
Chapter 15
FluidFM Applications in Single-Cell Biology
Orane Guillaume-Gentil1, Maximilian Mittelviefhaus1, Livie Dorwling-Carter2, Tomaso Zambelli2 and Julia A. Vorholt1
1ETH Zurich, Institute of Microbiology, Department of Biology, Vladimir-Prelog-Weg 4, CH 8093 Zurich, Switzerland
2ETH Zurich, Laboratory of Biosensors and Bioelectronics, Institute for Biomedical Engineering, Gloriastr. 35, CH 8092 Zurich, Switzerland
15.1 Introduction
Single-cell studies have the potential to substantially impact biology and medicine, providing greater details into cellular functions, cell–cell interactions, and cell responses to exogenous stimuli. However, innovative methods and technologies are required to enable controlled perturbation, isolation, and analyses with single-cell resolution. The fluidic force microscopy (FluidFM) technology provides a versatile force-sensitive nanopipette that can be operated in physiological environments (see previous chapter). The size and flexible design of the microchanneled cantilevers, in particular of their aperture, and the volumes it can handle, in the femto- to picoliter range, match the scale of individual eukaryotic and prokaryotic cells and their microenvironments (Table 15.1). The force range and sensitivity of the FluidFM is well suited both for gentle, nondestructive manipulations and for the measurements of mechanical properties. In addition, the technology allows for real-time monitoring by optical microscopy and for the study of live single cells under preservation of the cellular and physiological context. The FluidFM technology holds thus a great potential for single-cell biology, which is reflected by the panel of applications developed in the recent years. This chapter provides a synthesis of these complementary applications, starting with the spatial cell manipulation (Section 15.3) and controlled fluid delivery (Section 15.4), which allow for selective and well-defined cell perturbations, before describing single-cell analysis applications that comprise quantitative measurements of cell mechanical (Section 15.5) and electrical (Section 15.6) properties and the sampling and analysis of intracellular soluble molecules (Section 15.7).
Table 15.1 Comparative dimensions of cells and FluidFM cantilevers
| Cells | FluidFM | |||||
| HeLa (mammalian) | S. cerevisiae (yeast) | E. coli (bacterium) | Si3N4 | SU-8 | ||
| Size (diameter, µm) | 10–20 | 5–10 | 1–5 | Channel height (µm) | 0.5–1 | 3–22 |
| Volume (fL) | 1000–5000 | 50–500 | 0.1–5 | Aperture size (µm) | 2–8 (tipless) 0.01–2000 (pyramidal) | 4–10 |
| Young's modulus (Pa) | 104 | 108 | 107 | Stiffness (N/m) | 0.1–3 | 0.5–80 |
15.2 Nondestructive Cell Manipulations
FluidFM combines atomic force microscopy (AFM) with microfluidics [1]. A core feature of the technology is the hollow cantilever probe, which enables pressure-mediated handling of liquids and sensitive force detection simultaneously. In addition, the force monitoring is coupled to piezo-driven cantilever movement through a feedback loop. This coupling between the force encountered by the probe and its vertical displacement provides a powerful mean to ensure nondestructive interactions when physically contacting cells. Another critical issue during cell manipulation consists of biofouling, that is, the nonspecific adsorption of biomacromolecules to the probe surface. Upon physical contact with cells, fouling can damage cells, for example, through surface denaturation of membrane proteins, and it hampers cell release after manipulation. In addition, the excessive adsorption of biological material within the microchannel or at the probe aperture can lead to clogging or to the loss of material flown through the probe. Protein and cell adhesion to the cantilever surface thus limits the practical use of the microchanneled probes. In order to ensure nondestructive cell handling and allow for serial manipulations, the FluidFM probes are therefore functionalized with an antifouling coating prior to experimentation. The hydrophilic copolymer poly(l-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) has been most extensively applied to confer antifouling properties to the FluidFM probes. The polycationic PLL (poly(l-lysine)) backbone adsorbs onto negatively charged oxidized surfaces with the PEG chains extending to the aqueous environment into a brushlike configuration, forming an excellent protein-resistant interface. Coating of the FluidFM probes with the polymer can be achieved by plasma activation followed by immersion in an aqueous solution of the polymer [2–6]. The microchannel can be coated by flowing the polymer solution through the channel during immersion. The coating can be further improved by increasing the temperature during the coating process, which yields a higher polymer density on the surface [7]. Another coating approach for preventing protein adsorption onto FluidFM probes makes use of Sigmacote, a chlorinated organopolysiloxane that forms hydrophobic, microscopically thin films. Efficient coatings can be prepared upon dipping of the probe in a Sigmacote solution for a few minutes before rinsing with water, whereby the microchannel can be modified by flowing Sigmacote followed by water through it. Alternatively, the Sigmacote film can be applied by vapor phase coating, incubating the probe in the presence of the siliconizing agent in a desiccator under vacuum [8]. Microchannel can also be coated under these conditions by saturating the air with a syringe before flowing it through the probe, simultaneously, and the coating can then be stabilized by heat treatment [9].
15.3 Spatial Cell Manipulation
Within organs or tissues, mammalian cells are embedded into a highly structured microenvironment, which has a crucial influence on the function of individual cells and ultimately impacts the functional coherence of the entire tissue. Microorganisms not only rarely exist as solitary organisms in nature but also assemble into complex communities in which the interactions of the individual members govern the microbial life. Controlling the spatial organization of cells is therefore of critical importance to investigate cell physiology and the interactions between cells or with exogenous substances. Spatial cell manipulation also allows to isolate selected individual cells for downstream analysis or clonal expansion or to designate specific cell localization in cell-based sensors. Numerous approaches have been developed to control the spatial localization of cells. By creating cell adhesive patterns within non-fouling backgrounds, substrate micropatterning enables to assemble artificial cellular networks or single-cell arrays, arranging large numbers of cells into defined geometrical patterns. With the avenue of 3D printing technologies, cell printing has recently gained a tremendous interest, enabling in particular the creation of 3D tissue mimics. Fluidic technologies such as FACS (fluorescence-activated cell sorting) and microfluidic devices have been applied to efficiently sort and isolate suspended cells, providing extremely high throughput and offering different means for simultaneous cell analysis or perturbation. Lastly, diverse approaches (e.g., optical or magnetic tweezers, glass micropipettes) have been developed to directly trap and displace individual cells. These generally present relatively low throughput, but with the highest precision. The FluidFM provides various strategies for substrate micropatterning (Section 15.3.1), single-cell displacement (Section 15.3.2), and cell printing (Section 15.3.3).
15.3.1 Substrate Micropatterning
As described in Chapter 14, Section 4.1, FluidFM allows to perform lithographic patterning in liquid through the pressure-mediated release of a compound solution preloaded in the probe. Since the probe microchannel can be filled with virtually any solution, not only nanoparticles but also proteins or polymers can be deposited on surfaces with micrometric precision. Two different strategies have been developed that enable substrate micropatterning, with the advantages of offering unrestricted geometries and being achievable under physiological conditions, even in the presence of cells. In a pilot study, the FluidFM was used to generate well-defined streptavidin spots by dispensing a solution of the protein onto a substrate pre-functionalized with biotin-functionalized PLL-g-PEG [10]. The probe with a 1 µm aperture was brought in contact with the substrate using the force feedback system, and the streptavidin was released upon overpressure application. The highly reproducible dispensing process allowed for the creation of an array of 3 µm diameter streptavidin spots within a non-fouling background. This writing process based on the biotin–streptavidin system offers attractive prospects for the micropatterning of a wide range of biotinylated molecules, for example, proteins, peptides, nucleic acids, or antibodies.

Figure 15.1 (a) Confocal images of a glass bottom dish using Atto settings (top) and FITC settings (bottom left) after deposition of PLL-FITC dots with 50 kPa for 2 min. (b) Confocal image using Atto setting and bright field image of the neuron clusters 11 days after PLL line writing. In situ deposited lines are highlighted [11].
In another study, a writing process based on local polymer replacement was developed for cell patterning [11]. In this approach, cell adhesive patterns were produced through the displacement of physisorbed PLL-g-PEG brushes by a PLL homopolymer [12]. In the first step, 5–200 µm diameter spots of PLL were generated by delivering the polymer onto a PLL-g-PEG-coated substrate (Figure 15.1a). Rat embryonic hippocampal neurons seeded on the substrate adhered selectively on the created spots. Another lithographic step was implemented subsequently to trigger neurite outgrowth and the connection of the neuron clusters (Figure 15.1b). The fluidic probe was approached onto the substrate using the AFM force feedback, and PLL stripes were written in between neuron clusters by applying a pressure while moving the tip. The neurons effectively grew neurites along the PLL lines, bridging electrically active neuron clusters distant from about 250 µm. Beyond the ability to guide the axonal outgrowth of patterned neurons, this study evidenced the capability of FluidFM-based lithography to modify cell substrates in situ, which is of great interest for the modification of existing cell patterns and for the patterning of multiple cell types.

Figure 15.2 Representative examples of mammalian cells sorted in a microwell array (a) [7] and micropatterned yeast cells (b) and bacterial cells (c) [2].
15.3.2 Pick and Place
A more straightforward approach to control the location of a cell using FluidFM consists of capturing a selected cell at the probe aperture by suction, which allows for subsequent displacement and release at a desired location upon pressure reversal. The FluidFM force feedback allows gentle contact with the sedimented or adherent cell before applying negative pressure for reversible immobilization, whereas suspended cells can also be directly aspirated to the probe aperture without preceding contact. Once the cell is immobilized, the cantilever is lifted up and moved to a chosen position. The transported cell can then either be released into a confined microvessel (e.g., microwell) upon the application of a short overpressure pulse or let adhere on a surface. In the latter case, cell adherence at the new location can be promoted by surface modification with a cell adhesive. This pick-and-place procedure has been applied to both eukaryotic and prokaryotic cells, adapting the probe design to the targeted organism [2, 7]. Tipless cantilevers with a circular 8 µm diameter aperture were used to displace mammalian cells, typically 10–20 µm in diameter [7]. Adherent cells can be either physically detached from their substrate, overcoming adhesion forces of up to a few micronewton (see Section 15.5.2) [2, 4] or first detached by trypsin treatment [7]. In the latter case, the same cantilever can be used to dispense the enzyme and subsequently displace the cell. The detachment of a spread neuron has been achieved using a negative pressure of 30 kPa, whereas trypsinized HeLa cells were effectively immobilized with a negative pressure of 5 kPa. Selected HeLa cells were sequentially isolated from confluent cultures, sorted into individual microwells (as in Figure 15.2a), and assayed for viability, whereby more than 95% of the isolated cells remained viable [7]. Individual cells could also be transferred to a new substrate by pressing the cell onto a fibronectin-coated substrate with a gentle force up to initiation of cell spreading, upon which the probe was lifted off (unpublished data). Yeast cells were also successfully relocated using the same approach [2]. Saccharomyces cerevisiae, typically 8 µm in diameter, were immobilized to cantilevers with 4 µm diameter apertures and a negative pressure of ∼5 kPa. The yeasts were efficiently relocated onto a substrate to form a line pattern (Figure 15.2b). To handle bacteria, different probe designs have been used: either cylindrical probes [2] or cantilever with a pyramidal tip featuring a 300 nm wide aperture at the apex [5] or tipless probe with an aperture of 8 µm but with a decreased channel height of 0.5 µm [6]. Initial experiments demonstrated the possibility to relocate bacteria, creating a line pattern of Escherichia coli, typically 1.5 µm in diameter and 3 µm in length (Figure 15.2c). Pre-coating of the substrate with PLL helped preventing cell movement after cell deposition. The procedure was applied for isolating bacteria of interest from an environmental sample in a study aimed at the identification of aerobic anoxygenic phototrophic bacteria in phyllosphere communities. Such bacteria, recognizable by the characteristic fluorescence of bacteriochlorophyll in the infrared range, were isolated from complex leaf wash samples and identified by 16S rRNA gene amplification and sequencing. The study revealed that the small subset of phototrophic bacteria in the phyllosphere consists of different species of Methylobacterium. The pick-and-place approach has a relatively low throughput, but provides a high accuracy and offers the versatility to pattern, sort, or isolate mammalian cells, yeasts, or bacteria.
15.3.3 Cell Dispensing/Removal
Offering a new range of dimensions, microfabricated SU-8 cantilevers (see Chapter 14, Section 1.2) allowed for the development of an alternative cell patterning approach [13]. SU-8 hollow cantilevers integrating 22 µm thick channels enabled to flow cells into the fluidic channel instead of immobilizing single cells outside the probe. Such probes were applied to either dispense cells preloaded in the channel (positive patterning; Figure 15.3a) or remove cells from an undesired location upon suction (subtractive patterning; Figure 15.3b). Yeast cells (S. cerevisiae, typical diameter ∼8 µm) were readily flown through the large microchannel, and pressure-controlled flow allowed for dispensing the cells one by one into predefined microwells. Positive patterning was next demonstrated using a suspension of myoblast cells preloaded in the probe. The cantilever was approached onto an adhesive-coated surface, and a constant overpressure was applied. After initial contact with the surface, the cantilever was further bended in incremental vertical steps of 1 µm using stepper motors until the cells stopped floating away, and one stayed at the aperture. The overpressure was stopped, and the corresponding z-position, that is, the bending of the cantilever, typically 5 µm below the first contact, was recorded in order to reproduce the same physical confinement for the subsequent patterning events. After relocating the cantilever for the next spot by lifting it up and bending to the same extent, overpressure was applied again to deposit the next cell. Small patterns of cells could be designed, such as a grid of four cells with 5 µm resolution as displayed in Figure 15.3c. The mechanical robustness of the cantilevers enabled to contact the substrate without force feedback; the latter could however be implemented in order to facilitate and automate the approach. Subtractive patterning was demonstrated with pre-patterned hippocampal neurons. While microcontact printing techniques show limitations in yield and the impossibility for in situ modification of the existing patterns, the technology using thick hollow cantilevers can overcome these limitations. First, an adhesive PLL solution was dispensed on a glass surface. The dish was then backfilled with a repulsive PLL-g-PEG solution before seeding the neurons. While the cells mostly adhered to the adhesive patterns, some unwanted cells remained on the repulsive areas. Those were removed by suction into the cantilever placed in their vicinity. The obtained pattern remained intact for 12 days, as depicted in the fluorescing image in Figure 15.3d.
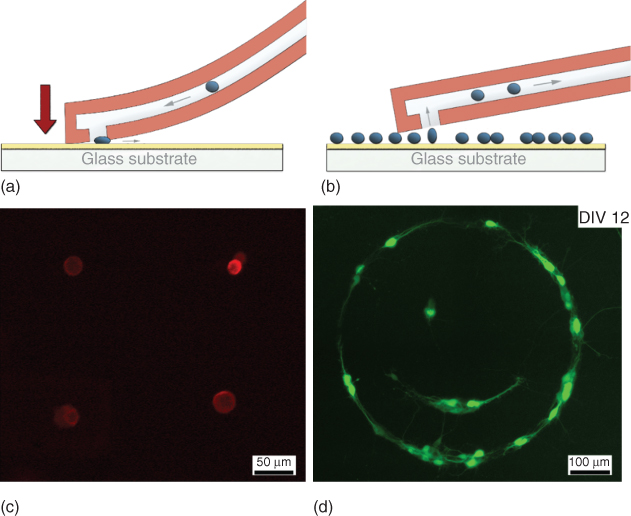
Figure 15.3 Configurations developed for single-cell patterning using thick SU-8 hollow cantilevers. Schematics illustrating (a) the controlled direct deposition by physical confinement and mechanical squeezing of cells and (b) the subtractive patterning with localized single-cell detachment in cells sitting on an adhesive surface or collective detachment from a repulsive surface. The bottom panel shows the corresponding outcome with (c) precise deposition of four myoblast cells and (d) subtractive patterning of primary hippocampal neurons from pre-patterned surfaces in the form of a smiley 12 days after removal. Myoblast cells are fluorescently tagged with a PKH26 dye. Hippocampal neurons have been transfected 4 days after deposition with the fluorescent calcium indicator GCaMP6s.
15.4 Controlled Fluid Delivery
The controlled delivery of bioactive agents to selected living cells is of high interest for a broad diversity of biological studies, facilitating the assessment of the influence of environmental cues and exogenous substances on the cell function and physiology. Moreover, the delivery of molecular probes also provides a mean to investigate specific intracellular processes. The FluidFM enables both extracellular (Section 15.4.1) and intracellular (Section 15.4.2) deliveries of biomolecules to selected cells within tissue culture samples, with defined dosage and high spatiotemporal control.
15.4.1 Extracellular Fluid Delivery
The extracellular fluid delivery with microscale resolution can be used to perturb individual cells by altering their microenvironment or by stimulation with cell-permeable compounds. Here, the FluidFM provides an efficient tool to deliver femto- to picoliter volumes of bioactive solutions to a selected cell. Controlled release of the preloaded solution through a nanometer- or micrometer-sized probe aperture can be achieved via diffusion- or pressure-mediated flow. Positioning of the probe with respect to the targeted cell before release is achieved with micrometric x–y resolution, and using force-controlled approach to position the probe aperture either in gentle contact with the cell membrane or retracted a few micrometer above it using the z-piezo. Meister et al. [1] presented the first strategy to perturb a single targeted cell by delivering biomolecules from the microchannel to the cell interior by diffusion. The microfluidic probe prefilled with a membrane-permeant fluorescent dye (CellTracker green or acridine orange) was brought in contact with the cell membrane using the force feedback system, and the probe aperture was maintained in contact for a few minutes to allow for the diffusion of the fluorescent molecules into the targeted neuroblastoma cell. Next, FluidFM was used for the pressure-controlled delivery of bioactive molecules in solution in a study aimed at isolating a selected cell from a near-confluent culture [7]. For this purpose, a solution of trypsin was released from 1 to 5 µm above the targeted cell under tight pressure regulation. The applied pressure pulse defined not only the volume of solution dispensed but also the cell culture area subjected to the solution. The selective trypsin-induced detachment of single HeLa cells from high density cultures was successfully achieved through the delivery of ∼30 pL of the protease solution (Figure 15.4).
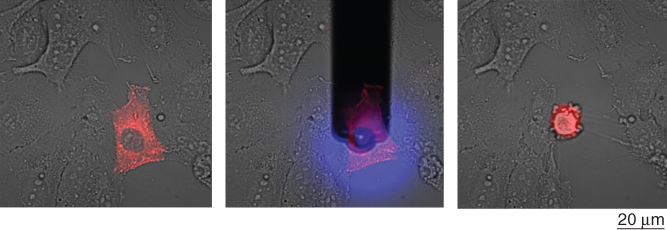
Figure 15.4 Controlled release of trypsin (30 pL) onto a selected cell (red) for selective cell detachment [7].
Beside biomolecules, FluidFM has also been applied for the controlled delivery of virus particles, enabling the study of viral infection at the single-cell level [14]. Vaccinia viruses (VACV; 360 × 250 nm) were suspended in physiological buffer and dispensed by FluidFM onto selected HeLa cells to investigate the initial step of the viral infection. Pressure-regulated flow enabled the consecutive release of single viruses through the probe aperture. Viral infectivity was then assessed in function of the number of deposited viruses, ranging from 1 to 12. The study resolved details of the early infection stages of the virus, revealing a cooperative behavior of the virus particles. Altogether, these studies showed the capacity of FluidFM for delivering controlled dosage of bioactive moieties onto single cells. While virtually any molecules and even viruses can be flown through the standard 1 µm high channel, the application range of controlled fluid delivery can be expanded to even larger entities such as bacteria or fungi using the recently developed SU-8 cantilevers, which feature channels up to 22 µm in height. As such, the FluidFM represents a powerful tool not only to investigate the effects of exogenous compounds on the cells but also to study cell–cell interactions.
15.4.2 Intracellular Fluid Delivery
While the local delivery of bioactive molecules for selective cell perturbation is highly attractive, many compounds of interest are not cell permeable and therefore require intracellular delivery. The injection of fluids directly inside a cell not only expands the spectrum of compounds that can be delivered but also enables circumventing diffusion of the bioactive molecules to neighboring or distant cells. In addition, injection makes it possible to deliver molecules directly into cell nuclei. The FluidFM provides an “infinite” reservoir for the active agents in solution and allows for an active, pressure-assisted delivery of fluids. As such, the dosage and timing of delivery can be precisely tuned. The force control and monitoring further provide the means to insert the tip of the probe inside the cell in a nondestructive fashion, with a real-time readout of cell contact, cell indentation, and cell membrane perforation. Intracellular injection by FluidFM has been achieved using cantilevers with a sharp pyramidal tip, by which a 300 nm triangular aperture was milled on the pyramidal face by focus ion beam [1, 3]. The tip was inserted inside a cell via a forward force spectroscopy. Once inserted, the tip was maintained inside the cell at a constant force, and the preloaded solution was injected into the cell upon application of positive pressure.
In the first experiments, a membrane-impermeable fluorescent dye was delivered into the cytosol of myoblasts and neuroblastoma cells, demonstrating confinement of the fluorophore in the cell cytoplasm and the resealing of the cell membrane upon probe withdrawal [1]. Besides monitoring cell contact and indentation, force spectroscopy data also revealed indentation peaks upon membrane perforation, providing a useful readout of the tip insertion. Next, the cell injection approach was extended to intranuclear injection [3]. As with cytoplasmic injection, fluorophore confinement and membrane resealing were observed. In addition, the injected volumes were quantified, demonstrating the ability to tune the amount delivered and showing that 50–800 fL could be injected without noticeable changes in cell morphology and preserving cell viability. Functional plasmid DNAs were then delivered directly into the cell nucleus, resulting in the subsequent transient expression of an encoded green fluorescent protein (Figure 15.5). Altogether, these injection studies using FluidFM showed the ability to deliver precise dosage of functional biomolecules into the cytoplasm or nucleus of mammalian cells while maintaining cell viability. In addition, such injection experiments were achieved directly in tissue culture samples, preserving cellular context and the physiological environment. Injected cells could thus be monitored for several days postinjection to assess the effect of the performed perturbation. Beyond fluorescent labeling and cell transfection, a broad range of biological applications can be envisioned for this injection procedure, allowing to selectively interact with and investigate cellular processes. In addition, current efforts are deployed to expand the procedure from animal cells to eukaryotic microorganisms (e.g., yeasts), making them challenging not only because of their smaller size but also because they are enveloped with a rigid cell wall. With most of the current molecular biology performed on yeasts, a successful adaptation of the intracellular injection procedure will be of tremendous interest to cell biologists.

Figure 15.5 Transfection of a HeLa cell with pmaxGFP by nuclear injection; the phase contrast images show the cell before (a) and during (b) injection. The fluorescence images show the cell immediately (c) and 2 days (d) after injection. DexTRITC co-injected with the plasmid remained localized in the nucleus (bright area in (c)), and 2 days postinjection the cell was producing GFP (bright area in (d)). The arrow in (a) indicates the targeted cell.
15.5 Mechanical Measurements
Living cells sense and respond not only to chemical signals but also to mechanical stimuli such as the forces externally applied or generated by cell–matrix and cell–cell contacts. The cells detect mechanical stimuli via mechanosensitive signaling pathways, convey the external forces to intracellular load-bearing structures, and generate internal forces to facilitate migration, undergo mitosis, or communicate with neighboring cells. Biomechanical studies, including investigations of cell stiffness and cell adhesion, have provided valuable insights into cellular physiology and pathology with significant implications for biotechnology and medicine.

Figure 15.6 Schematic overview of typical single-cell force spectroscopy measurements (SCFS) and the recorded force–distance curves. (a) The cell probe (small insets of cantilever with attached cell) starts at a distance from the surface (1) and is approached (upper curve), contacting the surface (2), until a certain force is reached (3), leading to compression of the cell. During the subsequent retraction (lower curve), the cell stretches (4), and the acting adhesive force restrains the cantilevers movement upwards, before the cell detaches from the surface and the cantilever is back in its starting position (5). (b) Representative force–distance curve depicting the information typically extracted from SCFS measurements. The elasticity (E) can be modeled from fitting the initial indentation. The detachment distance (d) is the distance required to completely detach the cell. The maximal adhesion force (Fadh) is characterized as the force minimum detected during retraction, and the detachment work (Wadh) can be calculated from the area between the retraction curve and the baseline.
Single-cell force spectroscopy (SCFS) by AFM is a powerful quantitative method to study both the viscoelastic and adhesive properties of cells, which combines high resolution and a large range of measurable forces (from 10 pN to 1 µN) [15, 16]. SCFS experiments exploit the precise force feedback of AFM, whereby the tip is approached toward a sample until a defined force setpoint is reached (forward curve) and maintained for a defined contact time, and the cantilever is then withdrawn with a given velocity (backward curve) to complete the force spectroscopy. The deflection of the cantilever during movement is recorded and translated into a force–distance curve, plotting the forces acting on the cantilever versus the distance traveled in z-direction (Figure 15.6). The cell elastic properties can be extracted from the forward curve by fitting to the Hertz model for the corresponding tip geometry, whereas the backward curve reflects the adhesive interactions between the cell and the substrate or the tip. The FluidFM technology retains the strength of AFM in SCFS experiments, whereas its ability for reversible cell fixation represents an invaluable additional strength. The next sections present the achievements and benefits of FluidFM for measurements of cell elasticity (Section 15.5.1) and cell adhesion (Section 15.5.2).
15.5.1 Quantification of Cell Elasticity
The stiffness is a sensitive phenotypical indicator of physiological and pathological changes in cells, with many potential applications in biology and medicine. In yeasts and bacteria, the cell wall is the primary load-bearing structure, and its synthesis, remodeling, and regulation are central to the cell physiology, with cell shape and size directly involved in a variety of critical functions including division, cell motility, cellular differentiation, and immunity [17]. In mammalian cells, the cytoskeleton is the major contributor to cellular elasticity and plays an important role in morphological changes of the cell during movement [18]. Cell elasticity measurements have provided valuable insights into multiple processes such as cell migration [19], cell division [20], mechanotransduction [21, 22], differentiation [23], tumor formation [24, 25], wound healing [26], or aging [27]. It has also been used as an indicator for cellular changes upon drug treatment [28].
FluidFM provides different strategies to measure cell elastic properties. One approach consists in immobilizing a cell at the probe aperture by suction, before performing SCFS on a hard substrate. Multiple force spectroscopies can therewith be performed with the fixed cell, before applying an overpressure pulse to free the cantilever for capturing the next cell. With this approach, McGrath et al. [29] investigated the elastic properties of Cryptosporidium, a protozoan pathogen that can contaminate treated water supplies. Serial measurements of individual oocysts allowed to distinguish a pathogenic Cryptosporidium species from a noninfectious one based on their height and deformability properties. In addition, measuring the effective spring constant enabled to further discriminate between viable and nonviable oocysts. Allowing to measure more than 50 cells/h, the proposed method represents a competitive alternative to the current water examination protocols.
Another approach for cell elasticity measurement with the FluidFM makes use of colloids as spherical indenters [30]. Like individual cells, the colloids can be immobilized at the cantilever aperture in a reversible fashion by pressure regulation, offering a powerful alternative to standard colloidal force spectroscopy. Dörig et al. showed the benefits of colloidal FluidFM for probing adhesive interactions and performing cell elasticity measurements. For the latter, the aperture of a tipless cantilever was brought in contact with single colloids pre-adsorbed onto HEK (human embryonic kidney) cells, and negative pressure was applied for fixation. The adsorbed bead was then pushed against the underlying cell for microindentation, before withdrawal and detachment of the bead. Employing the Hertz model for a spherical indenter, the apparent elastic modulus of the cells could be extracted from the forward force curves. Young's modulus resulted in a value of 0.77–0.68 kPa, in agreement with the values of ∼1 kPa reported by other groups. In this study, the colloids were pre-adsorbed onto the cells to allow for the measurement of long-term adhesive interactions simultaneously with the elasticity measurement. It is however also possible to first immobilize a colloid to the probe before performing the microindentation.
In both approaches, FluidFM enables to perform SCFS with the same accuracy as AFM while further allowing to exchange the cell or colloid immobilized at the probe aperture. This fast exchange enables to collect relatively large data set using a single cantilever, whereas traditional cell-probe and colloidal-probe preparations are time-consuming and usable only for a limited number of measurements.
15.5.2 Quantification of Single-Cell Adhesion Forces
Adhesion to biotic or abiotic surfaces is a crucial trait of all living cells, mediated by a complex interplay of specific and unspecific, long- and short-range interactions [31]. Microorganisms predominantly exhibit a surface-associated lifestyle, which forms the basis for efficient substrate utilization, the formation and maturation of biofilms, and a higher resistance to antibiotics [32]. Attachment to host cells is essential for commensal and mutualistic organisms and the initial step of infection by pathogens. Adhesion between individual cells was a prerequisite for the evolution of multicellular organisms and ultimately mammals. Besides holding our bodies together, cellular adhesion greatly influences mammalian cell physiology and governs cell differentiation, tissue development, or inflammation. Understanding the characteristics of adhesion and its underlying mechanisms is therefore important for a variety of applications. Several medical procedures, such as tissue grafting or implants, aim for a successful and sustainable cellular adhesion. On the other hand, biocompatible materials are sought after to minimize microbial biofilm formation on their surface. Likewise, there is a substantial interest in the food industry to minimize the binding of contaminating bacteria during production or packaging. Then again, many biotechnological processes depend on efficient bacterial adhesion in bioreactors to maximize production yields.
While AFM has been proven a powerful technique to quantify adhesion forces of both eukaryotic [33–35] and prokaryotic [36, 37] cells, it suffers from several limitations that are mostly related to the cell immobilization on the cantilever. Indeed, the cells need to be literally “glued” to cantilevers by chemical functionalization with an adhesive [36, 38], for example, lectins [33, 39], streptavidin [40, 41], or bioinspired wet adhesives such as polydopamine [42, 43]. Such chemical fixation harbors the risk of altering the cell physiology and ultimately influences the measured adhesion force [42, 44]. The cantilever functionalization is time and labor intensive and is irreversible, requiring the preparation and calibration of a separate cantilever for each cell to measure. This impedes the measurement of biological replicates and the generation of statistically relevant data, restricting current studies to a limited number of cells [38, 43]. Another limitation of chemical fixation is the maximal adhesion force measurable, defined by the immobilization strength of the cell on the cantilever. This problem can be circumvented by limiting the contact time, thereby effectively measuring only initial, weaker adhesion forces. In previous studies, the contact times for fungal [45], mammalian [46, 47], and bacterial cells [42, 43, 48] ranged from minutes to several tens of minutes, before adhesion forces exceeded the detectable range.
With the possibility for fast, stable, and reversible cell fixation to cantilevers by negative pressure, the FluidFM technology can solve these limitations and open the way for high throughput cell adhesion measurements [16]. The cell of interest, optically selected, is gently approached in a way that the cantilever aperture aligns on top of the cell (Figure 15.7). During a pause of several seconds, gentle contact is maintained, while an underpressure is applied to immobilize the cell, and the following retraction of the cantilever detaches the cell from the substrate. Recording the cantilevers, deflection allows to determine the force required for this detachment. Alternatively, cells can also be directly aspirated to the aperture from a solution. After completion of adhesion measurements, the immobilized cell can be released by application of a short overpressure pulse, rendering the cantilever free for immobilization of the next cell.
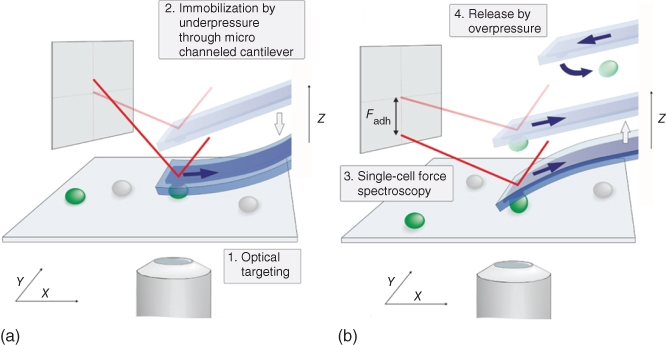
Figure 15.7 FluidFM-based single-cell force spectroscopy. Schematic view of the experimental setup. (a) Optical targeting of cell, followed by approach and immobilization to the cantilever through application of underpressure. (b) Single-cell force spectroscopy during retraction of the cantilever with immobilized cell and subsequent release of the cell by a short pulse of overpressure [4].
By generating functional cell probes through aspiration of yeast cells directly from solution to tipless cantilevers with 2 µm apertures, it was shown that chemical fixation can be replaced by physical immobilization altogether [4]. Furthermore, different yeast cells adhering to different substrates could be immobilized in situ and successfully detached with detachment forces between few nanonewton and several tens of nanonewton (Figure 15.8a). A single cantilever could be used to collect up to 200 force curves of different cells in a single day. Importantly, the method left cells unperturbed until the measurement, a prerequisite for recording dynamic adhesion behavior. Consequently, it could be used to resolve the temperature- and development-dependent changes in adhesion behavior observed in the dimorphic yeast Candida albicans, with forces ranging between 500 pN and 15 nN [4].

Figure 15.8 Representative force–distances curves recorded with (a) yeast and (b) mammalian cells during FluidFM-based SCFS. Upper curves depict the approach; lower curves represent the retraction movement. Arrows representing the calculated maximal adhesion force (Fadh) and the detachment distance (d) as well as the shaded area (Wadh) corresponding to the work performed during the detachment process are indicated [4].
Besides increasing the throughput of SCFS measurements, physical immobilization by underpressure also increases the upper limit of measurable forces, defined by the strength of the cell immobilization on the cantilever. In FluidFM, the immobilization force depends on the aperture size and the applied underpressure and can be estimated by the formula ![]() , with p corresponding to the underpressure in Pa and r corresponding to the radius of the aperture in m. With an applied underpressure of 80 kPa and aperture sizes of up to 8 µm, this yields maximal immobilization forces up to 4 µN, largely overcoming fixation by chemical fixation. This is mostly relevant for measuring the adhesion forces of mammalian cells, which spread on surfaces and are far more elastic than yeast cells [49], exhibiting adhesion forces often exceeding the strength of chemical fixation [50]. FluidFM using tipless cantilevers with 8 µm apertures enabled the serial measurement of adhesion forces of different mammalian cell types, even when grown overnight under standard cell culture conditions. Compared with those of yeast cells, measurements showed 35-fold longer detachment distances and 40-fold higher adhesion forces, up to 1600 nN (Figure 15.8b). Additionally, multiple distinct unbinding events could be identified from the force curves, highlighting the preserved high resolution of the force spectroscopy. Again, a single FluidFM probe could be used to collect force curves of multiple cells, ensuring statistical relevance and allowing to generate data on cell-to-cell variability [4]. This protocol has also found application in a study investigating successful endothelialization of cardiovascular devices [8]. Adhesion measurement of fully spread cells allowed the identification of defined surface topographies that maximize the normal adhesion force of human endothelial cells. Likewise, FluidFM-based SCFS was used to characterize the adhesive and mechanical properties of mammalian cells in its dependence to external electric current, exploring a possibility of cell manipulation for tissue engineering [51]. In addition to measuring adhesion forces, the elasticity of cells before and after electrical stimulation could be determined, demonstrating the method's versatility.
, with p corresponding to the underpressure in Pa and r corresponding to the radius of the aperture in m. With an applied underpressure of 80 kPa and aperture sizes of up to 8 µm, this yields maximal immobilization forces up to 4 µN, largely overcoming fixation by chemical fixation. This is mostly relevant for measuring the adhesion forces of mammalian cells, which spread on surfaces and are far more elastic than yeast cells [49], exhibiting adhesion forces often exceeding the strength of chemical fixation [50]. FluidFM using tipless cantilevers with 8 µm apertures enabled the serial measurement of adhesion forces of different mammalian cell types, even when grown overnight under standard cell culture conditions. Compared with those of yeast cells, measurements showed 35-fold longer detachment distances and 40-fold higher adhesion forces, up to 1600 nN (Figure 15.8b). Additionally, multiple distinct unbinding events could be identified from the force curves, highlighting the preserved high resolution of the force spectroscopy. Again, a single FluidFM probe could be used to collect force curves of multiple cells, ensuring statistical relevance and allowing to generate data on cell-to-cell variability [4]. This protocol has also found application in a study investigating successful endothelialization of cardiovascular devices [8]. Adhesion measurement of fully spread cells allowed the identification of defined surface topographies that maximize the normal adhesion force of human endothelial cells. Likewise, FluidFM-based SCFS was used to characterize the adhesive and mechanical properties of mammalian cells in its dependence to external electric current, exploring a possibility of cell manipulation for tissue engineering [51]. In addition to measuring adhesion forces, the elasticity of cells before and after electrical stimulation could be determined, demonstrating the method's versatility.

Figure 15.9 Adaptations of probe design and representative force–distance curve of bacterial SCFS. Scanning electron microscopy images of (a) a pyramidal tip with 900 nm opening and (b) side view of a pyramidal tip with hemicylindrical aperture design. Apertures were achieved by FIB treatment. Scale bars correspond to 1 µm. (c) Representative force–distance curve of SCFS of E. coli on glass surface using a cantilever with spring constant of 0.2 N/m (red: approach, blue: retraction curve).
(Adapted from Ref. [5].)
The approach was further adapted to the measurement of bacterial cell adhesion [5]. Several adjustments had to be made, owing to the variable shape, small size, and relatively weak adhesion forces of bacteria. Instead of tipless probes, cantilevers with a pyramidal tip were used, with a 900 nm aperture at the pyramidal apex or fitted with hemicylindrical apertures achieved by focused ion beam (FIB) drilling, to accommodate rod-shaped bacteria (Figure 15.9a and b). To enable high-resolution force curves, the cantilever thickness was reduced to shift the spring constant from approximately 0.2 to 2.5 nN/m. Bacterial SCFS was performed on different surfaces with both the rod-shaped Gram-negative model organism E. coli and the clinically relevant spherical Gram-positive pathogen Streptococcus pyogenes. The strong immobilization of cells by underpressure allowed for their detachment from glass surfaces coated with polydopamine, which is considered the most efficient wet adhesive commonly used for chemical fixation of cells onto cantilevers. Bacteria were detached after more than 2 h of unperturbed surface contact, yielding long-term adhesion forces up to 14 nN. Following SCFS, cells were deposited back onto the substrate, and cell division could be observed, underlining the non-interfering nature of the procedure. At the same time, the lowered cantilever spring constant showed a peak-to-peak noise of only 20 pN, enabling the measurement of forces as low as 120 pN, with the detection of distinct force plateaus and jumps in the piconewton range (Figure 15.9c). These distinct detachment force patterns are characteristic of cell appendages and taken together with the relatively long detachment distances can be accounted to fimbriae or pili that are involved in bacterial adhesion [5, 52, 53].
An alternative approach to quantify adhesive interactions consists of measuring cell-probe instead of cell-substrate interactions. For example, the FluidFM can be used to measure the interaction of a functionalized colloid with the cell surface. The approach has been used to measure the interaction of HEK cells with concanavalin A, which binds to mannose residues on the cell membrane in an irreversible fashion [30]. In this work, 50 µm colloids were functionalized with the lectin and left adsorbed on the cell surface for 1 h. A tipless cantilever was then used to contact the adsorbed colloid and immobilize it by underpressure, before pulling the probe away from the cell and measuring detachment forces involved between the colloid and the cell. The measured forces ranged between 10 and 60 nN, in good agreement with reported values. With the possibility to exchange the colloid at will, this strategy makes it possible to perform serial measurements and assess multiple bead functionalization within a single experiment.
In conclusion, FluidFM has proven to be a versatile and powerful tool for the quantification of single-cell adhesion forces. Not only does pressure-based immobilization of cells or colloids facilitate measurement of larger adhesion forces than before, but it also renders cell immobilization fully reversible and therefore allows rapid and serial collection of force curves from multiple cells. The universal applicability of the system has been impressively demonstrated with cell types ranging from mammalian to yeast and even bacterial cells. Additionally, fabrication of closed FluidFM probes without predefined apertures allows rapid customization of probe design and will further speed up adaptations for envisioned future experiments. Even though the upper physical limit is still restricted by the applicable underpressure, future users will be able to rely on a powerful technique that offers adhesion measurements from the low piconewton range up to several micronewton. Future applications will be able to focus on answering biologically relevant questions, which may include the characterization of materials with anti-adhesive properties to reduce problems of biofouling, or elucidating mechanisms of pathogen attachment to host tissue in the initial step of infection. Further analysis of, for example, bacterial adhesion on their natural substrates might explain differences observed in localization patterns or elucidate mechanisms leading to biofilm formation.
15.6 Ionic Current Measurements
The FluidFM has been further developed by integrating an electrode in its reservoir for measuring picoampere ionic currents flowing through the tip aperture. The setup was first applied in the field of electrophysiology, enabling force-controlled patch-clamp measurements in whole-cell configuration [54, 55], and, second, in the scanning probe imaging field by using the FluidFM to perform scanning ion conductance microscopy (SICM) [56, 57]. Using the same electrical system presented in Section 15.6.1, the patch clamp and the SICM potentials of the FluidFM are addressed in two different sections (Sections 15.6.2 and 15.6.3) for a better description of the strengths and weaknesses of the FluidFM for each technique.
15.6.1 Adaptation of the FluidFM Setup for Picoampere Current Measurements
Both the patch clamp and the SICM techniques require the possibility to measure ionic currents flowing through the probe aperture in the picoampere range (dotted path in Figure 15.10d). For this purpose, the FluidFM was modified by first integrating an Ag/AgCl electrode into its reservoir, drilling a hole in the cantilever probe holder to insert the electrode. Additional epoxy glue to fix the electrode and macroscopic coating of wax around the reservoir ensured its electrical isolation from the bath solution (Figure 15.10b). The integrated electrode was then connected to a custom full-metal amplifier connector (Figure 15.10a) that acts as a shielding interface between the AFM hardware featuring the FluidFM Cytoclip as a probe and an external patch-clamp amplifier brought as close as possible to the measurement site to ensure precise electrical measurements. Finally, a custom metallic dish holder (Figure 15.10c) allowed to shield the measurement site from electrical noise by acting as a Faraday cage. The electrical circuit was closed by an additional Ag/AgCl electrode in the bath solution that was connected to the ground. On the software side, LabVIEW was chosen to interface the different devices and provide simultaneous data acquisition for real-time analysis. To ensure the functionality and sufficient sensitivity of the electrical FluidFM setup, several experimental tests were performed. First, the resistance flowing from the electrode in the FluidFM reservoir to the bath electrode was monitored. Typical values for probe openings in the order of 200 nm–2 µm and an electrolyte solution of 150 mM KCl filling the cantilever and the bath were 25–35 MΩ. A higher resistance inferred a probe clogged either from particles or from air bubbles, while a lower resistance reflected a leak. Since the resistance mostly comes from the microchannel (90%) [55], this first test still did not guarantee that the very tip of the probe was actually functional (e.g., there could still be a leakage at the cantilever basis). To make sure that the ionic current was indeed flowing through the cantilever aperture, the tip was then approached onto a nonconductive substrate. At aperture–substrate distances below the probe aperture radius, the ion flow will be restricted, leading to a characteristic drop in current (Figure 15.10e, black curve) correlated to an increase in resistance. This squeezing effect gives the useful signal for SICM (drop in current monitoring) and patch clamp (increase in resistance monitoring). The setup is not functional when this key characteristic drop in current is not recorded at the moment of tip–substrate contact, identified by the simultaneous force recording from the deflection signal of the cantilever (Figure 15.10e, grey curve and dashed vertical line). The length of this section expresses the importance of this first crucial step in detecting picoampere current changes at the tip of the FluidFM probe before being able to achieve any patch-clamp or SICM experiments.

Figure 15.10 Modification of the FluidFM setup to measure pA currents. (a) Bottom view of the FluidFM hardware with its customized Cytoclip integrating – additionally to the conventional pressure tubing – an Ag/AgCl electrode in its reservoir and an amplifier connector interfacing the FluidFM to the proper electronics via a BNC connector. (b) Encapsulating the reservoir in wax ensures an electrically tight connection. (c) A custom metallic sample holder acts as a Faraday cage isolating the measurement site from electromagnetic noise. (d) Schematic illustrating the adaptation of the FluidFM to measure ionic currents flowing through the probe aperture (dotted path). Filled with and immersed in an electrolyte solution of 150 mM KCl, a FluidFM probe featuring a 300 nm cylindrical aperture shows an electrical resistance of ∼30 MΩ, mostly coming from the microchannel impedance (∼90%). (e) Simultaneous ionic (black curve) and force (gray trace) recording when approaching a FluidFM probe onto a glass substrate in a solution of 150 mM KCl (cantilever filled with the same solution). An electrical bias of V = 25 mV is applied. The moment of contact of the probe with the substrate is identified from the steep increase in the deflection signal. The characteristic drop in current at close working distances confirms that the setup is properly functional and current sensitive at its tip.
(Adapted from Ref. [55].)
15.6.2 Force-Controlled Patch Clamp with the FluidFM
Invented in the 1970s, the patch clamp [54] is the gold standard for recording the activity of ion channels that regulate transmembrane ionic flow and are involved in many pathologies. Using an electrolyte-filled glass micropipette with a 1–2 µm opening as an electrode, the conventional procedure consists of optically approaching the probe onto the cell of interest while monitoring the increase in resistance. Once in contact with the cell, negative pressure is applied to suck a cell membrane “patch” inside the pipette until a gigaohm seal is obtained. Further suction will rupture the membrane patch and lead to the whole-cell configuration of the patch clamp, allowing to accurately control the transmembrane potential and measure simultaneously the activity of multiple, usually voltage-gated channels spanning the entire cell membrane. Although powerful, the patch clamp remains a very labor-intensive and low throughput procedure, even for skilled operators, mainly due to the difficult positioning of the probe onto the cell. Of particular interest for pharmaceutical companies, several innovation efforts are investigating strategies to increase the efficiency of the technique, the most promising one being the automated patch clamp [58] in which microfabricated chips replace the conventional glass pipette. Yet, those systems are limited to the study of stable cell lines expressing the ion channel of interest.
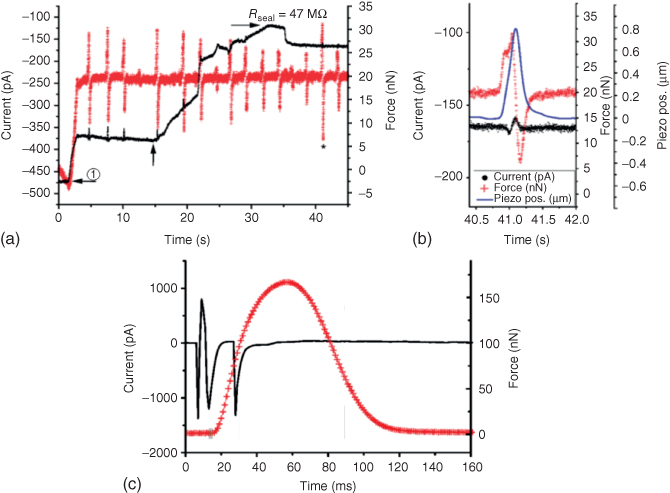
Figure 15.11 Simultaneous force and whole-cell current recordings on primary mouse cardiomyocytes. (a) Time evolution of the current and the forces acting on the cardiomyocyte when approaching the cell to create the seal and get to the whole-cell configuration. The tip is automatically approached with the AFM controller of the FluidFM until the proper force setpoint of 20 nN is reached and then maintained by the controller. The moment of contact of the probe onto the cell is identified by the characteristic drop in current and the increase in force to the setpoint (arrow 1). Negative suction at arrow 2 slowly creates the seal and finally ruptures the membrane at 35 s (increase in current), leading to the whole-cell configuration. The force peaks correspond to the spontaneous activity of the approached cardiomyocyte. (b) Zoom-in of the contraction marked with a star in (a), illustrating how the AFM controls the position of the piezo actuator to maintain the force at its setpoint and therefore minimize the mechanical stress onto the cardiomyocyte. (c) Simultaneous force and ion channels activity with a disable force controller: the electrophysiological recordings are enriched from the FluidFM using a force sensor, enabling to correlate with the very same probe the electrical activity of the cardiomyocyte (Na+ and Ca+ currents identified in the first 2 inward peaks; K+ in the slow outward current following the depolarization of the membrane) and its resulting contraction force.
(Adapted from Ref. [55].)
The FluidFM was used to address those hurdles by taking advantage of its force feedback to automatically and precisely position the probe onto the cell of interest [55]. The procedure fully used the FluidFM potential to act as a current recording pipette with the force sensitivity of an AFM (Figure 15.10e). After positioning optically the FluidFM aperture ∼100 µm above the cell, the probe reached cell contact automatically by engagement of the AFM force controller until the proper force setpoint was reached and maintained by the controller (Figure 15.11a). Following cell contact, membrane suction enabled to create the patch seal and then lead to the whole-cell configuration by rupturing the patch. By maintaining the force applied onto the cell as small as possible, the AFM controller rendered the patch-clamp procedure gentle and stable, as illustrated on the difficult system of contracting cardiac cells [55] (Figure 15.11). Indeed, after showing the potential of the FluidFM in recording fast kinetics via the study of the fast activated NaV1.5 voltage-gated ion channels of HEK cells [55], the force control capabilities of the FluidFM were exploited to study beating activity of cardiomyocytes, which are too challenging for conventional patch clamp. The force peaks in Figure 15.11a originated from the cardiomyocyte contractions that cumulate to a maximum of 35 nN instead of around 170 nN with a disengaged AFM force control. Figure 15.11b highlights the movement of the piezo actuator controlled by the AFM force controller to maintain the force to its constant setpoint, minimizing the mechanical stress on the cardiomyocyte during a contraction event by retracting the probe by the proper amount (∼800 nm). As seen in Figure 15.11b from the current stabilizing to its initial value after a contraction, the seal was undamaged. This illustrates how the force-controlled patch clamp allowed more stable seals that will resist vibrations, contractions, or cell-volume changes resulting from mechanical stress from which conventional patch clamp suffers. Moreover, by limiting the stress on the cell, serial patching and injection into cardiomyocytes could even be achieved with the FluidFM [55].
Furthermore, the FluidFM offered the possibility to simultaneously measure the transmembrane ionic current and the applied or generated forces with the same probe. Figure 15.11c shows the correlation between the cardiomyocyte membrane depolarization – recognizable in the Na+, Ca+, and K+ ion currents of the recorded current trace – and its resulting contraction force. The electrophysiological data of the patch clamp were thus enriched from the simultaneous force sensing of the FluidFM, which can turn particularly powerful in the field of mechanobiology whereby forces and electrophysiology are highly correlated. An interesting future application would be the study of mechanogated piezo ion channels [59, 60], which are a special family of mechanosensitive proteins. Also, thanks to the small aperture dimensions (routinely of 300–400 nm aperture for an averaged 30 MΩ resistance), the FluidFM probe makes it possible to study small cells without the prohibitive probe resistance that glass pipettes would face for such dimensions.
Nonetheless, before being able to reach such promises, the force-controlled patch clamp with the FluidFM suffers many challenges that need to be fixed. First of all, the current seal resistances achieved with the FluidFM are routinely in the 45–100 MΩ range and hardly, if ever, reach the gigaseal, which considerably deteriorates the measurements' quality (unstable seals, increased noise level and current leakage, imprecise transmembrane voltage control, performance loss of voltage controller electronics) [61]. Despite an optimization study with FIB to customize the FluidFM probe geometry for patch clamp [55], the pyramidal geometry of the FluidFM tip remains the limiting factor explaining the low seal resistances: its square section and small height depth left for the cell to invaginate inside the tip (max 600 nm) prevents the building up of a gigaohm seal. Ongoing efforts are focusing on obtaining the gigaseal by substituting the conventional pyramidal tips of the FluidFM by cylindrical probes better mimicking the dimensions of the conventional glass pipettes or by functionalizing the tip with a proper coating fusing the cell membrane with the probe. The ideal coating should enable the use of a FluidFM probe several times to enable serial patching and limit probe costs. The patch-clamp FluidFM setup would also benefit from minimizing the contribution of the microchannel in the recorded resistance by integrating directly the electrode in the cantilever instead of far in the reservoir. This would additionally offer the opportunity to design longer and therefore softer cantilevers aiming for more gentle and higher force-sensitive probes without a detrimental resistance as the microchannel resistance would no longer contribute to the probe resistance.
15.6.3 Scanning Ion Conductance Microscopy with the FluidFM
The same electrical setup used for patch clamp (Section 15.6.1) can be completed with a precise actuator to perform SICM [56]. Using glass pipettes with inner radius down to 13 nm for high-resolution SICM imaging [62], this scanning probe technique was specifically invented with the aim of imaging soft samples, including living cells in physiological conditions. It exploits the probe-sample distance dependence of the ionic current flowing through the probe aperture as feedback input (Figure 15.10e, black curve resulting from the squeezing effect described in Section 15.6.1). With a typical current setpoint of 1% drop of the initial current, the probe is still at one aperture radius distance from the sample, allowing noncontact topography imaging by raster scanning (DC mode of SICM) [56, 63]. Implementing the SICM on the FluidFM presents several advantages, including the possibility to record simultaneously mechanical and conductance images. AFM topography images are thus enriched from simultaneous ion conductance measurements, while SICM images benefit from a simultaneous force recording indicative of tip interactions with the sample (Figure 15.12A). The direct force recording offered by the FluidFM is especially valuable when scanning in SICM mode to ensure that no actual contact occurs with the sample. Indeed, even though the SICM is a noncontact method, it is highly dependent on the stability of its feedback control and setpoint that needs to be carefully chosen and updated in order to avoid damaging the sample (due to, e.g., drifts or partial blockage of the tip). After an aware definition of a proper setpoint compromising between the search for a maximal spatial resolution (small working distances) while seeking for minimal forces on the sample, Figure 15.12A illustrates the gentleness of a SICM scan with the FluidFM that leaves the delicate unfixed neurite network intact, despite a high raster scanning speed of 13 µm/s. This strongly contrasts with the use of stiff glass pipettes that would require to map the sample in the much slower hopping mode [64] to avoid damaging the soft sample. While the AC [65] and hopping modes [64] could successfully be implemented on the FluidFM [57], the challenging dynamics of the cantilever and the reduced 10 µm piezo actuator range limited the benefits of those two modes compared with the more straightforward DC SICM mode. This explains that no further work has been carried out on those two imaging modes yet. Simulations under COMSOL [57] inspired from the work of Rheinlaender and Schäffer on glass pipettes [66] showed that the FluidFM probes behave similarly to the SICM pipettes in terms of image formation and resolution, showing a similar theoretical lateral resolution of three times the inner aperture radius: the existing research knowledge acquired on the SICM glass pipettes can be exploited to understand the imaging results obtained with the FluidFM probes. Striving for higher resolution, the FluidFM probe aperture can be reduced to 10 nm by FIB without a prohibitive probe resistance (∼50 MΩ vs GΩ range for glass pipettes [62]) but to the expense of a bigger lateral thickness detrimental to probing deep into the sample [57].
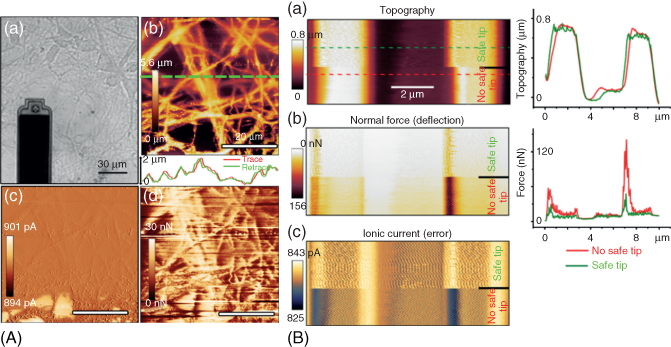
Figure 15.12 Scanning ion conductance microscopy with the FluidFM [57]. (A) 50 µm × 50 µm DC-SICM imaging of hippocampal neurons scanned with a 300 nm Apex tip. Scan speed: 13 µm/s, acquisition time: 16 min. (a) Optical image of the FluidFM tip over the neurites sample. (b) SICM topography showing the intricate neurite network. The trace and retrace in the lower inset highlight the gentleness of the scan that leaves the unfixed neurites in place. (c) Ionic current, representative of the error signal. The maximum extension of the piezo (10 µm) was reached in the lower part of the image, explaining the bigger error signal. (d) Simultaneous normal force recording showing mechanical interactions of the probe with the sample when striving for high resolution. The forces nonetheless do not damage the sample as shown in (b). (B) Comparison of imaging a PDMS sample calibration grid of 1 µm deep and 2 µm wide trenches with an additional force monitor (safe tip activated) and without (no safe tip). The force controller automatically disables the SICM and retracts the probe when the forces applied on the sample are too high, which reduces the forces (b) while keeping an accurate topography (a) (see the corresponding height profiles). The position of the force peaks also indicates how the SICM intrinsically fails when approaching high-aspect ratio obstacles (scanning direction from left to right) while achieving noncontact imaging on smooth surfaces.
While until now the systematic force recording offered by the FluidFM has only been used as an additional side information when scanning in SICM mode, the force can also be exploited to improve the control strategy of the SICM. Indeed, in “pure” DC-SICM scanning, the tip is blind to neighboring structures with too high-aspect ratio (>tip radius), resulting in unavoidable mechanical contact of the tip with the sample. Using the force sensitivity of the cantilever, the AFM controller can automatically disable the SICM controller to retract the probe by a certain distance when the measured forces on the sample exceed a given threshold. The success of this new combined control strategy is illustrated in Figure 15.12B, in which a direct comparison of the forces applied on a PDMS calibration grid sample shows how the forces are considerably reduced when the additional force monitor is present, while the topography is kept accurate. The safe tip strategy is nonetheless still elementary. One major limitation is that it relies on the definition of a proper force threshold to decide when the SICM controller should be disabled to retract the tip. This threshold is currently simply defined as a constant condition on the forces that does not take into consideration drifts in the recorded deflection signal and cannot discriminate between the forces due to an obstacle from the forces appearing from the natural bending of the cantilever above a higher feature. A better algorithm would, for example, use the derivative of the forces to define the threshold. An even better strategy would be to create a double controller that uses both the SICM and the AFM controllers to position the probe. Such a combination has been attempted as a weighted summation of the two controllers' outputs (Hybridin = a * AFMout + b * SICMout) and is presented in Ossola et al. [57], but instability issues quickly limited the obtaining of reproducible and comprehensive results. Future work should therefore focus on a better system modeling and controller design to implement the promising envisioned control strategy. Other ideas of improvement of the SICM-FluidFM setup would aim for better probe design that would enhance the resolution achievable with the FluidFM. With an enhanced resolution, the FluidFM, already able to achieve force-controlled patch clamp, could potentially also achieve the smart patch-clamp technique [67] consisting of a SICM pre-scan offering a high-resolution topography of the sample to position with nanometer precision the probe for patch clamp in a region of interest.
15.7 Molecular Analyses
The investigation of a cell at the molecular level, for example, nucleic acids, proteins, or metabolites, provides the most detailed and relevant information on cellular state and function. While a cell genotype is more or less stable, its transcriptome, proteome, and metabolome that reflect the cell function are highly dynamic, changing in the time scale of hours to milliseconds. In addition, the molecular content of each cell at one time point is unique, influenced by the random rate of a multitude of biochemical reactions and the specific changes in the microenvironment. Achieving single-cell resolution when performing molecular analyses is thus of tremendous interest in biology. However, the analysis of individual cells at the molecular level is highly challenging. Current approaches first isolate cells before lysing them to access the endogenous molecules. As a result, cells are removed from their physiological context and analyzed postmortem, and the process under investigation may be strongly influenced by the manipulations. In addition, while gene and transcript analyses benefit from the possibility to amplify nucleic acids to reach detectable signals, single-cell protein and metabolite monitoring is challenged by non-amplifiable, minute amount and a broad variety of analytes. Consequently, current investigations mostly focus on the cell genome or transcriptome, although the proteome and metabolome may arguably better reflect a cell phenotype.
A single-cell extraction approach was established using the FluidFM, which allows for the nondestructive sampling of soluble intracellular molecules and their dispensing on diverse substrates for analysis [9]. To extract the intracellular molecules, a pyramidal tip with a side aperture (400 nm) was inserted inside the nucleus or the cytoplasm of a selected cell by force spectroscopy, and negative pressure (>100 kPa) was applied to flow the cellular fluid into the probe. The cantilever was prefilled with oil to ensure the confinement of cellular extract within the channel, which also enabled the real-time monitoring of the extraction by optical microscopy, with the aqueous extract well distinguishable within the cantilever. Extraction was interrupted when desired by stopping the suction, before lifting the tip out of the cell. The known cantilever geometry enabled to quantify the volumes extracted with 0.1 pL resolution. Soluble molecules were retrieved selectively from either the cytoplasm or the nucleus, with the 400 nm aperture acting as a molecular sieve. Examination of the cell viability post-extraction showed the ability of most cells to survive the removal of nearly their entire cytoplasmic volume, whereas nucleoplasm removal was more critical. The picoliter samples of cytoplasm or nucleoplasm collected in the cantilever were then dispensed to different analytical substrates for analysis. Negative-stain electron microscopy imaging revealed the differential content of soluble molecules from the cytoplasm and the nucleus. Next, the activity of different endogenous enzymes was measured in picoliter wells using fluorogenic substrates. The developed enzymatic assays enabled to distinguish between transfected and non-transfected cells and to detect the activation of apoptosis upon chemical treatment, as well as its inhibition by viruses during infection. Targeted transcript analysis further demonstrated the integrity of the retrieved mRNAs and the potential for differential analysis of cytoplasmic and nuclear transcripts from a single cell. Altogether, the approach made it possible to sample soluble molecules from a single cell quantitatively and in a nondestructive fashion. The extraction was selective for the addressed cell compartment, collecting all the soluble molecules smaller than the probe aperture without compromising their structural and functional integrity.
15.8 Conclusion and Future Perspectives
In the past years, the FluidFM technology has been developed into a versatile and powerful tool for single-cell studies. The force monitoring coupled to precise vertical displacement of the probe ensures gentle and precise interaction with the manipulated cells, whereas the tight pressure regulation in the microchannel enables the handling of virtually any solution with femtoliter resolution. A variety of methods were established, which make it possible to perturb selected cells by physical manipulation or through the controlled delivery of bioactive compounds inside the cell or in its surroundings. The technology further gives the power to analyze the mechanical, electrical, and even molecular properties of single cells. In addition, all these manipulations can be performed directly within standard biological samples under physiological conditions, with a minimal impact on cell physiology. The technology has already moved beyond method development, with the first biological studies succeeding in, for instance, identifying a subset of bacteria isolated from a complex phyllosphere community or revealing new details in the early stages of a viral infection. In the future, a broad diversity of biological studies will be addressed, whereby the multifunctionality of the technology will prove helpful to adapt to specific requirements of the biological system under investigation. The different cell manipulations will be combined to assess natural changes in cell physiology and function, as well as cell responses to well-defined physical or chemical perturbations. In parallel, additional efforts will be invested for automation of the different procedures that have been explored, which will represent a major advancement for higher throughput.
References
- 1 Meister, A., Gabi, M., Behr, P., Studer, P., Voros, J., Niedermann, P. et al. (2009) FluidFM: combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett., 9, 2501–2507.
- 2 Dorig, P., Stiefel, P., Behr, P., Sarajlic, E., Bijl, D., Gabi, M. et al. (2010) Force-controlled spatial manipulation of viable mammalian cells and micro-organisms by means of FluidFM technology. Appl. Phys. Lett., 97, 023701.
- 3 Guillaume-Gentil, O., Potthoff, E., Ossola, D., Dorig, P., Zambelli, T., and Vorholt, J.A. (2013) Force-controlled fluidic injection into single cell nuclei. Small, 9, 1904–1907.
- 4 Potthoff, E., Guillaume-Gentil, O., Ossola, D., Polesel-Maris, J., LeibundGut-Landmann, S., Zambelli, T. et al. (2012) Rapid and serial quantification of adhesion forces of yeast and Mammalian cells. PLoS One, 7, e52712.
- 5 Potthoff, E., Ossola, D., Zambelli, T., and Vorholt, J.A. (2015) Bacterial adhesion force quantification by fluidic force microscopy. Nanoscale, 7, 4070–4079.
- 6 Stiefel, P., Zambelli, T., and Vorholt, J.A. (2013) Isolation of optically targeted single bacteria by application of fluidic force microscopy to aerobic anoxygenic phototrophs from the phyllosphere. Appl. Environ. Microbiol., 79, 4895–4905.
- 7 Guillaume-Gentil, O., Zambelli, T., and Vorholt, J.A. (2014) Isolation of single mammalian cells from adherent cultures by fluidic force microscopy. Lab Chip, 14, 402–414.
- 8 Potthoff, E., Franco, D., D'Alessandro, V., Starck, C., Falk, V., Zambelli, T. et al. (2014) Toward a rational design of surface textures promoting endothelialization. Nano Lett., 14, 1069–1079.
- 9 Guillaume-Gentil, O., Grindberg, R.V., Kooger, R., Dorwling-Carter, L., Martinez, V., Ossola, D. et al. (2016) Tunable single-cell extraction for molecular analyses. Cell, 166 (2), 506–516.
- 10 Meister, A., Polesel-Maris, J., Niedermann, P., Przybylska, J., Studer, P., Gabi, M. et al. (2009) Nanoscale dispensing in liquid environment of streptavidin on a biotin-functionalized surface using hollow atomic force microscopy probes. Microelectron. Eng., 86, 1481–1484.
- 11 Dermutz, H., Gruter, R.R., Truong, A.M., Demko, L., Voros, J., and Zambelli, T. (2014) Local polymer replacement for neuron patterning and in situ neurite guidance. Langmuir, 30, 7037–7046.
- 12 Gon, S., Fang, B., and Santore, M.M. (2011) Interaction of cationic proteins and polypeptides with biocompatible cationically-anchored PEG brushes. Macromolecules, 44, 8161–8168.
- 13 Martinez, V., Forro, C., Weydert, S., Aebersold, M.J., Dermutz, H., Guillaume-Gentil, O. et al. (2016) Controlled single-cell deposition and patterning by highly flexible hollow cantilevers. Lab Chip, 16 (9), 1663–1674.
- 14 Stiefel, P., Schmidt, F.I., Dorig, P., Behr, P., Zambelli, T., Vorholt, J.A. et al. (2012) Cooperative vaccinia infection demonstrated at the single-cell level using FluidFM. Nano Lett., 12, 4219–4227.
- 15 Helenius, J., Heisenberg, C.P., Gaub, H.E., and Muller, D.J. (2008) Single-cell force spectroscopy. J. Cell Sci., 121, 1785–1791.
- 16 Guillaume-Gentil, O., Potthoff, E., Ossola, D., Franz, C.M., Zambelli, T., and Vorholt, J.A. (2014) Force-controlled manipulation of single cells: from AFM to FluidFM. Trends Biotechnol., 32, 381–388.
- 17 Tuson, H.H., Auer, G.K., Renner, L.D., Hasebe, M., Tropini, C., Salick, M. et al. (2012) Measuring the stiffness of bacterial cells from growth rates in hydrogels of tunable elasticity. Mol. Microbiol., 84, 874–891.
- 18 Haga, H., Sasaki, S., Kawabata, K., Ito, E., Ushiki, T., and Sambongi, T. (2000) Elasticity mapping of living fibroblasts by AFM and immunofluorescence observation of the cytoskeleton. Ultramicroscopy, 82, 253–258.
- 19 Schulze, C., Muller, K., Kas, J.A., and Gerdelmann, J.C. (2009) Compaction of cell shape occurs before decrease of elasticity in CHO-K1 cells treated with actin cytoskeleton disrupting drug cytochalasin D. Cell Motil. Cytoskeleton, 66, 193–201.
- 20 Houchmandzadeh, B. and Dimitrov, S. (1999) Elasticity measurements show the existence of thin rigid cores inside mitotic chromosomes. J. Cell Biol., 145, 215–223.
- 21 Charras, G.T. and Horton, M.A. (2002) Single cell mechanotransduction and its modulation analyzed by atomic force microscope indentation. Biophys. J., 82, 2970–2981.
- 22 Zahn, J.T., Louban, I., Jungbauer, S., Bissinger, M., Kaufmann, D., Kemkemer, R. et al. (2011) Age-dependent changes in microscale stiffness and mechanoresponses of cells. Small, 7, 1480–1487.
- 23 Gonzalez-Cruz, R.D., Fonseca, V.C., and Darling, E.M. (2012) Cellular mechanical properties reflect the differentiation potential of adipose-derived mesenchymal stem cells. Proc. Natl. Acad. Sci. U.S.A., 109, E1523–E1529.
- 24 Faria, E.C., Ma, N., Gazi, E., Gardner, P., Brown, M., Clarke, N.W. et al. (2008) Measurement of elastic properties of prostate cancer cells using AFM. Analyst, 133, 1498–1500.
- 25 Plodinec, M., Loparic, M., Monnier, C.A., Obermann, E.C., Zanetti-Dallenbach, R., Oertle, P. et al. (2012) The nanomechanical signature of breast cancer. Nat. Nanotechnol., 7, 757–765.
- 26 Wagh, A.A., Roan, E., Chapman, K.E., Desai, L.P., Rendon, D.A., Eckstein, E.C. et al. (2008) Localized elasticity measured in epithelial cells migrating at a wound edge using atomic force microscopy. Am. J. Physiol. Lung Cell. Mol. Physiol., 295, L54–L60.
- 27 Lieber, S.C., Aubry, N., Pain, J., Diaz, G., Kim, S.J., and Vatner, S.F. (2004) Aging increases stiffness of cardiac myocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol. Heart Circ. Physiol., 287, H645–H651.
- 28 Li, M., Liu, L.Q., Xi, N., and Wang, Y.C. (2015) Nanoscale monitoring of drug actions on cell membrane using atomic force microscopy. Acta Pharmacol. Sin., 36, 769–782.
- 29 McGrath, J.S., Quist, J., Seddon, J.R., Lai, S.C., Lemay, S.G., and Bridle, H.L. (2016) Deformability assessment of waterborne protozoa using a microfluidic-enabled force microscopy probe. PLoS One, 11, e0150438.
- 30 Dorig, P., Ossola, D., Truong, A.M., Graf, M., Stauffer, F., Voros, J. et al. (2013) Exchangeable colloidal AFM probes for the quantification of irreversible and long-term interactions. Biophys. J., 105, 463–472.
- 31 Busscher, H.J. and Weerkamp, A.H. (1987) Specific and non-specific interactions in bacterial adhesion to solid substrata. FEMS Microbiol. Lett., 46, 165–173.
- 32 Hall-Stoodley, L., Costerton, J.W., and Stoodley, P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol., 2, 95–108.
- 33 Benoit, M., Gabriel, D., Gerisch, G., and Gaub, H.E. (2000) Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat. Cell Biol., 2, 313–317.
- 34 Bowen, W.R., Lovitt, R.W., and Wright, C.J. (2001) Atomic force microscopy study of the adhesion of Saccharomyces cerevisiae. J. Colloid Interface Sci., 237, 54–61.
- 35 Wojcikiewicz, E.P., Zhang, X., and Moy, V.T. (2004) Force and compliance measurements on living cells using atomic force microscopy (AFM). Biol. Proced. Online, 6, 1–9.
- 36 Emerson, R.J. 4th Bergstrom, T.S., Liu, Y., Soto, E.R., Brown, C.A., McGimpsey, W.G. et al. (2006) Microscale correlation between surface chemistry, texture, and the adhesive strength of Staphylococcus epidermidis. Langmuir, 22, 11311–11321.
- 37 Bowen, W.R., Fenton, A.S., Lovitt, R.W., and Wright, C.J. (2002) The measurement of Bacillus mycoides spore adhesion using atomic force microscopy, simple counting methods, and a spinning disk technique. Biotechnol. Bioeng., 79, 170–179.
- 38 Le, D.T., Guerardel, Y., Loubiere, P., Mercier-Bonin, M., and Dague, E. (2011) Measuring kinetic dissociation/association constants between Lactococcus lactis bacteria and mucins using living cell probes. Biophys. J., 101, 2843–2853.
- 39 Canale, C., Petrelli, A., Salerno, M., Diaspro, A., and Dante, S. (2013) A new quantitative experimental approach to investigate single cell adhesion on multifunctional substrates. Biosens. Bioelectron., 48, 172–179.
- 40 Panorchan, P., Thompson, M.S., Davis, K.J., Tseng, Y., Konstantopoulos, K., and Wirtz, D. (2006) Single-molecule analysis of cadherin-mediated cell–cell adhesion. J. Cell Sci., 119, 66–74.
- 41 Dobrowsky, T.M., Panorchan, P., Konstantopoulos, K., and Wirtz, D. (2008) Live-cell single-molecule force spectroscopy. Methods Cell Biol., 89, 411–432.
- 42 Kang, S. and Elimelech, M. (2009) Bioinspired single bacterial cell force spectroscopy. Langmuir, 25, 9656–9659.
- 43 Beaussart, A., El-Kirat-Chatel, S., Herman, P., Alsteens, D., Mahillon, J., Hols, P. et al. (2013) Single-cell force spectroscopy of probiotic bacteria. Biophys. J., 104, 1886–1892.
- 44 Friedrichs, J., Helenius, J., and Muller, D.J. (2010) Stimulated single-cell force spectroscopy to quantify cell adhesion receptor crosstalk. Proteomics, 10, 1455–1462.
- 45 Alsteens, D., Beaussart, A., Derclaye, S., El-Kirat-Chatel, S., Park, H.R., Lipke, P.N. et al. (2013) Single-cell force spectroscopy of Als-mediated fungal adhesion. Anal. Methods, 5, 3657–3662.
- 46 Taubenberger, A., Cisneros, D.A., Friedrichs, J., Puech, P.H., Muller, D.J., and Franz, C.M. (2007) Revealing early steps of α2β1 integrin-mediated adhesion to collagen type I by using single-cell force spectroscopy. Mol. Biol. Cell, 18, 1634–1644.
- 47 Selhuber-Unkel, C., Lopez-Garcia, M., Kessler, H., and Spatz, J.P. (2008) Cooperativity in adhesion cluster formation during initial cell adhesion. Biophys. J., 95, 5424–5431.
- 48 Thwala, J.M., Li, M., Wong, M.C., Kang, S., Hoek, E.M., and Mamba, B.B. (2013) Bacteria–polymeric membrane interactions: atomic force microscopy and XDLVO predictions. Langmuir, 29, 13773–13782.
- 49 Gigler, A., Holzwarth, M., and Marti, O. (2007) Local nanomechanical properties of HeLa-cell surfaces. Proc. Int. Conf. Nanosci. Technol., 61, 780–784.
- 50 Hosseini, B.H., Louban, I., Djandji, D., Wabnitz, G.H., Deeg, J., Bulbuc, N. et al. (2009) Immune synapse formation determines interaction forces between T cells and antigen-presenting cells measured by atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A., 106, 17852–17857.
- 51 Jaatinen, L., Young, E., Hyttinen, J., Voros, J., Zambelli, T., and Demko, L. (2016) Quantifying the effect of electric current on cell adhesion studied by single-cell force spectroscopy. Biointerphases, 11, 011004.
- 52 Wang, Y., Narain, R., and Liu, Y. (2014) Study of bacterial adhesion on different glycopolymer surfaces by quartz crystal microbalance with dissipation. Langmuir, 30, 7377–7387.
- 53 Xu, H., Murdaugh, A.E., Chen, W., Aidala, K.E., Ferguson, M.A., Spain, E.M. et al. (2013) Characterizing pilus-mediated adhesion of biofilm-forming E. coli to chemically diverse surfaces using atomic force microscopy. Langmuir, 29, 3000–3011.
- 54 Neher, E. and Sakmann, B. (1976) Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature, 260, 799–802.
- 55 Ossola, D., Amarouch, M.Y., Behr, P., Voros, J., Abriel, H., and Zambelli, T. (2015) Force-controlled patch clamp of beating cardiac cells. Nano Lett., 15, 1743–1750.
- 56 Hansma, P.K., Drake, B., Marti, O., Gould, S.A.C., and Prater, C.B. (1989) The scanning ion-conductance microscope. Science, 243, 641–643.
- 57 Ossola, D., Dorwling-Carter, L., Dermutz, H., Behr, P., Voros, J., and Zambelli, T. (2015) Simultaneous scanning ion conductance microscopy and atomic force microscopy with microchanneled cantilevers. Phys. Rev. Lett., 115, 238103.
- 58 Dunlop, J., Bowlby, M., Peri, R., Vasilyev, D., and Arias, R. (2008) High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov., 7, 358–368.
- 59 Martinac, B. (2012) Mechanosensitive ion channels: an evolutionary and scientific tour de force in mechanobiology. Channels, 6, 211–213.
- 60 Poole, K., Moroni, M., and Lewin, G.R. (2015) Sensory mechanotransduction at membrane-matrix interfaces. Pflugers Arch., 467, 121–132.
- 61 Sherman, A.J., Shrier, A., and Cooper, E. (1999) Series resistance compensation for whole-cell patch-clamp studies using a membrane state estimator. Biophys. J., 77, 2590–2601.
- 62 Shevchuk, A.I., Frolenkov, G.I., Sanchez, D., James, P.S., Freedman, N., Lab, M.J. et al. (2006) Imaging proteins in membranes of living cells by high-resolution scanning ion conductance microscopy. Angew. Chem. Int. Ed., 45, 2212–2216.
- 63 Korchev, Y.E., Milovanovic, M., Bashford, C.L., Bennett, D.C., Sviderskaya, E.V., Vodyanoy, I. et al. (1997) Specialized scanning ion-conductance microscope for imaging of living cells. J. Microsc., 188, 17–23.
- 64 Novak, P., Li, C., Shevchuk, A.I., Stepanyan, R., Caldwell, M., Hughes, S. et al. (2009) Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat. Methods, 6, 279–281.
- 65 Pastre, D., Iwamoto, H., Liu, J., Szabo, G., and Shao, Z. (2001) Characterization of AC mode scanning ion-conductance microscopy. Ultramicroscopy, 90, 13–19.
- 66 Rheinlaender, J. and Schäffer, T.E. (2009) Image formation, resolution, and height measurement in scanning ion conductance microscopy. J. Appl. Phys., 105, 094905.
- 67 Gorelik, J., Gu, Y., Spohr, H.A., Shevchuk, A.I., Lab, M.J., Harding, S.E. et al. (2002) Ion channels in small cells and subcellular structures can be studied with a smart patch-clamp system. Biophys. J., 83, 3296–3303.