
Chapter 5
Analysis and Prediction of Protein Posttranslational Modification Sites
5.1 Introduction
Protein has a wide range of essential functions in living cells, including building and repair of body tissues (structural or storage proteins), catalysis of biochemical reactions (enzymes), regulation of growth and metabolism (hormones), water balancing (membrane proteins), and nutrient transport (transporters or carriers). A protein synthesized immediately after translation from mRNA is typically called an immature protein, which is often not fully functional. In order to carry out specific functions in cells, new proteins usually undergo a process called posttranslational modification (PTM). PTM plays key roles in many cellular processes such as signaling, cellular differentiation, protein degradation, protein stability, gene expression regulation, protein function regulation, and protein interactions [1]. Irregular PTM activity is often a cause or consequence of many diseases such as cancer [2].
It has been estimated that there are more than 200 types of PTMs mediated by enzymes consisting of 5% of the proteome [3]. Many of these enzymes, such as transferases (e.g., kinases and phosphatases) and ligases, add or remove various types of chemical groups such as phosphate, acyl group, lipid, glycans, or even peptides covalently at amino acid sidechains. Many of these processes are reversible and can thereby change the protein confirmation back and forth or transduce the signal molecules. Other enzymes, such as proteases, perform proteolytic cleavage at specific points on protein backbones, which are involved in the posttranslational processes including terminal methionine removal, signal peptide cleavage, or zymogen activation. In this chapter, we focus on a few common covalent PTMs, listed in Table 5.1.
Table 5.1 PTMs Studied in This Chapter
| PTM | Modified | |
| Type | Amino Acid | Function |
| Phosphorylation | Phosphoserine Phosphothreonine Phosphotyrosine | Signaling pathway, protein degradation, cell cycle, growth, apoptosis |
| Glycosylation | O-GlcNAc-serine O-GlcNac-threonine | Protein folding, protein stability, protein activity |
| Methylation | N6-methyllysine Omega-N-methylarginine | Regulation of gene expression, protein stability |
| Acetylation | N6-acytyl-lysine | Regulation of gene expression, cellular localization |
| Palmitoylation | S-palmitoylcysteine | Cellular localization |
| SUMOylation | SUMOylatedlysine | Cellular localization, apoptosis |
| Sulfation | Sulfotyrosine | Protein–protein interaction |
More recently, proteomics data of various PTM sites are accumulating rapidly, thanks to high-throughput mass spectrometry studies [4–13] and associated web resources (Table 5.2). In particular, protein phosphorylation has been extensively characterized and annotated, especially in humans. Nevertheless, our knowledge of protein PTM is still limited. As a result, computational prediction of PTM sites is also becoming an increasingly active research area. Table 5.3 lists PTM site prediction tools.
Table 5.2 Summary of PTM Databases
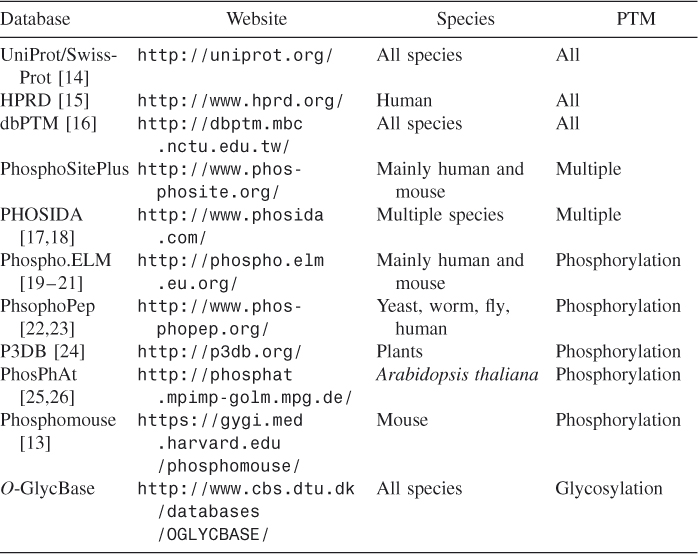
Table 5.3 Summary of PTM Site Prediction Tools
We developed a standalone bioinformatics tool, Musite, specifically designed for large-scale prediction of PTM sites. PTM site prediction was modeled as an unbalanced binary classification problem. High-quality proteomics data in multiple organisms were collected from several sources, listed in Table 5.2. Three sets of features [k-nearest-neighbor (KNN) scores, disorder scores, and amino acid frequencies] were extracted from the collected data. The extracted features were then combined using a support vector machine (SVM) to train prediction models for the PTMs in Table 5.1 by a comprehensive machine learning method called bootstrap aggregation. Cross-validation tests showed that our bootstrap aggregating method performed well. Besides the standalone version of Musite (http://musite.sourceforge.net/), a web server version named Musite.net was also implemented (http://musite.net).
5.2 Musite: A Machine Learning Approach
Posttranslational modification site prediction is a binary classification problem. For example, considering phosphoserine prediction, serine residues can be classified into two categories: phosphoserines or positive data and non-phosphoserines (serines that cannot be phosphorylated) or negative data. Note that our aim is to predict whether a residue can be modified by a specific PTM regardless of cell state or biological condition. Musite framework was designed for PTM site prediction in three processes: (1) data collection and preparation, (2) feature extraction, and (3) classifier/prediction model training and evaluation.
5.2.1 Data Collection
We collected high-quality experimentally verified data from several resources in Table 5.2. The numbers of sites for each PTM that we collected are listed in Table 5.5.
Table 5.5 Numbers of PTM Sites Collected
| Modified | Number of | |
| PTM | Residue Type | Modified Residues |
| Phosphorylation | Phosphoserine | 65,822 |
| Phosphothreonine | 14,478 | |
| Phosphotyrosine | 5,725 | |
| Acetylation | N6-acetyllysine | 4,121 |
| Methylation | N6-methyllysine | 230 |
| Omega-N-methylarginine | 60 | |
| Sulfation | Sulfotyrosine | 193 |
| SUMOylation | SUMOylated lysine | 299 |
| Palmitoylation | S-palmitoylcysteine | 212 |
| GlcNAcation | O-GlcNAc-serine | 137 |
| O-GlcNAc-threonine | 91 |
5.2.2 Feature Extraction
Feature selection and extraction is an important step in identifing patterns that characterize the commonalities within each class and differences between classes. Musite extracts three sets of features: k-nearest-neighbor (KNN) scores, protein disorder scores, and amino acid frequencies.
5.2.2.1 K-Nearest-Neighbor (KNN) Scores
KNN scores were extracted as follows to measure the local sequence similarity between query sites and known PTM sites:
5.2.2.2 Predicted Protein Disorder Scores
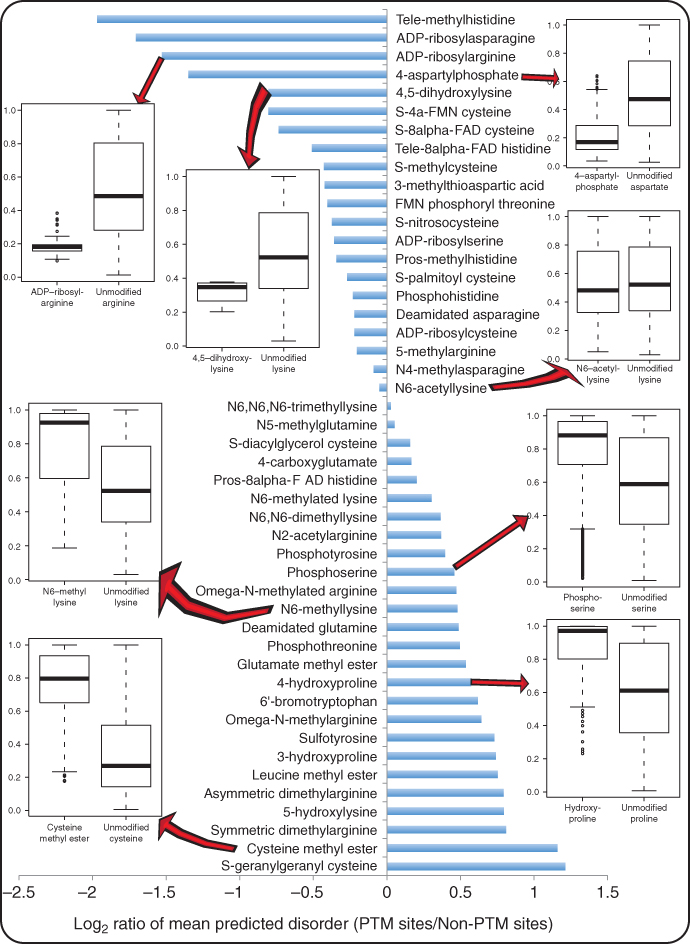
Protein disorder has been shown to be correlated with PTMs [39] [69]. The correlations between various PTMs and protein disorders are summarized in Figure 5.1 according to results that we previously reported [69]. Predicted protein disorder scores were extracted as features by the following procedure:
Figure 5.1 Correlation between PTM and protein disorder. The horizontal axis (abscissa) represents the log ratio of mean predicted disorder score of PTM sites versus that of non-PTM sites. A value below 0 means that the corresponding PTM is enriched in structure regions; otherwise, it is enriched in disordered regions. The vertical axis (ordinate) from top to bottom represents PTMs sorted from being most significantly enriched in structure regions to being most significantly enriched in disordered regions. Boxplots of predicted disorder scores of a few representative types of PTM sites and their non-PTM counterparts are also shown.
5.2.2.3 Local Amino Acid Frequency
It has been observed that amino acids around PTM sites have different compositions. For example, around phosphorylation sites, rigid, buried, neutral amino acids (W, C, F, I, Y, V, L) were significantly depleted, while flexible, surface-exposed amino acids (S, P, E, K) were significantly enriched [39]. We extracted amino acid frequencies around candidate sites with window size of 13 (6 residues from each side of a candidate site) as features.
5.2.3 Classifier Training
Using the extracted features, we trained classifies by a machine learning ensemble meta-algorithm called bootstrap aggregating or bagging [71] as follows:
5.2.4 Evaluation
To evaluate Musite, cross-validation tests were performed. Receiver operating characteristic (ROC) curves were calculated and plotted based on specificities [Eq. (5.1)] and sensitivities [Eq. (5.2)] by taking different thresholds:
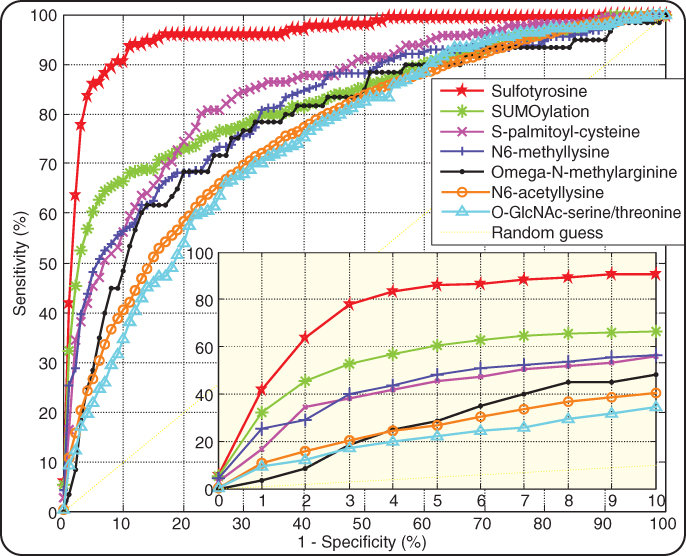
The results of cross-validation tests for phosphorylation site prediction were reported earlier [33]. For each of the other PTMs, we performed fivefold cross-validation tests according to the following procedure. The positive data were first divided into five groups. Each group was then combined with the same number of randomly selected negative data points, forming a sub-dataset. Each time, one of the five sub-datasets was retained as the validation data. All the remaining positive and negative data were used to train a prediction model. The validation data were then submitted to the trained model for prediction. The cross-validation process was repeated 5 times with each sub-dataset used exactly once as the validation data. Sensitivities at different specificity levels in each cross-validation run were calculated according to Equations (5.1) and (5.2). Figure 5.2 shows the receiver operating characteristic (ROC) curve by averaging the sensitivities at different specificities over five cross-validations.
Figure 5.2 ROC curves of Musite predictions of PTM sites. A curve for each PTM represents the average sensitivities and specificities for difference thresholds over five cross-validation runs. The bottom right figure is the zoomed-in region with high prediction specificities (0.9–1).
5.3 Musite Implementation
5.3.1 Core Modules of Musite Open-Source Framework
With its GNU GPL open-source license and extensible API, Musite provides an open framework for PTM site prediction applications. The Musite framework contains six core modules (more details on these modules are described in an earlier work [34]):
5.3.2 Musite.net: A Web Server
Musite.net is a web implementation and extension of Musite standalone application. Musite.net provides a uniform interface for submitting query sequences and analyzing prediction results for different PTMs in various species. Besides phosphorylation, it currently supports prediction of acetylation, methylation, sulfation, SUMOylation, palmitoylation, and GlcNAcation. As PTM data are accumulating rapidly, we will continue to update the prediction models and build new models for more PTMs and species.
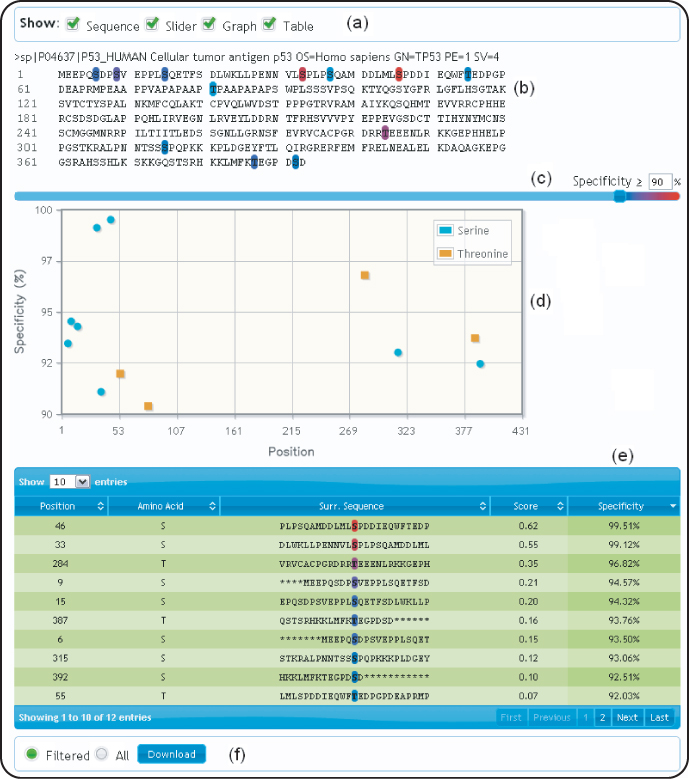
On Musite.net, a prediction request can be submitted using a web form. One can select a prediction model according to PTM types, organisms, or kinase types (for phosphorylation site prediction), and submit either sequences or accession numbers of the query proteins for prediction. The prediction result is presented in an interactive output page as illustrated in Figure 5.3. One can turn on or off different representations of the prediction result: the protein sequence with highlighted predicted sites, a plot of specificity levels against residue positions, and a table listing predicted sites. Using a slider, one can adjust the significance threshold to any specificity level. If multiple sequences are submitted, the prediction result for each individual sequence can be expanded or shrunk to help the user focus on predictions of interest. The prediction result can be exported to a tab-delimited text file for further analysis.
Figure 5.3 Example of a prediction result page; as an example, the query protein with UniProt accession P04637 was submitted for prediction of general phosphoserines/threonines. (a) different presentations of the prediction result can be selected (by default, sequence and slider are selected); (b) the query sequences with predicted PTM sites are highlighted in different colors on the basis of prediction confidence; (c) A slider is provided to control the stringency threshold (any threshold of specificity between 0% and 100% can be set); (d) a plot of specificities against residue positions is made available; (e) a table of predicted PTM sites is presented; (f) the prediction result can be downloaded as a tab-delimited text file.
Musite.net can also be accessed by direct URLs (e.g., http://musite.net/ ?acc=uniprot:P04637&model=Phosphorylation.H.sapiens.general .tyr). Similar to the web form submission, one should select the model and provide either the accession number or sequence of the query protein. Direct URLs enable other websites to hyperlink to Musite. For instance, P3DB [24] has linked to Musite in its phosphoprotein pages, to enable users who are interested in predicted phosphorylation sites to easily navigate from experimental phosphorylation data in P3DB to predicted results at Musite.net.
Musite.net also provides a RESTful web service (http://musite.net/ service) that can be accessed programmatically. Bioinformatics developers can utilize this web service to integrate the PTM site prediction capacity of Musite.net into their own bioinformatics applications.
5.4 Summary
By taking advantage of the large magnitude of experimentally verified PTM sites and utilizing a comprehensive machine learning method, Musite provides a useful bioinformatics software system for PTM site prediction. Musite has a few unique functionalities, including customized model training from users' own data and continuous adjustment of stringency levels of prediction results. Musite was also the first open-source project intended for PTM site prediction, making it very easy to add new features or explore new machine learning methods for PTM prediction. We provided standalone and web server versions of Musite, both with intuitive graphical user interface (GUI) to access the prediction models that we trained. The standalone version can be used for up to proteome-wide PTM site prediction in an automated fashion, while the web server is meant to provide quick access of PTM site prediction for light jobs. With the Musite project, we determined to build a platform for community use that enables both computational and experimental biologists to obtain a better prediction and understanding of protein PTMs.
Acknowledgments
We would like to thank Dr. Jay J. Thelen and Dr. A. Keith Dunker for helpful discussions. This work was supported by funding from the National Institute of Health (Grants R21/R33 GM078601 and R01 GM100701) and National Science Foundation (Grant DBI-0604439)
References
1. Seo J, Lee KJ, Post-translational modifications and their biological functions: Proteomic analysis and systematic approaches, J. Biochem. Mol. Biol. 37(1):35–44 (2004).
2. Krueger KE, Srivastava S, Posttranslational protein modifications: Current implications for cancer detection, prevention, and therapeutics, Mol. Cell. Proteomics 5(10):1799–1810 (2006).
3. Walsh C, Posttranslational Modification of Proteins: Expanding Nature's Inventory, Roberts Publishers, 2006.
4. Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP, Large-scale characterization of HeLa cell nuclear phosphoproteins, Proc. Natl. Acad. Sci. USA 101(33):12130–12135 (2004).
5. Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M, Global, in vivo, and site-specific phosphorylation dynamics in signaling networks, Cell 127(3):635–648 (2006).
6. Villen J, Beausoleil SA, Gerber SA, Gygi SP, Large-scale phosphorylation analysis of mouse liver, Proc. Natl. Acad. Sci. USA 104(5):1488–1493 (2007).
7. Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF, Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry, Proc. Natl. Acad. Sci. USA 104(7):2193–2198 (2007).
8. Munton RP, Tweedie-Cullen R, Livingstone-Zatchej M, Weinandy F, Waidelich M, Longo D, Gehrig P, Potthast F, Rutishauser D, Gerrits B, Panse C, Schlapbach R, Mansuy IM, Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations, Mol. Cell. Proteomics 6(2):283–293 (2007).
9. Sugiyama N, Nakagami H, Mochida K, Daudi A, Tomita M, Shirasu K, Ishihama Y, Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis, Mol. Syst. Biol. 4:193 (2008).
10. Zhai B, Villen J, Beausoleil SA, Mintseris J, Gygi SP, Phosphoproteome analysis of Drosophila melanogaster embryos, J. Proteome Res. 7(4):1675–1682 (2008).
11. Boersema PJ, Foong LY, Ding VM, Lemeer S, van Breukelen B, Philp R, Boekhorst J, Snel B, den Hertog J, Choo AB, Heck AJ, In depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immuno-affinity purification and stable isotope dimethyl labeling, Mol. Cell. Proteomics: 9(1):84–89 (2010).
12. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M, Lysine acetylation targets protein complexes and co-regulates major cellular functions, Science 325(5942):834–840 (2009).
13. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP, A tissue-specific atlas of mouse protein phosphorylation and expression, Cell 143(7):1174–1189 (2010).
14. Farriol-Mathis N, Garavelli JS, Boeckmann B, Duvaud S, Gasteiger E, Gateau A, Veuthey AL, Bairoch A, Annotation of post-translational modifications in the Swiss-Prot knowledge base, Proteomics 4(6):1537–1550 (2004).
15. Prasad TS, Kandasamy K, Pandey A, Human protein reference database and human proteinpedia as discovery tools for systems biology, Methods Mol. Biol. 577:67–79 (2009).
16. Lee TY, Huang HD, Hung JH, Huang HY, Yang YS, Wang TH, dbPTM: an information repository of protein post-translational modification, Nucleic Acids Res. 34(database issue):D622–D627 (2006).
17. Gnad F, Gunawardena J, Mann M, PHOSIDA 2011: The posttranslational modification database, Nucleic Acids Res. 39(database issue):D253–D260 (2011).
18. Gnad F, Ren S, Cox J, Olsen JV, Macek B, Oroshi M, Mann M, PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites, Genome Biol. 8(11):R250 (2007).
19. Diella F, Cameron S, Gemund C, Linding R, Via A, Kuster B, Sicheritz-Ponten T, Blom N, Gibson TJ, Phospho.ELM: A database of experimentally verified phosphorylation sites in eukaryotic proteins, BMC Bioinformatics 5:79 (2004).
20. Diella F, Gould CM, Chica C, Via A, Gibson TJ, Phospho.ELM: A database of phosphorylation sites—update 2008, Nucleic Acids Res. 36(database issue):D240–D244 (2008).
21. Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F, Phospho.ELM: A database of phosphorylation sites—update 2011, Nucleic Acids Res. 39(database issue):D261–D267 (2011).
22. Bodenmiller B, Campbell D, Gerrits B, Lam H, Jovanovic M, Picotti P, Schlapbach R, Aebersold R, PhosphoPep—a database of protein phosphorylation sites in model organisms, Nat. Biotechnol. 26(12):1339–1340 (2008).
23. Bodenmiller B, Malmstrom J, Gerrits B, Campbell D, Lam H, Schmidt A, Rinner O, Mueller LN, Shannon PT, Pedrioli PG, Panse C, Lee HK, Schlapbach R, Aebersold R, PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells, Mol. Syst. Biol. 3:139 (2007).
24. Gao J, Agrawal GK, Thelen JJ, Xu D, P3DB: A plant protein phosphorylation database, Nucleic Acids Res. 37(database issue):D960–D962 (2009).
25. Durek P, Schmidt R, Heazlewood JL, Jones A, MacLean D, Nagel A, Kersten B, Schulze WX, PhosPhAt: The Arabidopsis thaliana phosphorylation site database. An update, Nucleic Acids Res. 38(database issue):D828–D834 (2010).
26. Heazlewood JL, Durek P, Hummel J, Selbig J, Weckwerth W, Walther D, Schulze WX, PhosPhAt: A database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor, Nucleic Acids Res. 36(database issue):D1015–D1021 (2008).
27. Plewczynski D, Tkacz A, Wyrwicz LS, Rychlewski L, AutoMotif server: Prediction of single residue post-translational modifications in proteins, Bioinformatics 21(10):2525–2527 (2005).
28. Plewczynski D, Tkacz A, Wyrwicz LS, Rychlewski L, Ginalski K, AutoMotif server for prediction of phosphorylation sites in proteins using support vector machine: 2007 update, J. Mol. Model. 14(1):69–76 (2008).
29. Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DM, Ausiello G, Brannetti B, Costantini A, Ferre F, Maselli V, Via A, Cesareni G, Diella F, Superti-Furga G, Wyrwicz L, et al, ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins, Nucleic Acids Res. 31(13):3625–3630 (2003).
30. Balla S, Thapar V, Verma S, Luong T, Faghri T, Huang CH, Rajasekaran S, del Campo JJ, Shinn JH, Mohler WA, Maciejewski MW, Gryk MR, Piccirillo B, Schiller SR, Schiller MR, Minimotif Miner: A tool for investigating protein function, Nat. Methods 3(3):175–177 (2006).
31. Rajasekaran S, Balla S, Gradie P, Gryk MR, Kadaveru K, Kundeti V, Maciejewski MW, Mi T, Rubino N, Vyas J, Schiller MR, Minimotif miner 2nd release: A database and web system for motif search, Nucleic Acids Res. 37(database issue):D185–D190 (2009).
32. Schiller MR, Minimotif miner: A computational tool to investigate protein function, disease, and genetic diversity, Current Protocols in Protein Science, 2007, Unit 2.12, Chap. 2.
33. Gao J, Thelen JJ, Dunker AK, Xu D, Musite, a tool for global prediction of general and kinase-specific phosphorylation sites, Mol. Cell. Proteomics 9(12):2586–2600 2010.
34. Gao J, Xu D, The Musite open-source framework for phosphorylation-site prediction, BMC Bioinformatics 11(Suppl 12):S9 2010.
35. de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N, ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins, Nucleic Acids Res. 34(web server issue):W362–W365 (2006).
36. Gattiker A, Gasteiger E, Bairoch A, ScanProsite: A reference implementation of a PROSITE scanning tool, Appl. Bioinformatics 1(2):107–108 (2002).
37. Obenauer JC, Cantley LC, Yaffe MB, Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs, Nucleic Acids Res. 31(13):3635–3641 (2003).
38. Schwartz D, Chou MF, Church GM, Predicting protein post-translational modifications using meta-analysis of proteome scale data sets, Mol. Cell. Proteomics 8(2):365–379 (2009).
39. Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK, The importance of intrinsic disorder for protein phosphorylation, Nucleic Acids Res. 32(3):1037-1049 (2004).
40. Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X, GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy, Mol. Cell. Proteomics 7(9):1598–1608 (2008).
41. Blom N, Gammeltoft S, Brunak S, Sequence and structure-based prediction of eukaryotic protein phosphorylation sites, J. Mol. Biol. 294(5):1351–1362 (1999).
42. Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S, Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence, Proteomics 4(6):1633–1649 (2004).
43. Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li, S, Cesareni G, Pawson T, et al, Linear motif atlas for phosphorylation-dependent signaling, Sci. Signal. 1(35):ra2 (2008).
44. Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK, KinasePhos 2.0: A web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns, Nucleic Acids Res. 35(web server issue):W588–W594 (2007).
45. Gupta R, Jung E, Gooley AA, Williams KL, Brunak S, Hansen J, Scanning the available Dictyostelium discoideum proteome for O-linked GlcNAc glycosylation sites using neural networks, Glycobiology 9(10):1009–1022 (1999).
46. Caragea C, Sinapov J, Silvescu A, Dobbs D, Honavar V, Glycosylation site prediction using ensembles of support vector machine classifiers, BMC Bioinformatics 8:438 (2007).
47. Hamby SE, Hirst JD, Prediction of glycosylation sites using random forests, BMC Bioinformatics 9:500 (2008).
48. Julenius K, NetCGlyc 1.0: Prediction of mammalian C-mannosylation sites, Glycobiology 17(8):868–876 (2007).
49. Johansen MB, Kiemer L, Brunak S, Analysis and prediction of mammalian protein glycation, Glycobiology 16(9):844–853 (2006).
50. Julenius K, Molgaard A, Gupta R, Brunak S, Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites, Glycobiology 15(2):153–164 (2005).
51. Li S, Liu B, Zeng R, Cai Y, Li Y, Predicting O-glycosylation sites in mammalian proteins by using SVMs, Comput. Biol. Chem. 30(3):203–208 (2006).
52. Gupta R, Brunak S, Prediction of glycosylation across the human proteome and the correlation to protein function, Proc. Pacific Symp. Biocomputing 2002, pp. 310–322.
53. Eisenhaber B, Bork P, Eisenhaber F, Prediction of potential GPI-modification sites in proprotein sequences, J. Mol. Biol. 292(3):741–758 (1999).
54. Pierleoni A, Martelli PL, Casadio R, PredGPI: A GPI-anchor predictor, BMC Bioinformatics 9:392 (2008).
55. Bologna G, Yvon C, Duvaud S, Veuthey AL, N-Terminal myristoylation predictions by ensembles of neural networks, Proteomics 4(6):1626–1632 (2004).
56. Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F, Towards complete sets of farnesylated and geranylgeranylated proteins, PLoS Comput. Biol. 3(4):e66 (2007).
57. Ren J, Gao X, Jin C, Zhu M, Wang X, Shaw A, Wen L, Yao X, Xue Y, Systematic study of protein sumoylation: Development of a site-specific predictor of SUMOsp 2.0, Proteomics 9(12):3409–3412 (2009).
58. Xue Y, Zhou F, Fu C, Xu Y, Yao X, SUMOsp: A web server for sumoylation site prediction, Nucleic Acids Res. 34(web server issue):W254–W257 (2006).
59. Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X, CSS-Palm 2.0: An updated software for palmitoylation sites prediction, Protein Eng. Design Select. 21(11):639–644 (2008).
60. Zhou F, Xue Y, Yao X, Xu Y, CSS-Palm: Palmitoylation site prediction with a clustering and scoring strategy (CSS), Bioinformatics 22(7):894–896 (2006).
61. Xue Y, Chen H, Jin C, Sun Z, Yao X, NBA-Palm: Prediction of palmitoylation site implemented in Naive Bayes algorithm, BMC Bioinformatics 7:458 (2006).
62. Shao J, Xu D, Tsai SN, Wang Y, Ngai SM, Computational identification of protein methylation sites through bi-profile Bayes feature extraction, PLoS One 4(3):e4920 (2009).
63. Shien DM, Lee TY, Chang WC, Hsu JB, Horng JT, Hsu PC, Wang TY, and Huang HD, Incorporating structural characteristics for identification of protein methylation sites, J. Comput. Chem. 30(9):1532–1543 (2009).
64. Kiemer L, Bendtsen JD, Blom N, NetAcet: prediction of N-terminal acetylation sites, Bioinformatics 21(7):1269–1270 (2005).
65. Basu A, Rose KL, Zhang J, Beavis RC, Ueberheide B, Garcia BA, Chait B, Zhao Y, Hunt DF, Segal E, Allis CD, Hake SB, Proteome-wide prediction of acetylation substrates, Proc. Natl. Acad. Sci. USA 106(33):13785–13790 (2009).
66. Tung CW, Ho SY, Computational identification of ubiquitylation sites from protein sequences, BMC Bioinformatics 9:310 (2008).
67. Monigatti F, Gasteiger E, Bairoch A, Jung E, The Sulfinator: predicting tyrosine sulfation sites in protein sequences, Bioinformatics 18(5):769–770 (2002).
68. Chang WC, Lee TY, Shien DM, Hsu JB, Horng JT, Hsu PC, Wang TY, Huang HD, Pan RL, Incorporating support vector machine for identifying protein tyrosine sulfation sites, J. Comput. Chem. 30(15):2526–2537 (2009).
69. Gao J, Xu D, Correlation between posttranslational modification and intrinsic disorder in protein, Proc. Pacific Symp. Biocomputing 2012, Big Island, Hawaii, pp. 94–103.
70. Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK, Exploiting heterogeneous sequence properties improves prediction of protein disorder, Proteins 61(Suppl 7):176–182 (2005).
71. Breiman L, Bagging predictors. Machine Learn. 24(2):123–140 (1996).
72. Thorsten J, Learning to Classify Text Using Support Vector Machines: Methods, Theory and Algorithms, Kluwer Academic Publishers, 2002, p. 224.