
Chapter 15
Algorithmic Methodologies for Discovery of Nonsequential Protein Structure Similarities
15.1 Introduction
An increasing number of protein structures are becoming available that either have no known function or whose functional mechanism is unknown or incomplete. Using experimental methods alone to explore these proteins in order to determine their functional mechanism is unfeasible. For this reason, much research has been put into computational methods for predicting the function of proteins [5, 14, 31, 34, 44, 53]. One such computational method is functional inference by homology, where annotations from a protein with known function are transferred onto another protein on the basis of sequence or structural similarities.
Protein sequence comparisons have been used as a straightforward method for functional inheritance. If two proteins have a high level of sequence identity, frequently the two proteins have the same or related biological functions. This observation has been used as a basis for transferring annotations from a protein that is well characterized to a protein with unknown function when the two proteins have high sequence similarity [3, 4, 45]. Frequently, only the protein residues that are near the functional region of the protein are under evolutionary pressure for conservation. Therefore, the global sequence similarity may be relatively low while local regions within the two sequences maintain a higher level of sequence similarity. In this case, probabilistic models such as profiles have been constructed using only the local regions of high sequence similarity [3, 32, 28]. Sequence comparison methods have the advantage that large numbers of sequences are deposited into sequence databases such as Swiss-Prot [11], which provides adequate information for constructing probabilistic models. However, a relatively high level of sequence similarity is needed in order to accurately transfer protein function. In fact, problems begin to arise when the sequence identity between a pair of proteins is ![]() % [57].
% [57].
Because proteins often maintain structural similarities even when sequence identity falls as low as 30% [6], making protein structure more strongly correlated with protein function than protein sequence [24], many researchers have begun to compare the three-dimensional structure of proteins in an attempt to uncover more distant evolutionary relationships among proteins. The SCOP [40] and CATH [43] databases have organized protein structures hierarchically into different classes and folds on the basis of their overall similarity in topology and fold. Classification of protein structures relies heavily on the reliable protein structure comparison methods. Common structural comparison methods include DALI (see Section 14.3.4) [27] and CE [47]. However, structural alignment methods cannot guarantee optimal results and do not have an interpretability comparable to sequence alignment methods.
Several challenges arise when trying to compare protein structures:
In this chapter, we will discuss several issues of structural alignment and then discuss methods that we have implemented for sequence order–independent structural alignment at the global and local surface levels. We illustrate the utility of our methods by showing how our sequence order–independent global structural alignment method detects circular permuted proteins. We then show how our local surface sequence order–independent structural alignment method can be used to construct a basis set of signature pockets of binding surfaces for a specific biological function. The signature pocket representsstructurally conserved surface regions. A set of signature pockets can then be used to represent a functional family of proteins for protein function prediction.
15.2 Structural Alignment
Comparing the structure of two proteins is an important problem [23] that may detect evolutionary relationships between proteins even when sequence identity between two proteins is relatively low. A widely used method for measuring structural similarity is the root-mean-squared distance (RMSD) between the equivalent atoms or residues of two proteins. If the equivalence relationship is known, a rotation matrix ![]() and a translation vector
and a translation vector ![]() that when applied to one of the protein structures will minimize the RMSD can be found by solving the minimization problem
that when applied to one of the protein structures will minimize the RMSD can be found by solving the minimization problem
15.1 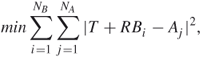
where ![]() is the number of points in structure
is the number of points in structure ![]() and
and ![]() is the number of points in structure
is the number of points in structure ![]() . If
. If ![]() , then the least-squares estimation of the parameters
, then the least-squares estimation of the parameters ![]() and
and ![]() in this equation can be found using singular value decomposition.
in this equation can be found using singular value decomposition.
The equivalence relationship is rarely known a priori when aligning to protein structures. In this case, the structural alignment method consists in minimizing RMSD while maximizing the number of aligned points. Heuristics must be used to solve this multiobjective optimization problem.
A number of heuristic methods have been developed [1, 22, 48, 49, 52, 56, 59] that can be divided into two main categories. Global methods are used to detect similarities between the overall fold of two proteins, and local alignment methods are used to detect similarities within local regions of interest within the two proteins. Most current methods are restricted to finding structural similarities only where the order of the structural elements within the alignment follows the order of the elements within the primary sequence. Sequence order–independent methods ignore the sequential ordering of the atoms or residues in primary sequence. These methods are better suited for finding more complex global similarities and can also be employed for finding all atoms local comparisons. We have implemented both sequence order–independent methods for both global and local structural alignments.
15.3 Global Sequence Order–Independent Structural Alignment
Looking for similarities between the overall fold can elucidate evolutionary or functional relationships between two proteins. However, most of the current methods for structural comparison are sequence order–dependent and are restricted to comparison of similar topologies between the two backbones. It has been discovered that throughout evolution, a genetic event can rearrange the topology of the backbone. One such example is regarded circular permutation. Conceptually, a circular permutation can be as a ligation of the ![]() and
and ![]() termini of a protein and cleavage somewhere else on the protein backbone. It has been observed that circular permutations often maintain a similar 3D spatial arrangement of secondary structures. In addition to circular permutations, research has shown that more complex topological rearrangements are possible [37]. Detection of these permuted proteins will be valuable for studies in homology modeling, protein folding, and protein design.
termini of a protein and cleavage somewhere else on the protein backbone. It has been observed that circular permutations often maintain a similar 3D spatial arrangement of secondary structures. In addition to circular permutations, research has shown that more complex topological rearrangements are possible [37]. Detection of these permuted proteins will be valuable for studies in homology modeling, protein folding, and protein design.
15.3.1 Sequence Order–Independent Global Structural Alignment
We have developed a sequence order–independent structural alignment algorithm for detecting structural similarities between two proteins that have undergone topological rearrangement of their backbone structures [17]. Our method is based on fragment assembly where the two proteins to be aligned are first exhaustively fragmented. Each fragment ![]() from protein structure
from protein structure ![]() is pairwise superimposed onto each fragment
is pairwise superimposed onto each fragment ![]() from protein structure
from protein structure ![]() . The result is a set of fragment pairs
. The result is a set of fragment pairs ![]() , where
, where ![]() and
and ![]() are the indices in the primary sequence of the first residue of the two fragments. The variable
are the indices in the primary sequence of the first residue of the two fragments. The variable ![]() is the length of the fragment. Each fragment pair is assigned a similarity score
is the length of the fragment. Each fragment pair is assigned a similarity score
15.2 ![]()
where cRMSD is the measured RMSD value after optimal superposition, ![]() and
and ![]() are two constants,
are two constants, ![]() is a scaling factor to the measured RMSD values that depends on the secondary structure of the fragments, and SCS is a BLOSUM (blocks substitution matrix)-like measure of similarity in sequence of the matched fragments [25]. Details of the scoring method can be found in an earlier study [17].
is a scaling factor to the measured RMSD values that depends on the secondary structure of the fragments, and SCS is a BLOSUM (blocks substitution matrix)-like measure of similarity in sequence of the matched fragments [25]. Details of the scoring method can be found in an earlier study [17].
The goal of the structural alignment is to find a consistent set of fragment pairs ![]() that minimizes the overall RMSD. Finding the optimal combination of fragment pairs is a special case of the well-known maximum-weight-independent set problem in graph theory. This problem is MAX-SNP-hard. We employ an approximation algorithm that was originally described for the scheduling of split-interval graphs [8] and is itself based on a fractional version of the local ratio approach.
that minimizes the overall RMSD. Finding the optimal combination of fragment pairs is a special case of the well-known maximum-weight-independent set problem in graph theory. This problem is MAX-SNP-hard. We employ an approximation algorithm that was originally described for the scheduling of split-interval graphs [8] and is itself based on a fractional version of the local ratio approach.
To begin, a conflict graph ![]() is created, where a vertex is defined for each aligned fragment pair. Two vertices are connected by an edge if any of the fragments
is created, where a vertex is defined for each aligned fragment pair. Two vertices are connected by an edge if any of the fragments ![]() or
or ![]() from the fragment pair is not disjoint, that is, if both fragments from the same protein share one or more residues. For each vertex representing aligned fragment pairs, we assign three indicator variables
from the fragment pair is not disjoint, that is, if both fragments from the same protein share one or more residues. For each vertex representing aligned fragment pairs, we assign three indicator variables ![]() , and
, and ![]() , and a closed neighborhood
, and a closed neighborhood ![]() indicates whether the fragment pair should be used (
indicates whether the fragment pair should be used (![]() or not (
or not (![]() ) in the final alignment.
) in the final alignment. ![]() and
and ![]() are artificial indicator values for
are artificial indicator values for ![]() and
and ![]() , which allow us to encode consistency in the selected fragments. The closed neighborhood of a vertex
, which allow us to encode consistency in the selected fragments. The closed neighborhood of a vertex ![]() of
of ![]() is
is ![]() , which is simply
, which is simply ![]() and all vertices that are connected to
and all vertices that are connected to ![]() by an edge.
by an edge.
The sequence order–independent structural alignment algorithm can be described as follows. To begin, initialize the structural alignment ![]() equal to the entire set of aligned fragment pairs. We then
equal to the entire set of aligned fragment pairs. We then
15.3 ![]()
15.4 ![]()
15.5 ![]()
15.6 ![]()
15.7 ![]()
15.8 ![]()
15.3.2 Detecting Permuted Proteins
This algorithm was implemented in a large-scale study to search for permuted proteins in the Protein Data Bank (PDB) [9]. A subset of 3336 protein structures taken from the PDBSELECT90 dataset [26] are structurally aligned in a pairwise fashion. From the subset of 3336 proteins, we aligned two proteins if they met the following conditions (see Ref. 17 for details):
Within the approximately 200,000 structural alignments performed, we found many known circular permutations and three novel circular permutations, as well as a more complex pair of noncyclic permuted proteins. Here we describe the details of the circular permutation that we found between a neucleoplasmin core and an auxin binding protein, as well as details of the more complex noncyclic permutation.
15.3.2.1 Nucleoplasmin Core and Auxin Binding Protein
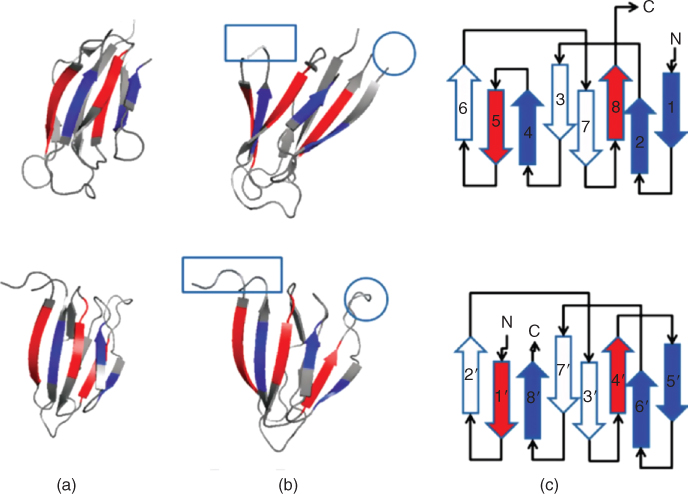
We found a novel circular permutation between the nucleoplasmin core protein in Xenopu laevis (PDB ID 1k5j, chain E) [19] and the auxin binding protein in maize (PDB ID 11rh, chain A, residues 37–127) [58]. The structural alignment between 1k5jE (Fig. 15.1, top) and 11rhA (Fig. 15.1, bottom) consisted of 68 equivalent residues superimposed with a RMSD of 1.36 Å. This alignment is statistically significant with a ![]() after Bonferroni correction. Details of the
after Bonferroni correction. Details of the ![]() value calculation can be found in our earlier study [17]. The short loop connecting two antiparallel strans in nucleoplasmin core protein (in circle, top of Fig. 15.1b) becomes disconnected in auxin binding protein 1 (in circle, bottom of Fig. 15.1b), and the N and C termini of the nucleoplasmin core protein (in square, top of Fig. 15.1b) are connected in auxin binding protein 1 (square, bottom of Fig. 15.1b). For details of other circular permutations we found, including permutations between microphage migration inhibition factor and the
value calculation can be found in our earlier study [17]. The short loop connecting two antiparallel strans in nucleoplasmin core protein (in circle, top of Fig. 15.1b) becomes disconnected in auxin binding protein 1 (in circle, bottom of Fig. 15.1b), and the N and C termini of the nucleoplasmin core protein (in square, top of Fig. 15.1b) are connected in auxin binding protein 1 (square, bottom of Fig. 15.1b). For details of other circular permutations we found, including permutations between microphage migration inhibition factor and the ![]() -terminal domain of arginine repressor, please see our earlier study [17].
-terminal domain of arginine repressor, please see our earlier study [17].
Figure 15.1 A newly discovered circular permutation between nucleoplasmin core [1k5j, chain E, top panel), and a fragment of auxin binding protein 1 (residues 37–127) (11rh, chain A, bottom panel). (a) These two proteins align well with a RMSD value of 1.36 Å over 68 residues, with a significant ![]() after Bonferroni correction. (b) The loop connecting strands 4 and 5 of nucleoplasmin core (in rectangle, top) becomes disconnected in auxin binding protein 1. The
after Bonferroni correction. (b) The loop connecting strands 4 and 5 of nucleoplasmin core (in rectangle, top) becomes disconnected in auxin binding protein 1. The ![]() and
and ![]() termini of nucleoplasmin core (in rectangle, top) become connected in auxin binding protein 1 (in rectangle, bottom). To facilitate visualization of the circular permutation, residues in the
termini of nucleoplasmin core (in rectangle, top) become connected in auxin binding protein 1 (in rectangle, bottom). To facilitate visualization of the circular permutation, residues in the ![]() -to-
-to-![]() direction before the cut in the nucleoplasmin core protein are colored by the lighter shade, and residues after the cut are colored by the darker shade. (c) The topology diagram of these two proteins. In the original structure of nucleoplasmin core, the electron density of the loop connecting strands 4 and 5 is missing in the PDB structure file. (This figure is modified from Hru
direction before the cut in the nucleoplasmin core protein are colored by the lighter shade, and residues after the cut are colored by the darker shade. (c) The topology diagram of these two proteins. In the original structure of nucleoplasmin core, the electron density of the loop connecting strands 4 and 5 is missing in the PDB structure file. (This figure is modified from Hru![]() ka et al. [17].)
ka et al. [17].)
15.3.2.2 Complex Protein Permutations
Because of their relevance in understanding the functional and folding mechanism of proteins, circular permutations have received much attention [37, 55]. However, the possibility of more complex backbone rearrangements were experimentally verified by artificially rearranging the topology of the ARC repressor and were found to be thermodynamically stable [50]. Very little is known about this class of permuted proteins, and the detection of noncyclic permutations is a challenging task [2, 15, 29, 46].
Our database search uncovered a naturally occurring noncyclic permutation between chain F of AML1/core binding factor (AML1/CBF, PDB ID 1e50, Fig. 15.2a, top) and chain A of riboflavin synthase (PDB ID 1pkv, Fig. 15.2a, bottom). The two structures align well with an RMSD of 1.23 Å at an alignment length of 42 residues, with a significant ![]() after Bonferroni correction.
after Bonferroni correction.
Figure 15.2 A noncyclic permutation discovered between AML1/core binding factor (AML1/CBF PDB ID 1e50, chain F, top) and riboflavin synthase (PDB ID 1pkv, chain A, bottom). (a) These two proteins structurally align with an RMSD of 1.23 Å over 42 residues and have a significant ![]() after Bonferroni correction. The residues that were assigned equivalences from the structural alignment are colored by the darker shade. (b) These proteins are structurally related by a complex permutation. The steps to transform the topology of AML1/CBF (top) to riboflavin (bottom) are as follows: (c) Remove the loops connecting strand 1 to helix 2, strand 4 to strand 5, and strand 5 to helix 6; (d) Connect the
after Bonferroni correction. The residues that were assigned equivalences from the structural alignment are colored by the darker shade. (b) These proteins are structurally related by a complex permutation. The steps to transform the topology of AML1/CBF (top) to riboflavin (bottom) are as follows: (c) Remove the loops connecting strand 1 to helix 2, strand 4 to strand 5, and strand 5 to helix 6; (d) Connect the ![]() -terminal end of strand 4 to the original
-terminal end of strand 4 to the original ![]() termini; (e) connect the
termini; (e) connect the ![]() -terminal end of strand 5 to the
-terminal end of strand 5 to the ![]() -terminal end of helix 2; (f) connect the original
-terminal end of helix 2; (f) connect the original ![]() -termini to the
-termini to the ![]() -terminal end of strand 5. The
-terminal end of strand 5. The ![]() -terminal end of strand 6 becomes the new
-terminal end of strand 6 becomes the new ![]() termini, and the
termini, and the ![]() -terminal end of strand 1 becomes the new
-terminal end of strand 1 becomes the new ![]() termini. We now have the topology diagram of riboflavin synthase. (This figure is modified from Hruska et al. [17]).
termini. We now have the topology diagram of riboflavin synthase. (This figure is modified from Hruska et al. [17]).
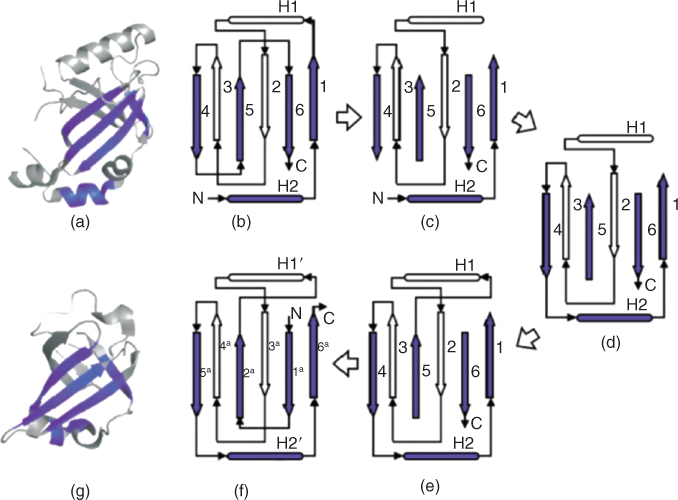
The topology diagram of AML1/CBF (Fig. 15.2b) can be transformed into that of riboflavin synthase (Fig. 15.2f) by the following steps. Remove the loops connecting strand 1 to helix 2, strand 4 to strand 5, and strand 5 to strand 6 (Fig. 15.2c). Connect the ![]() -terminal end of strand 4 to the original
-terminal end of strand 4 to the original ![]() terminal (Fig. 15.2d). Connect the
terminal (Fig. 15.2d). Connect the ![]() -terminal end of strand 5 to the
-terminal end of strand 5 to the ![]() -terminal end of helix 2 (Fig. 15.2e). Connect the original
-terminal end of helix 2 (Fig. 15.2e). Connect the original ![]() termini to the
termini to the ![]() -terminal end of strand 5. The
-terminal end of strand 5. The ![]() -terminal end of strand 6 becomes the new
-terminal end of strand 6 becomes the new ![]() termini, and the
termini, and the ![]() -terminal end of strand 1 becomes the new
-terminal end of strand 1 becomes the new ![]() termini (Fig. 15.2f).
termini (Fig. 15.2f).
15.4 Local Sequence Order–Independent Structural Alignment
Comparison of the global backbone can lead to discovery of distant evolutionary relationships between proteins. However, when attempting to detect similar functions or functional mechanisms between two proteins, global backbone similarity is not a robust indicator [20, 36, 41]. It can be assumed that the physicochemical properties of the local region where function takes place (i.e., substrate binding) is under more evolutionary pressure to be conserved. This assumption has been backed up by several studies [21, 30, 38, 42, 53, 54].
A typical protein contains many concave surface regions, commonly referred to as surface pockets. However, only a few of the surface pockets supply a unique physiochemical environment that is conducive to the protein carrying out its function. The protein must maintain this surface pocket throughout evolution in order to conserve its biological function. For this reason, shared structural similarities between functional surfaces among proteins may be a strong indicator of shared biological function. This has led to a number of promising studies, in which protein functions can be inferred by similarity comparison of local binding surfaces [7, 10, 21, 35, 39]
The inherent flexibility of the protein structure makes the problem of structural comparison of protein surface pockets challenging. A protein is not a static structure as represented by a PDB [9] entry. The whole protein as well as the local functionalsurface may undergo various degrees of structural fluctuations. The use of a single surface pocket structure as a representative template for a specific protein function can lead to many false negatives. This is due to the inability of a single representative to capture the full functional characteristics across all conformations of a protein.
We have addressed this problem by developing an algorithm that can identify the atoms of a surface pocket that are structurally preserved across a family of protein structures that have similar functions. Using a sequence order–independent local surface alignment method to pairwise-align the functional pockets across a family of protein structure, we automatically find the structurally conserved atoms and measure their fluctuations. We call these structurally conserved atoms the signature pocket. More than one signature pocket may result for a single functional class. In this case, our method can automatically create a basis set of signature pockets for that functional family. We can then use these signature pockets as representatives for scanning a structure database for functional inference by structural similarity.
15.4.1 Bipartite Graph Matching Algorithm for Local Surface Comparison
We have modified and implemented a sequence order–independent local structural alignment algorithm based on the maximum-weight bipartite graph matching formulation developed by Chen et al. [12].
As mentioned earlier, the structural alignment problem boils down to a problem of finding an equivalence relation between residues of a reference protein ![]() and a query protein
and a query protein ![]() that when applied will optimize the superposition of the two structures. The formulation here does this in an iterative two-step process: (1) an optimal set of equivalent atoms are determined under the current superposition using a bipartite graph representation and (2) the new equivalence relation is used to determine a new optimal superposition. The two steps are then repeated until a stopping condition is met.
that when applied will optimize the superposition of the two structures. The formulation here does this in an iterative two-step process: (1) an optimal set of equivalent atoms are determined under the current superposition using a bipartite graph representation and (2) the new equivalence relation is used to determine a new optimal superposition. The two steps are then repeated until a stopping condition is met.
The equivalence relationship is found between the two atoms of the functional pocket surfaces by representing the atoms the atoms of ![]() and
and ![]() as nodes in a graph. This graph is bipartite, meaning that edges exist only between atoms of
as nodes in a graph. This graph is bipartite, meaning that edges exist only between atoms of ![]() and atoms of
and atoms of ![]() . A directed edge is drawn between two nodes if a similarity threshold is met. In our implementation, the measure of similarity takes into account both spatial distances and the chemical property similarities between the two corresponding atoms.
. A directed edge is drawn between two nodes if a similarity threshold is met. In our implementation, the measure of similarity takes into account both spatial distances and the chemical property similarities between the two corresponding atoms.
Each edge ![]() connecting nodes
connecting nodes ![]() and
and ![]() is assigned a weight
is assigned a weight ![]() equal to the similarity score between the two corresponding atoms (see [12] for details). The optimal equivalence relationship given the current superposition is a subset of the edges within this bipartite graph that have maximum combined weight, where at most one edge can beselected per atom, making this a maximum-weight bipartite graph matching problem. The solution to this problem can be found using the Hungarian algorithm [33].
equal to the similarity score between the two corresponding atoms (see [12] for details). The optimal equivalence relationship given the current superposition is a subset of the edges within this bipartite graph that have maximum combined weight, where at most one edge can beselected per atom, making this a maximum-weight bipartite graph matching problem. The solution to this problem can be found using the Hungarian algorithm [33].
The Hungarian method is as follows. Initially, an overall score ![]() is set. Additionally, an artificial source node
is set. Additionally, an artificial source node ![]() and an artificial destination node
and an artificial destination node ![]() are added to the bipartite graph. A directed edge
are added to the bipartite graph. A directed edge ![]() with zero weight is added for each of the atom nodes
with zero weight is added for each of the atom nodes ![]() from
from ![]() and similarly, directed edges
and similarly, directed edges ![]() with zero weight are drawn from each of the atoms nodes of
with zero weight are drawn from each of the atoms nodes of ![]() . The algorithm then proceeds as follows:
. The algorithm then proceeds as follows:
15.9 ![]()
The iterative process of the Hungarian algorithm stops when either there is no path from ![]() to
to ![]() or the shortest distance from the source node to the destination node
or the shortest distance from the source node to the destination node ![]() is greater than the current overall score
is greater than the current overall score ![]() . At the end of the process, the graph will consist of a set of directed edges that have been reversed (they now point from nodes of
. At the end of the process, the graph will consist of a set of directed edges that have been reversed (they now point from nodes of ![]() to nodes from
to nodes from ![]() . These reversed edges represent the new equivalence relationships between the atoms of
. These reversed edges represent the new equivalence relationships between the atoms of ![]() and the atoms of
and the atoms of ![]() .
.
The equivalence relationship found by the bipartite matching algorithm can now be used to superimpose the two proteins using the singular value decomposition. After superpositioning the new equivalent atoms, a new bipartite graph is createdand the process is iterated until the change in RMSD on superposition falls below a threshold.
15.4.2 A Basis Set of Binding Surface Signature Pockets
The ability to compare structural similarities between to protein surface regions can provide insight into shared biological functions. As mentioned earlier, when dealing with local surface regions, one has to be careful when choosing a functional representative pocket because of the inherent flexibility of the binding surfaces. We have developed a method that automatically generates a set of functional pocket templates, called signature pockets of local surface regions that can be used as a representative a functional surface for structural comparison. These signature pockets contain broad structural information and have discriminating ability.
A signature pocket is derived from sequence order–independent structural alignments of precomputed surface pockets. Our signature pocket method does not require the atoms of the signature pocket to be present in all member structures. Instead, signature pockets can be created at varying degrees of partial structural similarity and can be hierarchically organized according to their structural similarity.
The input of our signature pocket algorithm is a set of functional pockets from the CASTp database [18]. All versus all pairwise sequence order–independent local surface alignment is performed on the input functional surface pockets. A distance is calculated on the basis of the RMSD and the chemistry of the paired atoms of the structural alignment [16]. The resulting distance matrix is used by an agglomerative clustering method. The signature of the functional pocket is then derived using a recursive process following the hierarchical tree.
The recursive process begins by finding the two closest siblings (pockets ![]() and
and ![]() ), and combining them into a single structure
), and combining them into a single structure ![]() . During the recursive process,
. During the recursive process, ![]() or
or ![]() may themselves already be a combination of several structures. When combining two structures, we follow these criteria:
may themselves already be a combination of several structures. When combining two structures, we follow these criteria:
A count of the number of times that an atom at the position ![]() was present in the underlying set of pockets (
was present in the underlying set of pockets (![]() ) is recorded during each step in the recursive process. A preservation ratio
) is recorded during each step in the recursive process. A preservation ratio ![]() is calculated for each atom of the signature pocket by dividing
is calculated for each atom of the signature pocket by dividing ![]() by the total number of constituent pockets. In addition, the mean distance of the coordinates of the aligned atoms to their geometric center is recorded as the location variation
by the total number of constituent pockets. In addition, the mean distance of the coordinates of the aligned atoms to their geometric center is recorded as the location variation ![]() . At the end of each step, the new structure
. At the end of each step, the new structure ![]() replaces the two structures
replaces the two structures ![]() and
and ![]() in the hierarchical tree and the process is repeated on the updated hierarchical tree.
in the hierarchical tree and the process is repeated on the updated hierarchical tree.
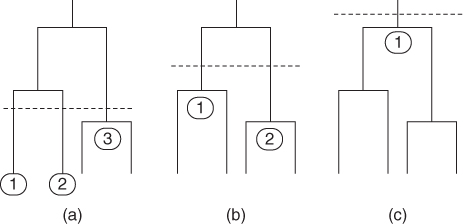
The recursive process can be stopped at any point during its traversal of the hierarchical tree by selecting a ![]() threshold. Depending on the choice of the
threshold. Depending on the choice of the ![]() threshold, a single or multiple signature pockets can be created. Figure 15.3a shows a low
threshold, a single or multiple signature pockets can be created. Figure 15.3a shows a low ![]() threshold that results in a set of three signature pockets. As the threshold is raised, fewer signature pockets are created (Fig. 15.3b). A single signature pocket representing all surface pockets in the dataset can be generated by raising the threshold even further (Fig. 15.3). The set of signature pockets from different clusters in the hierarchical tree form a basis set that represents an ensemble of differently sampled conformations of the surface pockets in the PDB. The basis set of signature pockets can be used to accurately classify and predict enzymatic function.
threshold that results in a set of three signature pockets. As the threshold is raised, fewer signature pockets are created (Fig. 15.3b). A single signature pocket representing all surface pockets in the dataset can be generated by raising the threshold even further (Fig. 15.3). The set of signature pockets from different clusters in the hierarchical tree form a basis set that represents an ensemble of differently sampled conformations of the surface pockets in the PDB. The basis set of signature pockets can be used to accurately classify and predict enzymatic function.
Figure 15.3 Different basis sets of signature pockets can be produced at different levels of structural similarity by raising or lowering the similarity threshold (vertical dashed line): (a) a low threshold will produce more signature pockets; (b) as the threshold is raised, fewer signature pockets will be created; (c) a single signature pocket can, in principle, be created to represent the full surface pocket dataset by raising the threshold.
15.4.2.1 Signature Pockets of NAD Binding Proteins
To illustrate how signature pockets and the basis set help identify structural elements that are important for binding and to show their accuracy in functional inference, we discuss a study performed on the nicotinamide adenine dinucleotide (NAD) binding proteins. NAD plays essential roles in metabolisms where it acts as a coenzyme in redox reactions, including glycolysis and the citric acid cycle.
We obtained a set of 457 NAD binding proteins of diverse fold and diverse evolutionary origin. We extracted the NAD binding surfaces from the CASTp database of protein pockets [18]. We obtained the hierarchical tree using the results of our sequence order–independent surface alignments. The resulting nine signature pockets of the NAD binding pocket form a basis set, shown in Figure 15.4.
Figure 15.4 Topology of the hierarchical tree and signature pockets of the NAD binding pockets: (a) the resulting hierarchical tree topology; (b–j) the resulting signature pockets of the NAD binding proteins, along with the superimposed NAD molecules that were bound in the pockets of the member proteins of the respective clusters. The NAD coenzymes have two distinct conformations. Those in an extended conformation are marked with an X, and those in a compact conformation are marked with a C.
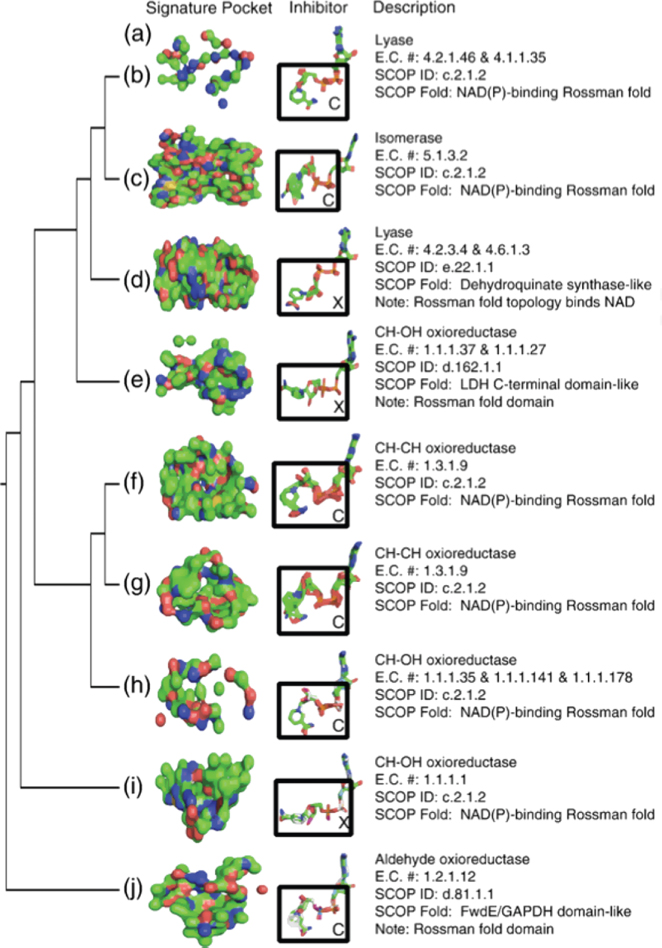
The signature pockets of NAD contain biological information. The signature pocket show in Figure 15.4j is based on a cluster of NAD binding proteins that act on the aldehyde group of donors, the signature pockets in (Fig. 15.4f,g) are for oxioreductases that act on the CH–CH group of donors, and the signature pockets of Figure 15.4e, h, and i are for clusters of alcohol oxioreductases that act on the CH–OH group of donors. The NAD–binding lyase family is represented in two signature pockets. The first represents lyases that cleave both C–O and P–O (Fig. 15.4d) and the second, containing lyases that cleave both C–O and CC bonds (Fig. 15.4b). These two signature pockets from two clusters of lyase conformations have a different class of conformations of the bound NAD cofactor (extended and compact).
In addition to the structural fold, the signature pockets are also determined by the conformation of the bound NAD cofactor (Fig. 15.4a). It can be seen in Fig. 15.4b–j that there are two general conformations of the NAD coenzyme. The coenzymes labeled C (Fig. 15.4b,c,f,g,h,j) have a closed conformation, while the conenzymes labeled X (Fig. 15.4d,e,i) have an extended conformation. This indicates that the binding pocket may take multiple conformations yet bind the same substrate in the same general structure. For example, the two structurally distinct signature pockets shown in Fig. 15.4f,g are derived from proteins that have the same biological function and SCOP fold. All of these proteins bind to the same NAD conformation.
We further evaluated the effectiveness of the NAD basis set by determining its accuracy at correctly classifying enzymes as either NAD binding or non-NAD-binding. We constructed a testing dataset of 576 surface pockets from the CASTp database [18]. This dataset is independent of the 457 NAD binding proteins that we used to create the signature pockets. We collected the 576 surface pockets by selecting the top three largest pockets by volume from 142 randomly chosen proteins and 50 proteins that have NAD bound in the PDB structure. We then structurally aligned each signature pocket against each of the 576 testing pockets. The testing pocket was assigned to be an NAD binding pocket if it structurally aligned to one of the nine NAD signature pockets with a distance under a predefined threshold. Otherwise it was classified as non-NAD-binding. The results show that the basis set of nine signature pockets can classify the correct NAD binding pocket with sensitivity and specificity of 0.91 and 0.89, respectively. We performed further testing to determine whether a single representative NAD binding pocket, as opposed to a basis set, issufficient for identifying NAD binding enzymes. We chose a single pocket representative from one of the nine clusters at random and attempted to classify our testing dataset by structural alignment. We used the same predefined threshold used in the basis set study. This was repeated 9 times using a representative from each of the nine clusters. We found that the results deteriorated significantly with an average sensitivity and specificity of 0.36 and 0.23, respectively. This strongly indicates that the construction of a basis set of signature pockets to be used as a structural template provides significant improvement for functional inference of a set of evolutionarily diverse proteins.
15.5 Conclusion
We have discussed methods that provide solutions to the problems that arise during functional inference by structural similarity at both the global and at local surface levels. Both of our methods disregard the ordering of residues in the protein's primary sequence, making them sequence order–independent. The global method can be used to address the challenging problem of detecting structural similarities even after topological rearrangements of the proteins backbone. The fragment assembly approach based on the formulation of a relaxed integer programming problem and an algorithm based on scheduling split-interval graphs is guaranteed by an approximation ratio. We showed that this method is capable of discovering circularly permuted proteins and other more complex topological rearrangements.
We also described a method for sequence order–independent alignment of local surfaces on proteins. This method is based on a bipartite graph matching problem. We further show that the surface alignments can be used to automatically construct a basis set of signature pockets representing structurally preserved atoms across a family of proteins with similar biological functions.
Acknowledgments
DasGupta and Liang was partially supported by NSF grant DBI-1062328.
References
1. Aghili AS, Agrawal D, El Abbadi A, PADS: Protein structure alignment using directional shape signatures, Proc. Database Systems for Advanced Applications (DASFAA) Conf., 2004.
2. Alexandrov NN, Fischer D, Analysis of topological and nontopological structural similarities in the PDB: New examples from old structures, Proteins 25:354–365 (1996).
3. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ, Gapped BLAST and PSI-BLAST: A new generation of protein database search programs, Nucleic Acids Res. 25:3389–3402 (1997).
4. Altschul SF, Warren G, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool, J. Mol. Biolo., 215:403–410 (1990).
5. Andrade MA, Brown NP, Leroy C, Hoersch S, de Daruvar A, Reich C, Franchini J, Tamames A, Valencia A, Ouzounis C, Sander C, Automated genome sequence analysis and annotation, Bioinformatics, 15:391–412 (1999).
6. Rost B, Twilight zone of protein sequence alignments, Protein Eng. 12:85–94 (1999).
7. Bandyopadhyay D, Huan J, Liu J, Prins J, Snoeyink J, Wang W, Tropsha A, Functional neighbors: Inferring relationships between nonhomologous protein families using family-specific packing motifs, IEEE Trans. Inf. Technol. Biomed. 14(5):1137–1143 (2010).
8. Bar-Yehuda R, Halldorsson MM, Naor J, Shacknai H, Shapira I, Scheduling split intervals, Proc. 14th ACM-SIAM Symp. Discrete Algorithms, 2002, pp. 732–741.
9. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The protein data bank, Nucleic Acids Res. 28:235–242 (2000).
10. Binkowski TA, Joachimiak A, Protein functional surfaces: Global shape matching and local spatial alignments of ligand binding sites, BMC Struct. Biol. 8:45 (2008).
11. Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M, The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003, Nucleic Acids Res. 31:365–370 (2003).
12. Chen L, Wu LY, Wang R, Wang Y, Zhang S, Zhang XS, Comparison of protein structures by multi-objective optimization, Genome Inform. 16(2):114–124 (2005).
13. Dayhoff MO, Schwartz RM, Orcutt BC, A model of evolutionary change in proteins, Atlas Protein Seq. Struct. 5(3):345–352 (1978).
14. Deng M, Zhang K, Mehta S, Chen T, Sun F, Prediction of protein function using protein-protein interaction data, J. Comput. Biol. 10(6):947–960 (2009).
15. Dror O, Benyamini H, Nussinov R, Wolfson HJ, MASS: Multiple structural alignment by secondary structures, Bioinformatics 19:i95–i104 (2003).
16. Dundas J, Adamian L, Liang J, Structural Signatures of Enzyme Binding Pockets from Order-Independent Surface Alignment: A Study of Metalloendopeptidase and NAD Binding Proteins, J. Mol. Biol. 406(5):713–729 (2011).
17. Dundas J, Binkowski TA, DasGupta B, Liang J, Topology independent protein structural alignment, BMC Bioinformatics 8:388 (2008).
18. Dundas J, Ouyang Z, Tseng J, Binkowski TA, Turpaz Y, Liang J, CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues, Nucleic Acids Res., 34:W116–W118 (2006).
19. Dutta S, Akey IV, Dingwall C, Hartman KL, Laue T, Nolte RT, Head JF, Akey CW, The crystal structure of nucleoplasmin-core implications for histone binding and neucleosome assemply, Mol. Cell 8:841–853 (2001).
20. Fischer D, Norel R, Wolfson H, Nussinov R, Surface motifs by a computer vision technique: Searches, detection, and implications for protein-ligand recognition, Proteins 16:278–292 (1993).
21. Binkowski TA, Adamian L, Liang J, Inferring functional relationship of proteins from local sequence and spatial surface patterns, J. Mol. Biol. 332:505–526 (2003).
22. Gold ND, Jackson RM, Fold independent structural comparisons of protein-ligand binding sites for exploring functional relationships, J. Mol. Biol. 355:1112–1124 (2006).
23. Hasegawa H, Holm L, Advances and pitfalls of protein structural alignment, Curr. Opin. Struct. Biol. 19:341–348 (2009).
24. Hegyi H, Gerstein M, The relationship between protein structure and function: A comprehensive survey with application to the yeast genome, J. Mol. Biol., 288:147–164 (1999).
25. Henikoff S, Henikoff JG, Amino acid substitution matrices from protein blocks, Proc. Natl. Acad. Sci. USA 89(22):10 915–10 919 (1992).
26. Hobohm U, Sander C, Enlarged representative set of protein structures, Protein Sci. 3:522 (1994).
27. Holm L, Sander C, Protein structure comparison by alignment of distance matrices, J. Mol. Biol. 233:123–138 (1993).
28. Hulo N, Sigrist CJA, Le Saux V, Recent improvements to the PROSITE database, Nucleic Acids Res. 32:D134–D137 (2004).
29. Ilyin VA, Abyzov A, Leslin CM, Structural alignment of proteins by a novel TOPOFIT method, as a superimposition of common volumes at a topomax point, Protein Sci. 13:1865–1874 (2004).
30. Jeffery C, Molecular mechanisms for multi-tasking; recent crystal structures of moon-lighting proteins, Curr. Opin. Struct. Biol. 14:663–668 (2004).
31. Jensen LJ, Gupta R, Blom N, Devos D, Tamames J, Kesmir C, Nielsen H, Staerfeldt HH, Rapacki K, Workman C, Andersen CAF, Knudsen S, Krogh A, Valencia A, Brunak S, Prediction of human protein function from post-translational modifications and localization features, J. Mol. Biol. 319:1257–1265 (2002).
32. Karplus K, Barret C, Hughey R, Hidden Markov models for detecting remote protein homologues, Bioinformatics 14:846–856 (1998).
33. Kuh HW, The Hungarian method for the assignment problem, Nav. Res. Logist. Q. 2:83–97 (1995).
34. Laskowski RA, Watson JD, Thornton JM, ProFunc: A server for predicting protein function from 3D structure, Nucleic Acids Res. 33:W89–W93 (2005).
35. Lee S, Li B, La D, Fang Y, Ramani K, Rustamov R, Kihara D, Fast protein tertiary structure retrieval based on global surface shape similarity, Proteins 72:1259–1273 (2008).
36. Lichtarge O, Bourne HR, Cohen FE, An evolutionary trace method defines binding surfaces common to protein families, J. Mol. Biol. 7:39–46 (1994).
37. Lindqvist Y, Schneider G, Circular permutations of natural protein sequences: Structural evidence, Curr. Opin. Struct. Biol. 7:422–477 (1997).
38. Meng E, Polacco B, Babbitt P, Superfamily active site templates, Proteins, 55:962–967 (2004).
39. Moll M, Kavraki L, LabelHash, A Flexible and Extensible Method for Matching Structural Motifs, available from Nature Precedings (http://dx.doi.org/10.1038/npre.2008.2199.1) (2008).
40. Murzin AG, Brenner SE, Hubbard T, Chothia C, SCOP: A structural classification of proteins database for the investigation of sequences and structures, J. Mol. Biol. 247:536–540 (1995).
41. Norel R, Fischer H, Wolfson H, Nussinov R, Molecular surface recognition by computer vision-based technique, Protein Eng. 7(1):39–46 (1994).
42. Orengo C, Todd A, Thornton J, From protein structure to function, Curr. Opin. Struct. Biol. 9:374–382 (1999).
43. Orengo CA, Michie AD, Jones DT, Swindells MB, Thornton JM, CATH: A hierarchical classification of protein domains structures, Structure 5:1093–1108 (1997).
44. Pal D, Eisenberg D, Inference of protein function from protein structure, Structure 13:121–130 (2005).
45. Shah I, Hunterm L, Predicting enzyme function from sequence: A systematic appraisal, Intell. Syst. Mol. Biol. 5:276–283 (1997).
46. Shih ES, Hwang MJ, Alternative alignments from comparison of protein structures, Proteins 56:519–527 (2004).
47. Shindyalov IN, Bourne PE, Protein structure alignment by incremental combinatorial extension (CE) of the optimal path, Protein Eng. 11(9):739–747 (1998).
48. Standley DM, Toh H, Nakamura H, Detecting local structural similarity in proteins by maximizing the number of equivalent residues, Proteins: Struct., Funct., Genet. 57:381–391 (2004).
49. Szustakowski JD, Weng Z, Protein structure alignment using a genetic algorithm, Proteins: Struct., Func., Genet. 38:428–440 (2000).
50. Tabtiang RK, Cezairliyan BO, Grant RA, Chochrane JC, Sauer RT, Consolidating critical binding determinants by noncyclic rearrangement of protein secondary structure, Proc. Natl. Acad. Sci. USA 7:2305–2309 (2004).
51. Teichert F, Bastolla U, Porto M, SABERTOOTH: Protein structure comparison basedon vectorial structure representation, BMC Bioinformatics 8:425 (2007).
52. Teyra J, Paszkowski-Rogacz M, Anders G, Pisabarro MT, SCOWLP classification: structural comparison and analysis of protein bindign regions, BMC Bioinformatics 9:9 (2008).
53. Tseng YY, Dundas J, Liang J, Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns, J. Mol. Biol. 387(2):451–464 (2009).
54. Tseng YY, Liang J, Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: A Bayesian Monte Carlo approach, Mol. Biol. Evol. 23:421–436 (2006).
55. Uliel S, Fliess A, Amir A, Unger R, A simple algorithm for detecting circular permutations in proteins, Bioinformatics 15(11):930–936 (1999).
56. Veeramalai M, Gilbert D, A novel method for comparing topological models of protein structures enhanced with ligand information, Bioinformatics 24(23):2698–2705 (2008).
57. Weidong T, Skolnick J, How well is enzyme function conserved as a function of pairwise sequence identity, J. Mol. Biol. 333:863–882 (2003).
58. Woo EJ, Marshall J, Bauly J, Chen JG, Venis M, Napier RM, Pickersgill RW, Crystal structure of the auxin-binding protein 1 in complex with auxin, EMBO J. 21:2877–2885 (2001).
59. Zhu J, Weng Z, A novel protein structure alignment algorithm, Proteins: Struct., Funct., Bioinform. 58:618–627 (2005).