of the single cell enzymology studies shows that the use of IR-B light locally, in conjunction with the BioPen, opens up a range of new experimental opportunities to investigate single cells. Temperature naturally plays an important role in many biochemical reactions. The biochemical activity of biopolymers has a very narrow temperature range. Thus, any temperature deviation in experimental setups can seriously affect the characteristics and rate of a reaction within a given environment and obscure or misrepresent the actual situation in the in vivo environment.
Accurate temperature determination remains a key issue in many experimental studies within the fields of applied life science. Microfluidics has clearly not only introduced an entire new range of possibilities in studies of single cells but also created a new set of technological challenges that are the result of miniaturization. Availability of temperature measurement instrumentation is one of them. A considerable number of applications that utilize microfluidics rely on temperature measurements. Some examples include various amplification techniques such as on-chip PCR of DNA [26], temperature gradient separation methods [27, 28], and fundamental studies of kinetics and thermodynamics of chemical and biochemical reactions at the microscale [29]. Temperature measurement and control in these applications is routinely performed on flows confined to fluidic channels of micro- to millimeter dimensions, using various solid-state probe concepts. However, in aqueous environments, which are not confined to channels, as in the case of hydrodynamically confined flow, local temperature measurement of a biological sample (e.g., single cells in culture or tissue) during microscopy experiments, is challenging.
Several techniques are available for temperature determination with a variety of physical phenomena that underlines them including thermoelectricity, temperature-dependent change of electrical conductor's resistance and optical (e.g., fluorescence) [30]. Most of these methods lack spatial resolution to be applied to small-scale devices. In Microsystems Technology (MST), a common way to measure temperature is by application of solid-state-sensing devices, thermistors [31], thermocouples [32], and resistance temperature detectors (RTDs) [33, 34]. Other methods that allow to determine the temperature at a microscale include conductometry through a narrow glass capillary [35], or the use of strongly thermally responsive fluorophores [36], or molecular beacons [37]. Thermistors and RTDs, both relying on a large change in resistance with a change in their body temperature, allow for high precision measurement with an excellent stability and repeatability. However, implementation of solid state sensors comes at the expense of relatively complicated fabrication and careful choice of materials. On the other hand, microthermocouples that rely on the formation of electrical junctions at differing temperatures have short response times, simple construction, and thus low cost. The measurements they provide are moderately precise with a requirement for interface junction potential compensation. These sensors can be obtained commercially for the following integration or can be microfabricated into a microfluidic chip/flow chamber. External microthermocouple probes [38] have good positional flexibility, but their implementation increases the required fabrication efforts further.
Alternatively, one can measure the temperature dependence of ion conductivity with microcapillaries to determine the temperature locally in a fluid environment at a microscale. Among the advantages of glass needles is an ability to be precisely positioned to a desired location in the sample using micromanipulators, along with a small footprint and relatively simple preparation, resulting in fast and highly localized measurements [36]. At the same time, these needles are, similar to the problems faced in fluid delivery applications, fragile and require an individual calibration before use due to the small variances in the tip geometry and its tendency to blockage.
The semi-invasive approach of utilizing an optical (fluorescence-based)-sensing principle can be a good substitute for solid-state devices [39]. It is based on the change in fluorescence intensity of the fluorophore that can be related to the variation of temperature via a previously obtained calibration curve [40]. Since many experiments in the life sciences utilize fluorescence or confocal microscopy as a standard technique, an additional intensity measurement of a fluorescence intensity of such sensor does not pose any additional challenge or cost. However, polydimethylsiloxane (PDMS) that is a common material used in microfluidics tends to absorb fluorescent dyes into the channel walls. Thus, inaccurate fluorescence measurements or the degradation of the fluorophore by photobleaching is a severe disadvantage of the technique [41]. This issue can be conveniently addressed by introducing an open-volume microfluidic device to deliver the temperature-sensing dye in solution to the measurement location. In this way a robust, moderately accurate optofluidic semi-contact thermometer compatible with microscopy experiments of biological and artificial cells and tissues can be constructed [42].
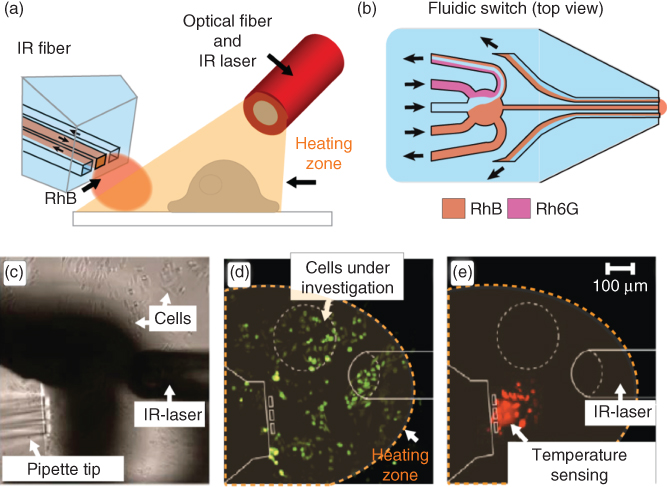
Figure 10.4 Concept of an open-volume optofluidic temperature sensing. (a) Schematic drawing of a typical setup, where the BioPen generates a hydrodynamically confined flow of a thermoresponsive dye solution in the environment, where the temperature is probed, while an optical fiber supplies IR-B light acting as a localized heat source. (b) The content of the flow switching at the tip of MFP, where the solution can be quickly changed between the strongly and weekly temperature-responsive dyes. (c) Bright-field micrograph of the actual experimental setup, showing the pipette and the optical fiber in close vicinity to TRPV-1 overexpressed CHO cells in a YO-PRO-1 DNA stain uptake study. (d) Fluorescence image of cells, measuring the intracellular fluorescence (λexc = 488 nm). (e) Fluorescence image of the same region recorded in the rhodamine channel (λexc = 543 nm). Note that the cells in the probing region are affected by the stain.
In the example shown in Figure 10.4, rhodamine B (RhB) was being employed as a temperature-responsive fluorophore that is recirculated by the MFP [43]. An aqueous solution of RhB is known to exhibit an inverse dependency of its fluorescence emission intensity on temperature [44]. On the other hand, rhodamine 6G (Rh6G) does not exhibit a strong dependence on temperature in the range applicable to biological systems. By using the switching capability of the MFP and alternating solutions of the two dyes, a comparative analysis of the ratio of fluorescence intensity of each rhodamine in response to temperature change can be made (Figure 10.4a,b). This allows eliminating all environmental factors such as pipette position, variations in the heating sources, and settings of microscope and detector (signal gain, confocal pinhole, etc.). Additionally, the setup allows to avoid complex fabrication and integration of the microfluidic device and a sensor. Once again, the resistant-to-accidental-breakage ability to be freely positioned close to the object of interest is a significant advantage that make this thermometer concept attractive. The virtual flow cell at the tip of the device is unaffected by absorption of the dye to the device walls [45], while fast exchange of rhodamine solutions prevents fluorescence photobleaching that is known to seriously affect the quantification of intensity. Dual dye and single wavelength intensity ratio measurement allows for a one-time calibration for a given pair of dye concentration values. Figure 10.4c shows a microscopy image of the experimental setup, comprising the BioPen thermometer and the IR-B laser fiber close to the surface of a culture dish with adherent cells. Figure 10.4d,e shows a YO-PRO-1 uptake experiment, similar to the one described in an earlier section, in which temperature increase activates TRPV1 receptors in CHO cells, and the respective measurement is demonstrated with the BioPen. The open-volume approach allows for safe application of the intrusive rhodamines as thermoprobes in a robust, facile setup, and with great positioning flexibility. It must be noted that rhodamine, due to the required proximity of the confined volume to the surface in order to reside within the focal range of the objective, is actively staining cells (Figure 10.4e). If the cells of interest are to be probed directly, or if studies on cell networks are intended, the effect of the dye uptake on the experimental outcome might need to be considered. However, this typically does not represent a problem in normal adherent cell cultures of relatively high confluency, where separate cell population in the field of view can be used for temperature sensing, provided that the extent of the heating zone covers the cells of interest and the cells in the temperature measurement region equally.
10.5.4 Intercellular Communication
In 2004, Rustom et al. [46] reported for the first time on highly sensitive membrane tubular connections between cultured rat pheochromocytoma (PC12) cells, which were referred to as tunneling nanotubes (TNTs). Since then, these membrane intercellular connections, which are 50–200 nm in diameter and tens of micrometers in length, were recognized as a novel mode of cell-to-cell communication allowing for exchange of molecular cargo, membrane components, and organelles between the mammalian cells [47]. The later studies have also shown that TNTs are involved in cell signaling and provide a transportation route for spreading of the human immunodeficiency virus (HIV), as well as prions [47–50]. Formation and growth of the TNTs is often compared with filopodia, due to the observed structural similarities [51]. To obtain new insights into the growth of membrane protrusions and their ability to form intercellular connections, Zhang et al. [52] designed a study, where the growth and connectivity of cellular protrusions were guided by surface-fabricated glass microlanes. The microlanes were intersected by cytophobic Teflon® AF microgaps of various lengths ranging from 2 to 16 µm. It could be demonstrated that the protrusions originating from two adjacent cells do form tubular connections bridging the Teflon® AF microgap (Figure 10.5a), where the frequency of tubular connections was found to be strongly dependent on the microgap size. The short microgaps (2 and 4 µm long) resulted in higher frequency of formed connections compared with longer microgaps (8 and 16 µm). The authors also determined that these connections vary in diameter. Both nano- (0.5–0.9 µm in diameter) and microsized (>1 µm) connections were formed.

Figure 10.5 Probing of intercellular connections with respect to their ability to transport molecular cargo. (a) Schematic drawing of the investigated network consisting of two cells, interconnected by a micro/nanotube connection spanning a hydrophobic microgap in the hydrophilic substrate surface. (b) Selective exposure of one cell with a solution containing the TRPV8 ion channel agonist menthol and 10 mM Ca2+, using the BioPen, which leads to the internalization and subsequent transport of Ca2+ through the interconnection to the adjacent cell. (c,d) Bright-field and confocal fluorescence image of the two-cell network 30 min after exposure. The false colored (light grey) fluorescence signal indicates transport of the ions through the connection. The BioPen use prevents direct exposure of the second cell by agonist and calcium ions.
The intercellular connections were examined further with respect to their ability to transport molecular cargo, by monitoring the diffusive transport of Ca2+ ions from one cell to another through the joined protrusions. The study required delivery of calcium ions to one of the adherent cells without affecting the adjacent cell. This challenge was met with the BioPen, which was utilized for on-demand stimulation of the selected cell by local superfusion (Figure 10.5b,c). The experiments were performed on HEK-293 cells with overexpressed TRPM8 ion channels. The cells were first incubated with a cell-penetrating fluorescent calcium indicator dye, calcium green-1, which was used for sensing intercellular Ca2+ concentration changes. After the incubation, the selected cell was exposed with a solution of 50 µM menthol (one of the various agonists for the TRPM8 channel), supplemented with 10 mM Ca2+, in order to activate ion channel and deliver calcium ions inside the cell. The BioPen was applied to the selected cell until the fluorescence intensity of the calcium ion indicator fluorescence increased to its maximum. After that, all solutions were switched off and migration of calcium ions from the superfused to the non-exposed cell was monitored by detecting the change of fluorescence intensity of the non-exposed cell.
The analysis of fluorescence intensity of calcium green-1 indicated that the average diffusion time of Ca2+ between the cells was clearly dependent on the diameter of the tubular connections. For the nanoconnections, this time was ∼30 min, while microconnections exhibited approximately 50 times faster diffusion of Ca2+, with an average diffusion time of 35 s. In addition to calcium ion exchange, it could be shown that the formed connections can either be open (tunnel) or closed (junction) in both nano- and microsized cell connections.
The experiments demonstrated the utility of the BioPen on simple cellular networks, where it supported investigation of transport phenomena in intercellular connections. Such a study would be highly challenging to perform by using conventional superfusion techniques. Even the puffer needle as an alternative, which has only a comparatively limited range of exposure, would introduce a considerable risk of contaminating the second cell with diffusing dye during exposure, due to the close proximity of the two cells in this particular arrangement.
10.5.5 Single-Cell Viability Test
Microfluidic devices have led to a rapid development of methods and techniques for single-cell investigations. One important related aspect, the determination of the viability of the addressed cell or cell group, has so far not yet been considered, most likely due to the unavailability of a suitable experimental setup. The establishment of cell viability is critical in most cell studies, for example, drug discovery and toxicology. The intrusive nature of many procedures, foremost due to the chemical compounds used for superfusion at higher concentrations, electroporation for drug delivery, manipulation of ion channels, or mechanical perturbation by patch clamp needles, makes a method for instant viability determination highly desirable. In the single-cell context, of course only the affected cell should be subjected to the test, ideally as final step in the experimental sequence. A viable cell has an intact plasma membrane and should proliferate at least over two generation. Either of these aspects is commonly used for the establishment of cell survival. Confirming the ability to divide is by far the most reliable test, since other tests do not consider the possibility of recovery from temporary damage. However, it requires prolonged sustenance of growth conditions for several hours, and periodic monitoring after the experiment has been concluded. Examination of selected cellular functions is a practical alternative. Those functions include genomic or proteomic assays related to cell stress pathways, mitochondrial activity assays, oxygen consumption measurement, and membrane integrity determination. The latter is typically achieved by assays for exclusion or the uptake and retention of indicator dyes. A classic, simple test is the trypan blue assay, in which cells lacking membrane integrity take up the dye and can be colorimetrically identified. Other dye systems include the use of propidium iodide (PI)/fluorescence diacetate (FDA) in combination, which is widely available and among the various commercial assay kits. FDA is a non-fluorescent molecule that can traverse the cell plasma membrane, while PI is a nucleic acid-binding dye that cannot penetrate the intact cell membrane. Once a cell membrane loses integrity, PI molecules enter the cell and bind to DNA. Adherent cells are typically subjected to both dyes in the culture dish and imaged using microscopy. As this test involves the entire culture and renders it unusable for further experiments, a cell by cell viability test would be useful in many instances where prolonged use of a cell culture is required.
This has been achieved with the BioPen, using double staining with FDA and PI for distinguishing cells with intact and non-intact membranes in both a stand-alone assay, and as an integrated step of a multistage poration/enzyme activity determination experiment. Single target cells (HEK-293, NG108-15, PC12, and CHO) were first temporarily permeabilized by the pore-forming glycoside digitonin (50 µM) for up to 5 s. The plasma membrane was subsequently permitted to heal prior to exposure to the viability assay stains. The poration agent reacts with cholesterol in the plasma membrane and forms pores 8–10 nm in diameter. The degree level of poration was controlled by careful timing of digitonin exposure, depending on cell types and on differences between individual cells. If an exposed cell recovers and remains viable, the membrane recovers, typically, within 1 min. Cell viability was afterward established by simultaneous exposure to FDA/PI solution by means of the BioPen. To establish the possibility of integration of this assay in a cell manipulation experiment, the first poration step was followed by exposure of the cell to 100 µM of the AP substrate FDP for 20 s, after which the assay was instantly performed. Figure 10.6a–c shows schematically the poration/substrate delivery/viability assay sequence. In panels (a–c) the results of the test are displayed. A low intensity fluorescence signal, arising from the conversion of the pre-fluorescent substrate, confirms successful poration and substrate delivery. After a waiting period of 1 min for membrane recovery, application of the viability test compounds lead to bright fluorescence in the FDA channel (488 nm excitation, 505 nm emission), indicating a healthy recovered cell, whereas no signal is detectable in the PI channel (594 nm excitation, 610 nm emission) (Figure 10.6d). In contrast, a nonviable cell produces an increasing fluorescence signal in the PI channel, while the intensity in the FDA channel increases only briefly, as the membrane is unable to recover and hold the internalized FDA (Figure 10.6e).
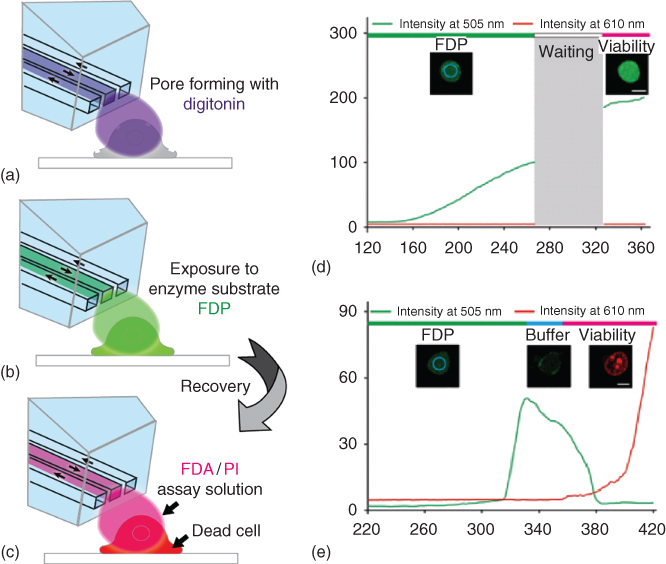
Figure 10.6 Chemical single-cell poration with integrated fluorescein diacetate (FDA)/propidium iodide (PI) membrane integrity assay for viability testing. (a–c) Experimental single cell exposure sequence with digitonin poration step (a), delivery of alkaline phosphatase substrate FDP (b), and subsequent exposure to the viability test reagents FDA/PI (c). Panel (c) depicts the response of a nonviable cell. (d) Fluorescence intensity measurement results for a viable cell. Displayed is the fluorescence intensity development over time for the FDA channel (488/505 nm), and the PI channel (495/610 nm). (e) Time-dependent fluorescence intensity plot for a nonviable cell, using the same conditions as in the measurement shown in (d). A buffer washing step included in the sequence accelerates the removal of FDA from the cell.
10.5.6 Single Muscle Fiber Physiology
The BioPen has to date largely been deployed for analysis of adherent cell cultures. However, the hydrodynamic confinement has specific utility for larger cell systems and is also scalable with respect to channel size and transported volume. Taking advantage of the characteristic localized delivery coupled with high temporal resolution enables targeting of test solutions to small regions of a cell, without any significant leakage to other parts of the same cell, or to the bulk solution. This embodiment was utilized to probe the extent of mitochondrial connectivity within mouse skeletal muscle fibers (Figure 10.7a).

Figure 10.7 Probing the mitochondrial connectivity within a mouse skeletal muscle fiber. (a) Schematic drawing of the general experimental procedure, whereby a skeletal muscle fiber is fluidically exposed at an isolated section, using the BioPen, and probed in multiple regions along the fiber. (b) Image of the BioPen tip positioned close to the end of a muscle fiber during an experiment. The arrows (left to right, an, right to left) indicate the flow directions, which define the delivery zone, with the end of a fiber sitting in front of the central channel. (c) Shows the sites at which changes in the TMRE signal were measured (numbered 1–4) during an experiment. The confocal micrograph is false colored to easier represent the mitochondrial potential. The panel width represents 200 µm. (d) Graph of the response within the regions highlighted in (c), demonstrating that in mitochondria close to the site of FCCP application, the TMRE signal falls to a minimum of 0.3 after 110–120 s. In contrast, at region 2, 40 µm from the site of FCCP application, the TMRE signal actually increases during the period of FCCP application. At regions 2 and 3, distant from the FCCP application site, there is no significant change in the TMRE signal. The black bar indicates the 120 s period of FCCP application.
In mammalian cells, mitochondria exist in a variety of forms from the almost universally assumed ovoid structure, to the long thread-like branching structures found in human fibroblasts [53, 54]. In adult skeletal and cardiac muscle, little is known about the extent to which functional networks of mitochondria exist [55–57]. The densely arranged filaments of actin and myosin compose the majority (80%) of the cell volume, leaving little free space for mitochondria to move around. Within skeletal muscle fibers, the majority of mitochondria are located in pairs on the z-line side of t-tubules [58–60]. However, high-resolution electron microscopy studies have shown additional subsarcolemmal and longitudinal columns of mitochondria are often present. These connections extend transversely and to other adjacent mitochondria, at distances up to 7 µm over several myofibrils [61–63]. In skeletal muscle, for short time periods, mitochondria show no discernible movement when fatigued or subjected to mild osmotic shock [55, 64, 65]. However, the number of mitochondria does increase in response to periods of endurance training, indicating that fission and linkage between mitochondria can occur [66, 67]. It has also been shown that disappearance or fusion of mitochondria occurs if muscles are immobilized, highlighting the adaptable nature of mitochondrial structure [68].
Investigation of mitochondrial coupling in skeletal muscle is a challenging endeavor and has largely been limited by inherent fluidic coupling. It was recently shown that the BioPen can be utilized to deliver FCCP (Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone – an uncoupling agent, which depolarizes mitochondria) to the end or side of single adult mouse skeletal muscle fibers, to test whether changes in mitochondrial membrane potential are transmitted to mitochondria away from the application region. Intracellular changes were monitored using fluorescent probes; mitochondrial membrane potential was monitored using tetramethylrhodamine ethyl ester (TMRE), and cytosolic-free Ca2+ was monitored with the calcium indicator dye fluo-3.
The BioPen was positioned close to a muscle fiber (Figure 10.7b), and a pulse of FCCP (100 µM) was applied to a small area on the fiber (30 µm in diameter). This produced a rapid decrease in the mitochondrial TMRE signal (indicative of depolarization), typically to approximately 35% of its initial value. A buffer solution, Tyrode, was then recirculated over the fiber to wash out FCCP from the region, at which time the TMRE signal partially recovered. The muscle fibers were probed at various distances away from the FCCP application site (Figure 10.7c). The exposed area was monitored by spiking the delivered Tyrode solution with the cell-impermeant dye, sulforhodamine B, known to have no negative influence upon the fibers [64]. Experimental confocal micrographs were recorded at either regular intervals, with the size and position of the area to be exposed first being checked using a sulforhodamine B-spiked Tyrode solution. This spiked solution was washed off the fiber prior to application of FCCP using clean Tyrode solution. Confocal micrographs of large fiber sections were continuously recorded during and after FCCP exposure, to observe the deoplarization response and recovery of the targeted region, in addition to any possible contamination effects. After application FCCP, depolarizing a small region of mitochondria, cytosolic Ca2+ levels monitored using the fluo-3 indicator, were unchanged and fibers contracted in response to electrical stimulation.
This experimental implementation of the BioPen platform successfully demonstrated that minute amounts of a chemical can indeed be applied to a small region of a single cell, without exposing or contaminating the whole. Through application of FCCP, only mitochondria that lie directly in the recirculation volume became depolarized. These mitochondria released the TMRE indicator, which could then be taken up by adjacent mitochondria. The size of the exposure area did not appear to affect the stability, and only mitochondrial subunits in the exposed area were affected.
Mitochondrial depolarization was not detected in mitochondria greater than 50 µm away from the FCCP application region (Figure 10.7d). This localized depolarization was found to occur for single or multiple exposures on the same fiber. Mitochondria within this 50 µm zone may have been subjected to FCCP freely diffusing within in the myoplasm. In addition, FCCP might have flowed through the short (<10 µm) branching structures that have been reported to link nonadjacent mitochondria in mammalian muscle [61–63]. However, FCCP effects through these avenues must be minimal as a clear border was always apparent during the FCCP application. Upon ceasing the FCCP exposure and subsequent washing, there was a limited recovery of the mitochondrial TMRE signal (Figure 10.7d). This is not surprising given that the majority of TMRE released into the myoplasm, from the depolarized mitochondria during the FCCP application, would likely diffuse out of the cell or be uptaken by adjacent mitochondria, a phenomenon observed by brighter TMRE bands appearing directly adjacent to the exposed regions.
Localized delivery of FCCP to either the end or side of a muscle fiber did not result in any major irreversible change in Ca2+ homeostasis. The resting fluo-3 fluorescent intensity (reflecting [Ca2+]i) showed a slight increase during FCCP exposure, but rapidly stabilized and returned the resting value upon termination. Previous studies have shown that bathing muscle fibers in FCCP result in a significant increase in resting Ca2+, which cannot be reversed after removal of FCCP [69, 70]. This Ca2+ imbalance is unlikely to come from depolarized mitochondria, since mitochondrial [Ca2+] is low [71] and that mitochondria in mouse FDB muscle fibers do not accumulate Ca2+ [59]. A possible source of the rise is decreased activity of the Ca2+ ATPase in the sarcoplasmic reticulum membrane due to decreased availability of ATP upon deactivating the mitochondrial potential.
The hydrodynamically confined compounds, delivered by the BioPen, successfully probed the extent of mitochondrial connectivity along skeletal muscle fibers. Our findings, using FCCP as a mitochondrial depolarizing agent, indicate that communication between mitochondria is limited and likely short ranging. It also demonstrated that the limited and reversible effects of targeted FCCP exposure do not affect calcium homeostasis, highlighting the advantage of localized over bulk exposure.
10.5.7 Single-Cell Electroporation
SCE is a technique for the delivery of molecules into individual cells, including cells in in vivo tissue preparations. The electroporation, or electropermeabilization technique, utilizes high external electric fields in order to temporarily generate nanopores in the plasma membrane of exposed cells, leading to increased transmembrane conductivity for molecules and ions. The technique has been widely applied to deliver chemical and biochemical compounds, such as DNA, RNA, antibodies, or dyes to the interior of the cell. Electric pulses at high field strengths induce a potential across the cell membrane, which is proportional to the cell size and the field strength. At a threshold transmembrane potential of 1–2 V, hydrophilic pores are generated in the membrane, either reversibly or irreversibly, depending on the strength of the applied field and the pulse length. If the applied field exceeds a critical value, the membrane pores cannot reseal, and permanent cell damage occurs. For effective drug delivery studies, membrane regeneration is required in order to maintain cell viability. In today's typical electroporation experiments, bulk exposure of large cell populations with a single electrode pair is most common, which suffers from a number of shortcomings. For example, the large electrode distance requires high DC voltages to be applied, which requires additional safety precautions. The field affects individual cells differently over the exposed large area, causing differences in permeabilization in the population. Miniaturization of electroporation technology has shown to be beneficial in this context. Several different concepts for SCE have been developed in the past. Cells can be supplied one by one by a microflow to a channel-integrated microelectrode pair and individually treated, where the short electrode distances make high voltages unnecessary. Other concepts involve trapping of cells in predefined positions on top of electrodes and the confinement of cells in droplets, which are either transported on a surface to a poration electrode pair (digital microfluidics) or flown in oil suspension past the electrodes. These single-cell concepts ensure that each cell is effectively porated and thus achieve a higher efficiency of delivery of material to the cell interior as compared with bulk electroporation.
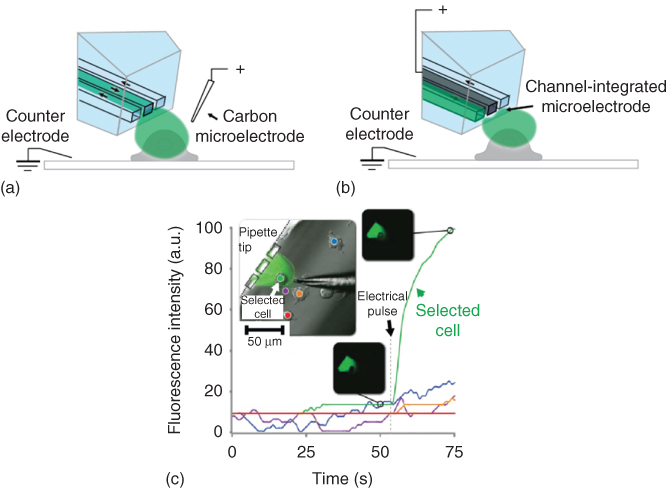
Figure 10.8 Single-cell electroporation in combination with fluidic interrogation. (a) Schematic picture of the superfusion/poration experiment in the presence of an external carbon fiber microelectrode. (b) Representation of the same experiment, utilizing the internalized Field's metal electrode. (c) Fluorescence measurement data of the single-cell electroporation experiment with an external carbon fiber electrode and simultaneous superfusion with 25 mM fluorescein solution in PBS (phosphate-buffered saline). The regions of interest and the corresponding intensity versus time graphs are coded with matching colors on the bright-field inset image. The insets on black background show fluorescence micrographs of the cell of interest at time points before and after electroporation.
However, closed channel or droplet microfluidic devices are not useful for adherent cell cultures. For that reason, the BioPen was combined with either an internal or an external microelectrode, which enabled simultaneous SCE and superfusion for content delivery to cells in their culture environment. A micro carbon fiber served as an external electrode in one configuration (Figure 10.8a), while in the other configuration a metal filling of the center channel of the device was used (Figure 10.8b). The low-melting point Field's metal was pressed into the channel at elevated temperature in a post-fabrication step and interfaced through the device-integrated well. In the experiments designed to demonstrate the utility of the combination of superfusion and poration, the selected adherent cell (NG-108-15 preloaded with fluo-3 calcium indicator) was electroporated by the internal microelectrode while being superfused through the BioPen with 1 mM Ca2+ solution. In experiments with the external electrode, 25 µM fluorescein solution was delivered. Figure 10.8c shows the results of the confocal microscopy-based measurements of fluorescence intensity of ROIs comprising the selected cell, and several reference areas, obtained from the experiments with the external electrode. A bright field image of the arrangement is shown as inset along the graphs, as well as before/after images of the affected cell. The viability of the treated cell was finally tested with the trypan blue assay, also supplied through the BioPen. It was established that the delivered solutions did not penetrate the cell membrane before application of an electroporation pulse of up to 100 V for 1–10 ms. Critical poration parameters were established by gradually increasing voltage and pulse length. Data were collected from 50 cells, with almost 100% electroporation success rate. Poration conditions were adapted to each individual cell. Note that some of the cells showed a slight residual response to external calcium changes for as long as 2.5 min after the electroporation pulse. This passage of calcium, but not trypan blue, suggests that the cells remain viable, but membrane is only slowly restored to its original state.
The addition of electroporation functionality significantly extends the versatility and performance of the BioPen as a multifunctional single-cell manipulation tool, in particular in combination with an integrated electrode. The use of Field's metal is an unconventional approach, which is most likely not suitable for field use. A recently published new fabrication route for BioPen tip structures offers the possibility to directly integrate electrode structures [72].
10.5.8 Local Superfusion of Tissue Slices
In vitro brain slices are a valuable experimental model system for studying the effects of drugs on neurons and astrocytes in an environment, which preserves the native cellular network, that is, the organization in cell layers, processes, and synapses. Brain slice preparations have the distinct advantage that single cells are more easily accessible by probes and imaging techniques, which enables precise pharmacological and physiological studies of properties and functions of neuronal networks. Key aims of such studies are the effect of drugs as well as the improved understanding of communication between the cells.
There are several examples of micro-perfusion devices designed for extracellular delivery of biologically active substances to brain slices. Among them are glass micropipettes, which allow applying active compounds via a laminar flow through their tiny tip, but have limitations in terms of solution confinement and control of flow dynamics. Microfluidic technology provides increasingly powerful instruments for fluidic control in neuropharmacological studies using brain slices. As discussed further earlier, such devices overcome several limitations of conventional submerged slice chambers, leading to better spatiotemporal control over delivery of drugs to specific regions in a slices, while reducing the dead volumes and limiting reagent consumption and providing improved oxygen and nutrient delivery. Specific designs have been reported, for example, chambers, which support interstitial flow for better gas penetration of thicker tissue slices. Focal perfusion is used within conventional chambers for targeting selected slice regions. Microfluidic technology provides solutions for one or more of the persistent problems, yet none of the present devices is ideal for tissue slice applications. Open-volume microfluidics promises new improvements, particularly interesting is the ability of hydrodynamically confined flow devices to provide local exposure in selected areas. The free-standing BioPen with its distinct ability to be rapidly repositioned to regions of interest has the potential to improve the situation further. The BioPen was evaluated in two recent brain slice studies and its capabilities compared with conventional superfusion techniques [73, 74]. Firstly, the administration of the glutamate receptor agonist α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and its antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) to pyramidal cells within rat hippocampal and cortical brain slices was investigated, and the response was recorded by electrophysiological techniques (intracellular recording). This study intended to evaluate the achievable spatial and temporal resolution. Figure 10.9a shows schematically the experimental setup with co-located stimulation and recording electrodes. The graphs in Figure 10.9b show the time dependence of intracellularly recorded AMPA-induced currents after administration of the agonist by the BioPen and in a conventional perfusion chamber. The gain in solution exchange time is nearly 100-fold. The substantial decrease in response time in comparison with conventional superfusion allows drug uptake kinetics to be probed in greater detail. Multiple sites in a slice could be accessed without contamination of the surrounding tissue. Consequently, a larger number of experiments can be performed on a single slice, minimizing the amount of tissue required for a series of experiments, for example, the collection of data for a dose–response curve. This amounts to a cost decrease per experiment and can lead to reduced animal use in such studies.
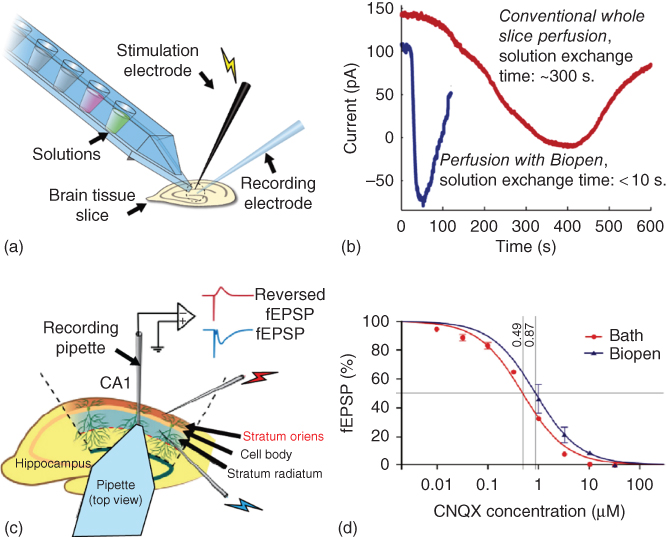
Figure 10.9 The use of the BioPen for local administration of biologically active substances to cells in the brain slices in vitro. (a) Experimental setup (schematic) for electrophysiological measurements of brain tissue (intracellular recording) in combination with BioPen superfusion with CNQX for blocking electrically stimulated excitatory postsynaptic potential (fEPSP) in the CA1 region of a hippocampal brain slice, or with AMPA to induce response currents from cells in layers V and VI of the medial prefrontal cortex (mPFC). (b) Comparison of solution exchange times (development of the AMPA-induced current over time) upon local administration of AMPA by the BioPen to intracellularly recorded pyramidal cells of the rat mPFC. (c) Schematic drawing of the experimental setup for fEPSP measurements in rat hippocampus slices with a three-electrode setup and simultaneous perfusion with the BioPen. Stimulating electrode 1 (S1) is located in stratum radiatum and stimulating electrode 2 (S2) in stratum oriens. A recording pipette is located in stratum radiatum. Different concentrations of the AMPA receptor antagonist CNQX are applied to the tissue surface by means of the BioPen (and for comparison by conventional perfusion). The fEPSP in stratum radiatum induced by S1 is directly recorded by the recording pipette, and the field potential in stratum oriens induced by S2 is also recorded by the (same) recording pipette but in reversed form (reversed fEPSP). (d) Concentration–response curves for bath perfusion and BioPen. Each data point represents the percentage of the fEPSP slope (with respect to the value measured for drug-free buffer) after application of a certain concentration of CNQX, in the range between 10 nM and 30 µM in equal logarithmic steps (data from 8 bath and 4 BioPen experiments, error bars; standard error of mean). The determined IC50 values (0.49 and 0.87 µM, respectively) are also shown.
Secondly, a more detailed study on the performance of the BioPen in drug delivery for dose–response measurements was subsequently commenced. The device was applied to selectively perfuse one dendritic layer in the CA1 region of hippocampus with CNQX, while not affecting the other layers in this region (Figure 10.9c). The changes in field excitatory postsynaptic potential (fEPSP) in response to the applied dose were measured by intracellular recording. In electrophysiological experiments, independent activation of axons in stratum oriens and stratum radiatum (Figure 10.9c) is often used to trigger a pair of spatially separated synaptic inputs to the pyramidal cell population. In the experiments, two stimulating electrodes (S1 and S2) were placed in stratum oriens and stratum radiatum, respectively, providing activation of axon collaterals projecting from the CA3 region to CA1, which form synapses with both basal dendrites in stratum oriens and apical dendrites in stratum radiatum of the pyramidal cells in the CA1 region. A recording pipette was inserted into stratum radiatum (Figure 10.9c) at a distance of 200–500 µm from S1, for recording of the field fEPSPs.
The concentration–response measurements showed that the ability of the BioPen to control local drug concentration is comparable with that of whole slice perfusion, while in comparison the required amounts of active compounds can be reduced by several orders of magnitude (Figure 10.9d). Four concentrations of CNQX were administered sequentially via solution switching. Each new (higher) concentration was applied after a steady state was established, that is, when the previous dose had resulted in a maximum effect on the fEPSP. The half maximal inhibitory concentration (IC50) values of fEPSP inhibition by CNQX delivered by either perfusion method were determined after fitting the data with sigmoid curves, yielding values of 0.49 and 0.87 µM (p < 0.001), respectively, implying a ratio of 56%. For curve fitting, a reverse Hill equation with slope constrained to 1, y = 100/(1 + (c/IC50) was used. The graphs have sigmoidal shape when plotted semi-logarithmically. These findings demonstrate that the efficiency of the drug delivery by the BioPen is just slightly lower than with bath perfusion. However, a ∼ 100 times smaller amount is required. The experimental setup with the device also revealed that the accessible space for other probing and number of benefits compared with conventional perfusion. It decreases the solution exchange time dramatically and has the ability to deliver drugs selectively to regions of interest, such as the hippocampus CA1 region. The degree of selectivity was found to be critically related to the hydrodynamically confined flow, which is the key feature of the device.
10.6 Future Technology
The application examples show conclusively that the BioPen is a versatile multifunctional research tool that facilitates and enhances a variety of experimental studies on single adherent cells and can even enable new experimental concepts. It is a medium throughput device, which can certainly not compete with the high-throughput technologies used for suspended cells, but compares in many aspects very favorably with the techniques currently established for adherent cells. The full potential of the device, however, has not yet been reached. To date, only the superfusion, that is, solution delivery, angle of the BioPen has been explored. Apart from incremental technological improvements, such as reduction of the channel size to the physical limit, new materials and fabrication processes, which would further reduce the footprint of the device tip, or a larger number of switchable solutions, which can further facilitate the use and eliminate weaknesses, several pathways for future development of the device can be envisioned. Most interesting are two aspects: the capture and on-chip processing of compounds released by the superfused cells in response to the stimuli and the integration of sensors and assays. These developments are challenging, in particular the isolation and analysis of cellular release, given that only minute amounts of material can be expected, and that the intake is further diluted at least 20-fold due to the unavoidable inflow of external bath solution. Studies are required to determine the loss of material due to adsorption to the channel walls, and effective means of capturing and concentrating the molecules of interest inside the chip need to be explored. A good initial approach might be lysis of whole cells in the confined volume [75] and aspiration of the products into a dedicated on-chip collection well (Figure 10.10a).
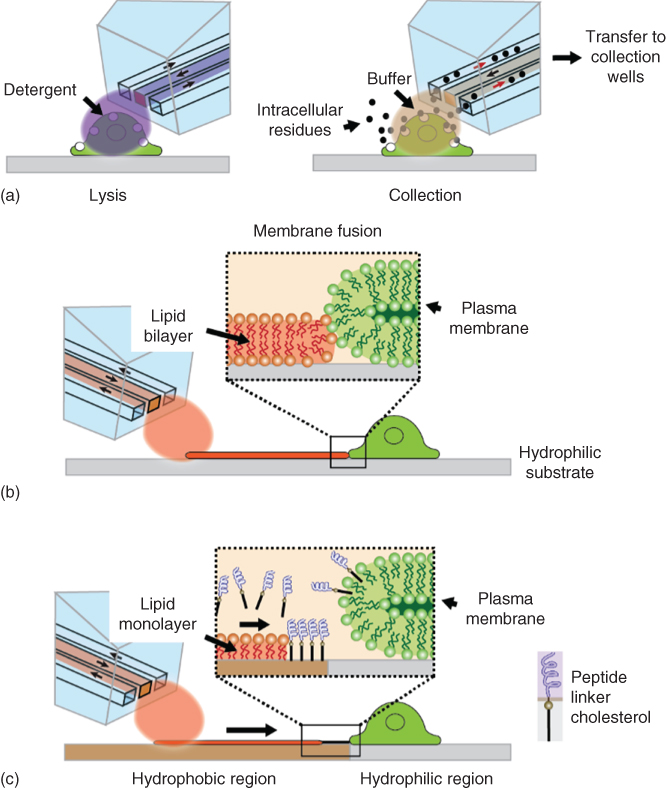
Figure 10.10 Examples of envisioned future developments and applications of the BioPen. (a) Cell lysis and collection of the lysis products via the inflow channels of the device. (b) Interfacing of cells by a spontaneously spreading lipid bilayer is generated and sustained by deposition and fusion of small unilamellar vesicles through the BioPen. (c) Transfer of finely controlled amounts of active compounds from a hydrophobic surface patch by a self-spreading surfactant monolayer also supplied through the device.
This would provide sufficient material for mass spectrometric analysis of some of the cell content. Immediately feasible is the incorporation of on-chip electrodes or analytical sensing devices, for example, surface acoustic wave or electrochemical sensors, which can be fabricated on flat surfaces by means of clean room processes and bonded to the bottom of the device. Interfacing and readout is merely an engineering problem, while cost of production for the sensor-enhanced consumable chip is most likely the limiting factor. Other future developments might include the exploitation of the lab on a membrane concept, that is, the rapid prototyping of patterned molecular films. Spreading phospholipid layers, deposited by the BioPen, could potentially be used to interface to adherent cells via membrane fusion, allowing for transfer of membrane components under exploitation of the benefits of 2D transport (Figure 10.10b). Much finer control over the amounts of delivered material can be expected. A related concept is the release of surface-adhered active material from the surface by displacement (Figure 10.10c), which has been demonstrated a few years ago utilizing the spontaneous wetting of a polymer surface by a lipid monolayer for the release of cholesterol-anchored DNA.
In summary, the BioPen has clearly brought the hydrodynamically flow confinement concept closer to real-life single-cell applications. Since the problems of addressing adherent cells in experimental studies are persisting, new technological solutions are indeed urgently required. The device has high potential beyond the demonstrated superfusion applications and can become a user-friendly, versatile, and affordable µTAS (micro total analysis system) platform for the investigation of adherent cells in the not-too-distant future.
Acknowledgments
We acknowledge support from the Swedish Strategic Research Foundation (SSF) and the European Union Horizon 2020 program and thank Dr. Alar Ainla for providing some of the graphic artwork.
References
- 1 Ainla, A. et al. (2010) A microfluidic pipette for single-cell pharmacology. Anal. Chem., 82 (11), 4529–4536.
- 2 Ainla, A. et al. (2012) A multifunctional pipette. Lab Chip, 12 (7), 1255–1261.
- 3 Dueck, H. et al. (2015) Deep sequencing reveals cell-type-specific patterns of single-cell transcriptome variation. Genome Biol., 16, 122.
- 4 Greenwald, I. and Rubin, G.M. (1992) Making a difference: the role of cell–cell interactions in establishing separate identities for equivalent cells. Cell, 68 (2), 271–281.
- 5 Hodne, K. and Weltzien, F.A. (2015) Single-cell isolation and gene analysis: pitfalls and possibilities. Int. J. Mol. Sci., 16 (11), 26832–26849.
- 6 Gross, A. et al. (2015) Technologies for single-cell isolation. Int. J. Mol. Sci., 16 (8), 16897–16919.
- 7 Huang, H.L. et al. (2010) Trypsin-induced proteome alteration during cell subculture in mammalian cells. J. Biomed. Sci., 17, 36.
- 8 Antoni, D. et al. (2015) Three-dimensional cell culture: a breakthrough in vivo. Int. J. Mol. Sci., 16 (3), 5517–5527.
- 9 Ravi, M. et al. (2015) 3D cell culture systems: advantages and applications. J. Cell. Physiol., 230 (1), 16–26.
- 10 Feinerman, O. and Moses, E. (2003) A picoliter “fountain-pen” using co-axial dual pipettes. J. Neurosci. Methods, 127 (1), 75–84.
- 11 Juncker, D., Schmid, H., and Delamarche, E. (2005) Multipurpose microfluidic probe. Nat. Mater., 4 (8), 622–628.
- 12 Chen, D. et al. (2008) The chemistrode: a droplet-based microfluidic device for stimulation and recording with high temporal, spatial, and chemical resolution. Proc. Natl. Acad. Sci. U.S.A., 105 (44), 16843–16848.
- 13 Laster, S.M. and Mackenzie, J.M. (1996) Bleb formation and F-actin distribution during mitosis and tumor necrosis factor-induced apoptosis. Microsc. Res. Tech., 34 (3), 272–280.
- 14 Bauer, B., Davidson, M., and Orwar, O. (2006) Direct reconstitution of plasma membrane lipids and proteins in nanotube-vesicle networks. Langmuir, 22 (22), 9329–9332.
- 15 Nauta, A.J. et al. (2002) Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur. J. Immunol., 32 (6), 1726–1736.
- 16 Baumann, N.A. et al. (2000) Cell surface display and intracellular trafficking of free glycosylphosphatidylinositols in mammalian cells. J. Biol. Chem., 275 (10), 7378–7389.
- 17 Levine, J.D. and Alessandri-Haber, N. (2007) TRP channels: targets for the relief of pain. Biochim. Biophys. Acta, Mol. Basis Dis., 1772 (8), 989–1003.
- 18 Caterina, M.J. et al. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature, 389 (6653), 816–824.
- 19 Jansson, E.T. et al. (2013) Effect of cholesterol depletion on the pore dilation of TRPV1. Mol. Pain, 9, 1.
- 20 Trkulja, C.L. et al. (2014) Probing structure and function of ion channels using limited proteolysis and microfluidics. J. Am. Chem. Soc., 136 (42), 14875–14882.
- 21 Neher, E. and Sakmann, B. (1976) Agonist-induced discrete conductance changes in frog muscle. Biophys. J., 16 (2), A154.
- 22 Ihenetu, K. et al. (2003) Pharmacological characterisation of cannabinoid receptors inhibiting interleukin 2 release from human peripheral blood mononuclear cells. Eur. J. Pharmacol., 464 (2–3), 207–215.
- 23 Colquhoun, D., Jonas, P., and Sakmann, B. (1992) Glutamate receptor channel activation following fast agonist application to patches from rat hippocampus CA1 and CA3 pyramidal cells. J. Physiol. (London), 446, P183.
- 24 Sinclair, J. et al. (2002) A cell-based bar code reader for high-throughput screening of ion channel-ligand interactions. Anal. Chem., 74 (24), 6133–6138.
- 25 Xu, S.J. et al. (2015) A heating-superfusion platform technology for the investigation of protein function in single cells. Anal. Chem., 87 (1), 381–387.
- 26 Kopp, M.U., de Mello, A.J., and Manz, A. (1998) Chemical amplification: continuous-flow PCR on a chip. Science, 280 (5366), 1046–1048.
- 27 Vigolo, D. et al. (2010) Thermophoresis: microfluidics characterization and separation. Soft Matter, 6 (15), 3489–3493.
- 28 Bargiel, S., Gorecka-Drzazga, A., and Dziuban, J.A. (2004) A micromachined system for the separation of molecules using thermal field-flow fractionation method. Sens. Actuators, A, 110 (1–3), 328–335.
- 29 Chabala, L.D. et al. (1985) A microscope stage temperature controller for the study of whole-cell or single-channel currents. Pflugers Arch., 404 (4), 374–377.
- 30 Childs, P.R.N., Greenwood, J.R., and Long, C.A. (2000) Review of temperature measurement. Rev. Sci. Instrum., 71 (8), 2959–2978.
- 31 Beakley, W.R. (1951) The design of thermistor thermometers with linear calibration. J. Sci. Instrum., 28 (6), 176–179.
- 32 Allen, P.B. et al. (2003) Selective electroless and electrolytic deposition of metal for applications in microfluidics: fabrication of a microthermocouple. Anal. Chem., 75 (7), 1578–1583.
- 33 Tsutsui, M., Kawai, T., and Taniguchi, M. (2012) Unsymmetrical hot electron heating in quasi-ballistic nanocontacts. Sci. Rep., 2, 217.
- 34 Barber, C.R. (1950) Platinum resistance thermometers of small dimensions. J. Sci. Instrum., 27 (2), 47–49.
- 35 Yao, J., Liu, B.Y., and Qin, F. (2009) Rapid temperature jump by infrared diode laser irradiation for patch-clamp studies. Biophys. J., 96 (9), 3611–3619.
- 36 Zeeb, V., Suzuki, M., and Ishiwata, S. (2004) A novel method of thermal activation and temperature measurement in the microscopic region around single living cells. J. Neurosci. Methods, 139 (1), 69–77.
- 37 Barilero, T. et al. (2009) Fluorescent thermometers for dual-emission-wavelength measurements: molecular engineering and application to thermal imaging in a microsystem. Anal. Chem., 81 (19), 7988–8000.
- 38 Markstrom, M. et al. (2007) Dynamic microcompartmentalization of giant unilamellar vesicles by sol gel transition and temperature induced shrinking/swelling of poly(N-isopropyl acrylamide). Soft Matter, 3 (5), 587–595.
- 39 Ross, D., Gaitan, M., and Locascio, L.E. (2001) Temperature measurement in microfluidic systems using a temperature-dependent fluorescent dye. Anal. Chem., 73 (17), 4117–4123.
- 40 Shah, J.J., Gaitan, M., and Geist, J. (2009) Generalized temperature measurement equations for rhodamine B dye solution and its application to microfluidics. Anal. Chem., 81 (19), 8260–8263.
- 41 Toepke, M.W. and Beebe, D.J. (2006) PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip, 6 (12), 1484–1486.
- 42 Wegrzyn, I. et al. (2013) An optofluidic temperature probe. Sensors, 13 (4), 4289–4302.
- 43 Gui, L. and Ren, C.L. (2008) Temperature measurement in microfluidic chips using photobleaching of a fluorescent thin film. Appl. Phys. Lett., 92 (2), 024102.
- 44 Sakakibara, J. and Adrian, R.J. (1999) Whole field measurement of temperature in water using two-color laser induced fluorescence (vol 26, pg 7, 1999). Exp. Fluids, 27 (1), U1.
- 45 Robinson, T. et al. (2009) Removal of background signals from fluorescence thermometry measurements in PDMS microchannels using fluorescence lifetime imaging. Lab Chip, 9 (23), 3437–3441.
- 46 Rustom, A. et al. (2004) Nanotubular highways for intercellular organelle transport. Science, 303 (5660), 1007–1010.
- 47 Gerdes, H.H., Rustom, A., and Wang, X. (2013) Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev., 130 (6–8), 381–387.
- 48 Sowinski, S. et al. (2008) Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol., 10 (2), 211–219.
- 49 Gurke, S., Barroso, J.F.V., and Gerdes, H.H. (2008) The art of cellular communication: tunneling nanotubes bridge the divide. Histochem. Cell Biol., 129 (5), 539–550.
- 50 Gurke, S. et al. (2008) Tunneling nanotube (TNT)-like structures facilitate a constitutive, actomyosin-dependent exchange of endocytic organelles between normal rat kidney cells. Exp. Cell. Res., 314 (20), 3669–3683.
- 51 Sherer, N.M. et al. (2007) Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol., 9 (3), 310–315.
- 52 Zhang, H.J. et al. (2015) Cellular communication via directed protrusion growth: critical length-scales and membrane morphology. Nano Commun. Networks, 6 (4), 178–182.
- 53 Collins, T.J. et al. (2002) Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J., 21 (7), 1616–1627.
- 54 Amchenkova, A.A. et al. (1988) Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol., 107 (2), 481–495.
- 55 Boncompagni, S. et al. (2009) Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell, 20 (3), 1058–1067.
- 56 Dahl, R. et al. (2014) Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol., 213 (1), 145–155. doi: 10.1111/apha.12289
- 57 Kirkwood, S.P., Munn, E.A., and Brooks, G.A. (1986) Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol., 251 (3 Pt 1), C395–C402.
- 58 Bruton, J. et al. (2003) Mitochondrial and myoplasmic [Ca2+] in single fibres from mouse limb muscles during repeated tetanic contractions. J. Physiol., 551, 179–190.
- 59 Lännergren, J., Westerblad, H., and Bruton, J.D. (2001) Changes in mitochondrial Ca2+ detected with Rhod-2 in single frog and mouse skeletal muscle fibres during and after repeated tetanic contractions. J. Muscle Res. Cell Motil., 22 (3), 265–275.
- 60 Reipert, S. et al. (1999) Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp. Cell. Res., 252 (2), 479–491.
- 61 Ogata, T. and Yamasaki, Y. (1985) Scanning electron-microscopic studies on the three-dimensional structure of mitochondria in the mammalian red, white and intermediate muscle fibers. Cell Tissue Res., 241 (2), 251–256.
- 62 Ogata, T. and Yamasaki, Y. (1997) Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec., 248 (2), 214–223.
- 63 Picard, M., White, K., and Turnbull, D.M. (2013) Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J. Appl. Physiol., 114 (2), 161–171.
- 64 Lännergren, J., Bruton, J.D., and Westerblad, H. (1999) Vacuole formation in fatigued single muscle fibres from frog and mouse. J. Muscle Res. Cell Motil., 20 (1), 19–32.
- 65 Lännergren, J., Bruton, J.D., and Westerblad, H. (2000) Vacuole formation in fatigued skeletal muscle fibres from frog and mouse: effects of extracellular lactate. J. Physiol., 526, 597–611.
- 66 Kirkwood, S.P., Packer, L., and Brooks, G.A. (1987) Effects of endurance training on a mitochondrial reticulum in limb skeletal muscle. Arch. Biochem. Biophys., 255 (1), 80–88.
- 67 Tarnopolsky, M.A. et al. (2007) Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol., 292 (3), R1271–R1278.
- 68 Staron, R.S. et al. (1984) Human skeletal muscle fiber type adaptability to various workloads. J. Histochem. Cytochem., 32 (2), 146–152.
- 69 Caputo, C. and Bolanos, P. (2008) Effect of mitochondria poisoning by FCCP on Ca2+ signaling in mouse skeletal muscle fibers. Pflugers Arch., 455 (4), 733–743.
- 70 Zima, A.V. et al. (2013) Effects of mitochondrial uncoupling on Ca2+ signaling during excitation-contraction coupling in atrial myocytes. Am. J. Physiol., 304 (7), H983–H993.
- 71 Rizzuto, R. et al. (1995) Photoprotein-mediated measurement of calcium ion concentration in mitochondria of living cells. Methods Enzymol., 260, 417–428.
- 72 Kim, A. et al. (2017) SU-8 free-standing microfluidic probes. Biomicrofluidics, 11 (1), 014112.
- 73 Ahemaiti, A. et al. (2013) A multifunctional pipette for localized drug administration to brain slices. J. Neurosci. Methods, 219 (2), 292–296.
- 74 Ahemaiti, A. et al. (2015) Spatial characterization of a multifunctional pipette for drug delivery in hippocampal brain slices. J. Neurosci. Methods, 241, 132–136.
- 75 Sarkar, A. et al. (2014) Microfluidic probe for single-cell analysis in adherent tissue culture. Nat. Commun., 5, 3421.