
Chapter 10
Protein Contact Order Prediction: Update
10.1 Introduction
Contact order (CO) is the most widely adopted property used to measure the topological complexity of a protein structure. More specifically, contact order quantitatively measures the nonadjacent amino acid proximity within a folded protein. A contact between two distinct amino acid residues in a protein is formed when there is a pair of heavy atoms (C, O, S, or N), one from each residue, whose physical (Euclidean) distance is within a defined threshold [22, 36]. The absolute contact order (denoted as Abs_CO in this chapter) of a protein is defined as the average number of residues separating the contacts inside the protein (where two sequentially adjacent residues are separated by one residue). The relative contact order, or simply the contact order (denoted as CO), is the Abs_CO normalized over the protein length.
Mathematically, given a protein with a primary sequence of length ![]() , we use
, we use ![]() to denote its
to denote its ![]() th amino acid residue. For two distinct residues
th amino acid residue. For two distinct residues ![]() and
and ![]() , if there are two heavy atoms (C, O, S, or N), one from each residue, within
, if there are two heavy atoms (C, O, S, or N), one from each residue, within ![]() Å, then
Å, then ![]() and
and ![]() form a contact. Let
form a contact. Let ![]() denote the number of residues,
denote the number of residues, ![]() , separating this contact. Assuming that there are a total of
, separating this contact. Assuming that there are a total of ![]() contacts in the protein, the Abs_CO of this protein is defined as
contacts in the protein, the Abs_CO of this protein is defined as
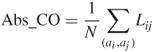
where the summation goes over all contacting residue pairs ![]() in the protein [22, 36]. The CO is defined as
in the protein [22, 36]. The CO is defined as
10.2 ![]()
Essentially, Abs_CO measures the average separation between contacting residues in the native state of a protein, while CO is the normalized variant. Abs_CO (CO as well) increases with the proportion of interacting atoms that are far away in the protein sequence.
10.2 Correlated protein properties
Since the early 1980s, considerable computational and experimental efforts have been devoted to learning about or predicting how proteins fold. Bulk properties such as protein folding rates [26, 32], free energies of folding [43], and hydrogen exchange rates [13] can be measured experimentally to provide insights into protein folding mechanisms. These folding mechanisms are correlated with molecular properties such as secondary structure [23], molecular topology [36], and solvent accessibility [33]. In the past few decades, there were remarkable observations that protein folding rates vary over many orders of magnitude, from microseconds [32] to hours [26]. In combination with theoretical studies, these experimental observations have led to a general agreement that protein folding mechanisms and folding landscapes are determined largely by the topology of the protein native state and are relatively insensitive either to the details of the interatomic interactions [1, 7, 16, 22, 36] or to protein length [36].
To quantify the topological complexity and the stability of protein native states, various measurements pertaining to the contacts between amino acid residues in protein 3D structure have been proposed [12, 15, 20, 22, 27, 29, 36–39, 48], where the relative contact order CO is the most robust representative. Both positive and negative correlations have been found between CO and several bulk protein properties such as protein folding rate and transition state placements [5, 17, 18, 21, 22, 31, 36].
Using a test dataset containing (only) ![]() proteins, Plaxco et al. showed that there is a statistically significant relation between protein folding kinetics and native state topological complexity [36]. As it is proportional to the height of the transition state barrier, the logarithm of the intrinsic refolding rate was found to be well correlated with CO with a coefficient of
proteins, Plaxco et al. showed that there is a statistically significant relation between protein folding kinetics and native state topological complexity [36]. As it is proportional to the height of the transition state barrier, the logarithm of the intrinsic refolding rate was found to be well correlated with CO with a coefficient of ![]() and an associated
and an associated ![]() value of
value of ![]() . It was also observed that the correlation coefficient between estimates of folding transition state placement
. It was also observed that the correlation coefficient between estimates of folding transition state placement ![]() and CO is
and CO is ![]() with an associated
with an associated ![]() value of
value of ![]() [36]. Here
[36]. Here ![]() is computed from the ratio of the denaturant dependences of the relative free energy of the native folding and folding transition states. It is thought to reflect the fraction of solvent-accessible surface buried in the native state that is also buried in the transition state. Plaxco et al. also found a high correlation coefficient between CO and the helical content of the protein [36]. This observation is not surprising because helices have numerous close contacts characterized by a three-residue periodicity. However, the correlations between helical content or protein folding rate and transition state placement were shown to be much less significant; likewise, relationships between the size or stability of the proteins in the dataset and their refolding kinetics were found to be weak or nonexistent.
is computed from the ratio of the denaturant dependences of the relative free energy of the native folding and folding transition states. It is thought to reflect the fraction of solvent-accessible surface buried in the native state that is also buried in the transition state. Plaxco et al. also found a high correlation coefficient between CO and the helical content of the protein [36]. This observation is not surprising because helices have numerous close contacts characterized by a three-residue periodicity. However, the correlations between helical content or protein folding rate and transition state placement were shown to be much less significant; likewise, relationships between the size or stability of the proteins in the dataset and their refolding kinetics were found to be weak or nonexistent.
Contact order has also been shown to have certain utility in ab initio protein structure prediction [7], in addition to its application in predicting protein folding kinetic properties. Bonneau et al. observed that protein “decoy” (i.e., candidate) structures with higher topological complexity are more likely to be undersampled during the candidate structure generation stage in ab initio structure prediction programs, especially among larger proteins [7]. Such a bias can be alleviated by normalizing the CO distribution of candidate structures, and subsequently better protein structure predictions were generally achieved [7]. CO filtering is now an integral part of the Rosetta protein structure prediction package [10].
10.3 Other contact measurements
As shown in early studies, Abs_CO exhibits a weaker correlation with two-state protein folding kinetics than CO does [16, 36]. More recently, however, Ivankov et al. showed that Abs_CO is a more appropriate parameter for predicting the folding rate of proteins as it actually spans a wider range of folding state kinetics (i.e., two-state, multistate, and short peptides) [22]. Ivankov et al. summarized CO and protein length (![]() ) into a general parameter called the size-modified contact order (SMCO):
) into a general parameter called the size-modified contact order (SMCO):
Apparently, from Eq. (10.3), SMCO reduces to CO when ![]() , and reduces to Abs_CO when
, and reduces to Abs_CO when ![]() . It was observed that any
. It was observed that any ![]() results in approximately the same correlation for the totality of proteins and peptides collected [22], with the best correlation achieved at
results in approximately the same correlation for the totality of proteins and peptides collected [22], with the best correlation achieved at ![]() , that is, when SMCO
, that is, when SMCO ![]() Abs_CO. This hints that the more promising applications of CO prediction or calculation lie in the prediction of protein folding rates, folding transition state placements, and other folding properties.
Abs_CO. This hints that the more promising applications of CO prediction or calculation lie in the prediction of protein folding rates, folding transition state placements, and other folding properties.
In the literature, there are several other well-studied concepts on residue contacts, such as residuewise contact order (RWCO) [25, 28, 30, 42], effective contact order (ECO) [39], contact number [CN; also known as residue contact number or residue coordination number (RCN)] [12, 15, 20, 27, 29, 37, 38, 48], and Kendall's tau nearest-neighbor topology (![]() -NN) [39]. These measurements are used largely to characterize the topology or topological complexity of protein native structure, but unlike CO, they are not directly correlated with certain global protein properties such as protein folding rate and folding transition state placements.
-NN) [39]. These measurements are used largely to characterize the topology or topological complexity of protein native structure, but unlike CO, they are not directly correlated with certain global protein properties such as protein folding rate and folding transition state placements.
The residuewise contact order (RWCO) describes the sequence separations between the residues of interest and its contacting residues in a protein sequence [42]. RWCO provides important information for reconstructing a protein 3D structure from a set of one-dimensional structural properties. RWCO can also assist in protein 3D structure prediction and protein folding rate prediction, as well as providing insights into protein sequence–structure relationships. The discrete RWCO value of the ![]() th residue in a length
th residue in a length ![]() protein sequence is defined by
protein sequence is defined by
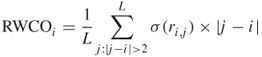
where ![]() if
if ![]() and
and ![]() otherwise.
otherwise. ![]() is the Euclidean distance between the
is the Euclidean distance between the ![]() atoms of the
atoms of the ![]() th and the
th and the ![]() th residues in the protein sequence. Note that a sequential separation of at least two residues is required. By replacing the step function
th residues in the protein sequence. Note that a sequential separation of at least two residues is required. By replacing the step function ![]() with a sigmoid function, one obtains the continuous RWCO, or simply RWCO [42]. Song et al. developed a (continuous) RWCO prediction method based on PSI-BLAST and support vector regression from protein primary sequences, and achieved a correlation coefficient of
with a sigmoid function, one obtains the continuous RWCO, or simply RWCO [42]. Song et al. developed a (continuous) RWCO prediction method based on PSI-BLAST and support vector regression from protein primary sequences, and achieved a correlation coefficient of ![]() and a root-mean-square error of
and a root-mean-square error of ![]() on a well-curated dataset containing
on a well-curated dataset containing ![]() protein sequences [42].
protein sequences [42].
The effective contact order (ECO), as an alternative measurement to contact order, was proposed by Dill and coworkers [11, 14] who adjusted the number of residues between a contacts as the number of residues on the shortest path between the two contacting residues. For example, when residues ![]() and
and ![]() (
(![]() ) form a contact with
) form a contact with ![]() , and in between them residues
, and in between them residues ![]() and
and ![]()
![]() form an inner contact with
form an inner contact with ![]() , then in ECO
, then in ECO ![]() is adjusted to be
is adjusted to be ![]() instead of
instead of ![]() , as used in Equation (10.1). The shortest path between residues
, as used in Equation (10.1). The shortest path between residues ![]() and
and ![]() in this case is
in this case is ![]() . Because of the presence of existing covalent or topological links, ECO operationalizes contacts and scope in terms of shortest pathlengths between residues. ECO is assumed to relate to protein folding rate because it attempts to capture loop size, which is related to the size of the conformational search space necessary to form a conditional contact based on preexisting links.
. Because of the presence of existing covalent or topological links, ECO operationalizes contacts and scope in terms of shortest pathlengths between residues. ECO is assumed to relate to protein folding rate because it attempts to capture loop size, which is related to the size of the conformational search space necessary to form a conditional contact based on preexisting links.
The contact number (CN) measures how amino acid residues are spatially arranged. It is defined as the number of ![]() atoms in other residues and within a sphere centered at the
atoms in other residues and within a sphere centered at the ![]() atom of interest [48]. Therefore, CN is a residuewise measure. The same as for RWCO, Yuan et al. defined two kinds of contact number in their study: the discrete and the continuous. The discrete CN of the
atom of interest [48]. Therefore, CN is a residuewise measure. The same as for RWCO, Yuan et al. defined two kinds of contact number in their study: the discrete and the continuous. The discrete CN of the ![]() th residue
th residue ![]() in a length
in a length ![]() protein is defined as
protein is defined as
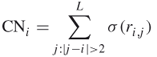
where ![]() is the same step function used in the definition of
is the same step function used in the definition of ![]() , as well as
, as well as ![]() and the radius
and the radius ![]() . Note again that a sequential separation of at least two residues is required. Also, if one replaces the step function
. Note again that a sequential separation of at least two residues is required. Also, if one replaces the step function ![]() with a sigmoid function, the continuous CN of the
with a sigmoid function, the continuous CN of the ![]() th residue is defined. CN can be used to assist in protein fold recognition [24], to describe conserved solvent exposure of similar folds without a common evolutionary origin [19], to determine the energy function allowing molecular dynamics simulations of protein structures [27], and to partly characterize protein 3D structure [48]. Yuan et al. proposed a CN prediction method using PSI-BLAST and support vector regression, and achieved a correlation coefficient of
th residue is defined. CN can be used to assist in protein fold recognition [24], to describe conserved solvent exposure of similar folds without a common evolutionary origin [19], to determine the energy function allowing molecular dynamics simulations of protein structures [27], and to partly characterize protein 3D structure [48]. Yuan et al. proposed a CN prediction method using PSI-BLAST and support vector regression, and achieved a correlation coefficient of ![]() ; furthermore, they showed that if residues are classified as being either “contacted” or “non-contacted” then the correlation coefficient can reach
; furthermore, they showed that if residues are classified as being either “contacted” or “non-contacted” then the correlation coefficient can reach ![]() .
.
Most recently, Segal et al. proposed a new topological measurement based on a means for operationalizing 3D proximity with respect to the underlying chain [39]. Specifically, the euclidean distances between all pairs of residues are computed and recorded in a distance matrix using their 3D coordinates. Then, for each residue, its Euclidean distances are mapped to a nearest-neighbor ranking. With this ranking, cycle structure can be used to capture topology with respect to the underlying chain. Such a ranking-based approach is insensitive to noise, which is a known concern with regard to structure determination experiments. For each residue a reference ranking is generated to capture the denatured random coil and the Kendall's tau nearest-neighbor (![]() -NN) approach is used to measure the difference between the ranking of the folded structure's residue and the ranking of the reference residue. To measure the topology of the whole folded structure, Segal et al. took the average over all residues'
-NN) approach is used to measure the difference between the ranking of the folded structure's residue and the ranking of the reference residue. To measure the topology of the whole folded structure, Segal et al. took the average over all residues' ![]() -NN values. Compared to CO, the
-NN values. Compared to CO, the ![]() -NN measurement needs no tuning parameters during the computation; moreover, the chain deformation/structural information between the reference and contacting residues captured by the
-NN measurement needs no tuning parameters during the computation; moreover, the chain deformation/structural information between the reference and contacting residues captured by the ![]() -NN measurement is ignored in computing CO. When tested on a set of two-state proteins under standardized conditions, this measurement showed an improved correlation coefficient with folding and unfolding rates. On a selected dataset containing
-NN measurement is ignored in computing CO. When tested on a set of two-state proteins under standardized conditions, this measurement showed an improved correlation coefficient with folding and unfolding rates. On a selected dataset containing ![]() proteins, Segal et al. showed that the correlation coefficient between the folding and the unfolding rates of the proteins and the
proteins, Segal et al. showed that the correlation coefficient between the folding and the unfolding rates of the proteins and the ![]() -NN values are
-NN values are ![]() and
and ![]() , respectively, while the correlation coefficient between the two rates and the CO are only
, respectively, while the correlation coefficient between the two rates and the CO are only ![]() and
and ![]() , respectively.
, respectively.
10.4 Contact order calculation
Here and in Sections 10.5 and 10.6, we introduce our work on contact order calculation and prediction. Because of the trivial relationship between Abs_CO and CO, where one can be directly computed from the other, in the sequel, we often refer to Abs_CO and CO interchangeably, unless otherwise explicitly specified.
While a large number of contact order algorithms have been described, the limited accessibility or availability of these algorithms (i.e., lack of downloadable programs or web servers) has prevented their widespread use by protein chemists or structural biologists. To increase the utility and accessibility of contact order calculations for experimentalists, we have developed a web server that both calculates Abs_CO (and CO) from 3D coordinate data and predicts Abs_CO (and CO) from protein sequence data. This public web server (http://www.copredictor.ca) calculates Abs_CO using Equation (10.1), where two distinct residues form a contact if there are two heavy atoms (C, O, S, or N), one from each residue, within ![]() Å. It is worth pointing out that in the literature, several different distance thresholds other than
Å. It is worth pointing out that in the literature, several different distance thresholds other than ![]() Å have been tested with no significant difference found [48]; besides, some constraints on contacting residue sequential separation (such as at least three residues apart from each other) and/or heavy atoms (such as Cβ only) have been suggested [7], but again no essential difference exists as the underlying idea of using CO to quantify the topology of a protein's native state 3D structure remains the same.
Å have been tested with no significant difference found [48]; besides, some constraints on contacting residue sequential separation (such as at least three residues apart from each other) and/or heavy atoms (such as Cβ only) have been suggested [7], but again no essential difference exists as the underlying idea of using CO to quantify the topology of a protein's native state 3D structure remains the same.
The input to our Abs_CO calculator is a three-dimensional structure (uploading either the PDB [6] coordinate file or the PDB ID). The calculator typically returns the Abs_CO value within a few seconds. We note that there is an earlier published CO calculation server [36], and these two calculators returned nearly identical Abs_CO values with a correlation coefficient of ![]() . However, this earlier server failed (tested on May 2, 2007) to recognize 61% of the
. However, this earlier server failed (tested on May 2, 2007) to recognize 61% of the ![]() monomeric PDB files that were successfully processed by our server.
monomeric PDB files that were successfully processed by our server.
10.5 Contact order prediction by homology
Many protein properties, including tertiary structure, secondary structure, and solvent accessibility, can be predicted via homology [35]. In other words, the properties of a query sequence can be predicted by directly transferring the properties or features of a homologous protein to the query protein. Since CO is a property that is a function of structure, we hypothesized that the calculated CO of known 3D structures could be used to predict the CO of homologous proteins.
This approach has been implemented in our public CO web server http://www.copredictor.ca.
More specifically, the Abs_CO for 16,499 nonredundant proteins obtained from the PDB were calculated. These proteins were selected using the PDB culling/filtering service called PISCES [45]. Structures were initially selected using a 95% identity sequence redundancy cutoff and a requirement for better than 3 Å resolution (for X-ray structures). Structures were further processed by removing disordered structures (i.e., total secondary structure content <10%) as well as all membrane proteins (i.e., membrane ![]() barrel and trans-membrane helix proteins). These 16,499 sequences with their Abs_CO values form the CO database that the server uses for its homology search, via a local copy of BLAST [2].
barrel and trans-membrane helix proteins). These 16,499 sequences with their Abs_CO values form the CO database that the server uses for its homology search, via a local copy of BLAST [2].
When the input to our web server is the primary sequence of a query protein, the BLAST search finds a homolog from the web server's CO database of 16,499 sequences. If this homolog is not an exact match to the query sequence but exhibits more than 20% sequence identity (which is computed as the number of identical residues divided by the query sequence length) and the query sequence is ![]() of the length of the homolog, the pre-computed Abs_CO of the homolog is used as the predicted Abs_CO of the query sequence [40].
of the length of the homolog, the pre-computed Abs_CO of the homolog is used as the predicted Abs_CO of the query sequence [40].
We performed tests through a modified five-fold cross-validation using a random sample of 1,000 sequences on the CO database, using a variety of sequence identity cutoffs and sequence length thresholds to assess their influence on both the accuracy and the coverage (coverage refers to the percentage of query sequences that could be predicted by this homology-based method) [40]. Among other tested settings, the 20% sequence identity cutoff and the 40% length threshold provided the best overall accuracy–coverage tradeoff. Under this specific setting, on average ![]() (
(![]() standard deviation) of sample sequences found homologs in the CO database. CO prediction by sequence homology turns out to be a surprisingly accurate prediction method, with a correlation coefficient of
standard deviation) of sample sequences found homologs in the CO database. CO prediction by sequence homology turns out to be a surprisingly accurate prediction method, with a correlation coefficient of ![]() between the
between the ![]() pairs of true absolute contact order and predicted absolute contact order values. These Abs_CO predictions are on average
pairs of true absolute contact order and predicted absolute contact order values. These Abs_CO predictions are on average ![]() correct (
correct (![]() standard deviation) [40].
standard deviation) [40].
10.6 Contact order prediction from sequence
Obviously not every protein can have its CO calculated from coordinate data or predicted via sequence homology. In the above experiment with 1,000 sequences, ![]() of them failed to find a homolog. In order to deal with the situation where no homolog can be found for CO prediction, we have implemented a regression-based prediction method in the http://www.copredictor.ca CO web server [40]. As Abs_CO is observed to correlate well with a linear combination of the percentage of residues in
of them failed to find a homolog. In order to deal with the situation where no homolog can be found for CO prediction, we have implemented a regression-based prediction method in the http://www.copredictor.ca CO web server [40]. As Abs_CO is observed to correlate well with a linear combination of the percentage of residues in ![]() -helices
-helices ![]() , the percentage of residues in
, the percentage of residues in ![]() strands
strands ![]() and the protein length
and the protein length ![]() , a linear regression to optimize the correlation between Abs_CO and the protein primary and secondary structures was developed
, a linear regression to optimize the correlation between Abs_CO and the protein primary and secondary structures was developed
where ![]() ,
, ![]() , are the coefficients of the three factors
, are the coefficients of the three factors ![]() ,
, ![]() , and
, and ![]() , and
, and ![]() is a constant. During prediction, proteins with unknown three-dimensional structure can have their secondary structures predicted by Proteus [35] (or any other similar programs such as PSIPRED [34]). Proteus is a secondary structure predictor that achieves highly accurate predictions (
is a constant. During prediction, proteins with unknown three-dimensional structure can have their secondary structures predicted by Proteus [35] (or any other similar programs such as PSIPRED [34]). Proteus is a secondary structure predictor that achieves highly accurate predictions (![]() accuracy score of
accuracy score of ![]() ) based on VADAR [46] and the PPT-Database [47]. To train the regressor, that is, to determine the values for
) based on VADAR [46] and the PPT-Database [47]. To train the regressor, that is, to determine the values for ![]() ,
, ![]() ,
, ![]() , a set of
, a set of ![]() monomeric proteins with an X-ray resolution <1.5 Å were extracted from the PDB [6]. Readers may refer to the study by shi et al. [40] for more detailed statistics on these proteins, such as their SCOP classification [4] and length distribution. Using these
monomeric proteins with an X-ray resolution <1.5 Å were extracted from the PDB [6]. Readers may refer to the study by shi et al. [40] for more detailed statistics on these proteins, such as their SCOP classification [4] and length distribution. Using these ![]() high-resolution three-dimensional protein structures through a five-fold cross-validation, the optimal parameters in Equation (10.4) localize at
high-resolution three-dimensional protein structures through a five-fold cross-validation, the optimal parameters in Equation (10.4) localize at ![]() ,
, ![]() ,
, ![]() , and
, and ![]() .
.
In addition to this three-factor linear CO predictor, denoted as F3-LR, several other linear regressors have also been developed to include more factors that might be strongly correlated to Abs_CO. These factors are the number of ![]() hairpins (two adjacent
hairpins (two adjacent ![]() -strand segments form a hairpin if they are separated by two to five residues), the number of distant beta strands (two adjacent
-strand segments form a hairpin if they are separated by two to five residues), the number of distant beta strands (two adjacent ![]() -strand segments are considered “distant” if they are separated by at least five residues), the amino acid frequencies, and the hydrophobicity frequencies [40]. Besides linear regression, support vector regression (SVR) [41] and neural network (NN) [3] methods have also been implemented on our web server.
-strand segments are considered “distant” if they are separated by at least five residues), the amino acid frequencies, and the hydrophobicity frequencies [40]. Besides linear regression, support vector regression (SVR) [41] and neural network (NN) [3] methods have also been implemented on our web server.
On the training dataset of ![]() proteins, the five-fold cross-validation in the study by shi et al. [40] shows that the simplest regression model (F3-LR) actually performed remarkably well, achieving a correlation coefficient of
proteins, the five-fold cross-validation in the study by shi et al. [40] shows that the simplest regression model (F3-LR) actually performed remarkably well, achieving a correlation coefficient of ![]() . Using more factors was shown to improve the correlation coefficient by a small amount. For instance, using four more factors, the F7-LR model improved the correlation coefficient by
. Using more factors was shown to improve the correlation coefficient by a small amount. For instance, using four more factors, the F7-LR model improved the correlation coefficient by ![]() ; and the F27-LR achieved a correlation coefficient of
; and the F27-LR achieved a correlation coefficient of ![]() .
.
10.7 The public contact order web server
A contact order calculator, the homology-based contact order predictor, and the linear regression–based contact order predictors are implemented as a public web server http://www.copredictor.ca. The input to the server can be either a three-dimensional structure (either the PDB coordinate file or the PDB Id) or the primary sequence of the query protein. When the input is a sequence, our server will first use BLAST to identify a sequence in our CO database that is either identical or the most homologous to the query. There are three possible scenarios: (1) if the input is a 3D structure, or the query sequence matches exactly a known structure in our database of 16,499 proteins, our server will calculate its Abs_CO; (2) if the input is a sequence and the BLAST search finds a homolog that is not an exact match but satisfies certain criteria, the precomputed Abs_CO of the homolog is used as the predicted Abs_CO of the query sequence; (3) if the input is a sequence and has no BLAST match that falls into scenario 2, our server will call Proteus to predict the secondary structure content for the query protein, and then report its Abs_CO predicted by Equation (10.4).
We have used our program to predict the Abs_COs and the derived protein folding rates [22] for all the proteins collected in TrEMBL (http://www.uniprot.org) [44], as of July 21, 2011. The result is available as a downloadable file from the server website.
10.8 Conclusions
Contact order (CO) is the most widely used approach to quantitatively measure the topological complexity of protein structures. CO can be used to accurately predict protein folding rates and to assist in de novo protein structure prediction/generation. However, the utility of the CO method, especially for experimentalists, has been limited by the lack of availability of programs or web servers that either support CO calculation (from coordinate data) or allow CO prediction (from sequence data). For proteins with solved three-dimensional structures, we have developed a public web server (http://www.copredictor.ca) that accurately calculates COs, thereby overcoming the limited functionality of an earlier web server. In addition, this server also offers a very effective method for predicting protein contact order from primary sequence data. This latter function is particularly important because of the 3D structure only a tiny fraction of known proteins is known. Many factors, in particular the percentage of residues in alpha helices, the percentage of residues in beta strands, and sequence length, are known to be strongly correlated with the absolute contact order. Tests using a large dataset of high-resolution monomeric proteins showed that our method achieved a correlation coefficient of 0.857–0.870. In addition, we have shown that it is possible to use sequence homology to accurately predict the contact order for proteins for which no 3D structure exists, with a high correlation coefficient of ![]() . This web server has been recognized or used to help in a number of studies in protein folding [8, 9, 39, 49] and to demonstrate the effectiveness of
. This web server has been recognized or used to help in a number of studies in protein folding [8, 9, 39, 49] and to demonstrate the effectiveness of ![]() -NN [39].
-NN [39].
References
1. Alm E, Baker D, Matching theory and experiment in protein folding, Curr. Opin. Struct. Biol. 9:189–196 (1999).
2. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool, J. Mol. Biol. 215:403–410 (1990).
3. Anderson JA, An Introduction to Neural Networks, MIT Press, 1995.
4. Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG, SCOP database in 2004: Refinements integrate structure and sequence family data, Nucleic Acids Res. 32:D226–D229 (2004).
5. Baker D, A surprising simplicity to protein folding, Nature 405:39–42 (2000).
6. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The protein data bank, Nucleic Acids Res. 28:235–242 (2000).
7. Bonneau R, Ruczinski I, Tsai J, Baker D, Contact order and ab initio protein structure prediction, Protein Sci. 11:1937–1944 (2002).
8. Campbell K, Kurgan L, Sequence-only based prediction of beta-turn location and type using collocation of amino acid pairs, Open Bioinformatics J. 2:37–49 (2008).
9. Chen K, Stach W, Homaeian L, Kurgan L, iFC2: An integrated web-server for improved prediction of protein structural class, fold type, and secondary structure content, Amino Acids 40:963–973 (2011).
10. Chivian D, Kim DE, Malmstrom L, Schonbrun J, Rohl CA, Baker D, Prediction of CASP6 structures using automated Robetta protocols. (Available at http://robetta.bakerlab.org/pub/dylan/).
11. Dill KA, Fiebig KM, Chan HS, Cooperativity in protein-folding kinetics, Proc. Nat. Acad. Sci. USA 90:1942–1946 (1993).
12. Fariselli P, Casadio R, RCNPRED: prediction of the residue co-ordination numbers in proteins, Bioinformatics 17:202–204 (2001).
13. Fezoui Y, Braswell EH, Xian W, Osterhout JJ, Dissection of the de novo designed peptide alpha-t-alpha: Stability and properties of the intact molecule and its constituent helices, Biochemistry 38:2796–2804 (1999).
14. Fiebig KM, Dill KA, Protein core assembly process, J. Chem. Phys. 98:3475–3487 (1993).
15. Flöckner H, Braxenthaler M, Lackner P, Jaritz M, Ortner M, Sippl MJ, Progress in fold recognition, Proteins 23:376–386 (1995).
16. Grantcharova V, Alm EJ, Baker D, Horwich AL, Mechanisms of protein folding, Curr. Opin. Struct. Biol. 11:70–82 (2001).
17. Gromiha MM, Selvaraj S, Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction, J. Mol. Biol. 310:27–32 (2001).
18. Gromiha MM, Thangakani AM, Selvaraj S, Fold-rate: Prediction of protein folding rates from amino acid sequence, Nucleic Acids Res. 34:W70–W74 (2006).
19. Hamelryck T, An amino acid has two sides: A new 2d measure provides a different view of solvent exposure. Proteins 59:38–48 (2005).
20. Ishida T, Nakamura S, Shimizu K, Potential for assessing quality of protein structure based on contact number prediction, Proteins 64:940–947 (2006).
21. Ivankov DN, Finkelstein AV, Prediction of protein folding rates from the amino acid sequence-predicted secondary structure, Proc. Nat. Acad. Sci. USA 101:8942–8944 (2004).
22. Ivankov DN, Garbuzynskiy SO, Alm E, Plaxco KW, Baker D, Finkelstein AV, Contact order revisited: Influence of protein size on the folding rate, Protein Sci. 13:2057–2062 (2003).
23. Kabsch W, Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features, Biopolymers 22:2577–2637 (1983).
24. Karchin R, Cline M, Karplus K, Evaluation of local structure alphabets based on residue burial, Proteins 55:508–518 (2004).
25. Kihara D, On the effect of long range interactions on secondary structure formation in proteins, Protein Sci. 14:1955–1963 (2005).
26. Kim PS, Baldwin RL, Intermediates in the folding reactions of small proteins, Annu. Rev. Biochem. 59:631–660 (1990).
27. Kinjo AR, Horimoto K, Nishikawa K, Predicting absolute contact numbers of native protein structure from amino acid sequence, Proteins 58:158–165 (2005).
28. Kinjo AR, Nishikawa K, Predicting secondary structures, contact numbers, and residue-wise contact orders of native protein structure from amino acid sequence using critical random networks, Biophysics 1:67–74 (2005).
29. Kinjo AR, Nishikawa K, Recoverable one-dimensional encoding of three-dimensional protein structures, Bioinformatics 21:2167–2170 (2005).
30. Kinjo AR, Nishikawa K, CRNPRED: Highly accurate prediction of one-dimensional protein structures by large-scale critical random networks, BMC Bioinformatics 7:401 (2006).
31. Koga N, Takada S, Roles of native topology and chain-length scaling in protein folding: A simulation study with a Go-like model, J. Mol. Biol. 313:171–180 (2001).
32. Kubelka J, Hofrichter J, Eaton WA, The protein folding “speed limit,” Curr. Opin. Struct. Biol. 14:76–88 (2002).
33. Lee B, Richards FM, The interpretation of protein structures: Estimation of static accessibility, J. Mol. Biol. 55:379–380 (1971).
34. McGuffin LJ, Bryson K, Jones DT, The PSIPRED protein structure prediction server, Bioinformatics 16:404–405 (2000).
35. Montgomerie S, Sundararaj S, Gallin W, Wishart DS, Improving the accuracy of protein secondary structure prediction using structural alignment, BMC Bioinformatics 7:301 (2006).
36. Plaxco KW, Simons KT, Baker D, Contact order, transition state placement and the refolding rates of single domain proteins, J. Mol. Biol. 227:985–994 (1998). (Available at http://depts.washington.edu/bakerpg/contact_order/).
37. Pollastri G, Baldi P, Fariselli P, Casadio R, Improved prediction of the number of residue contacts in proteins by recurrent neural networks, Bioinformatics 17:S234–S242 (2001).
38. Pollastri G, Baldi P, Fariselli P, Casadio R, Prediction of coordination number and relative solvent accessibility in proteins, Proteins 47:142–153 (2002).
39. Segal MR, A novel topology for representing protein folds, Protein Sci. 18:686–693 (2009).
40. Shi Y, Zhou J, Arndt D, Wishart DS, Lin G, Protein contact order prediction from primary sequences, BMC Bioinformatics 9:255 (2008). (Available at http://www.copredictor.ca/).
41. Smola AJ, Schölkopf B, A tutorial on support vector regression, Stat. Comput. 14:199–222 (2003).
42. Song J, Burrage K, Predicting residue-wise contact orders in proteins by support vector regression, BMC Bioinformatics 7:425 (2006).
43. Tanaka S, Scheraga HA, Model of protein folding: inclusion of short-, medium-, and long-range interactions, Proc. Nat. Acad. Sci. USA 72(10): 3802–3806 (1975).
44. The UniProt Consortium, Ongoing and future developments at the Universal Protein Resource, Nucleic Acids Res. 39:D214–D219 (2011). (Available at http://www.uniprot.org/).
45. Wang G, Dunbrack RL Jr, PISCES: A protein sequence culling server, Bioinformatics 19:1589–1591 (2003).
46. Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS, VADAR: A web server for quantitative evaluation of protein structure quality, Nucleic Acids Res. 31:3316–3319 (2003).
47. Wishart DS, Arndt D, Berjanskii M, Guo AC, Shi Y, Shrivastava S, Zhou J, Zhu Y, Lin G, PPT-DB: The protein property prediction and testing database, Nucleic Acids Res. 36:D222–D229 (2008).
48. Yuan Z, Better prediction of protein contact number using a support vector regression analysis of amino acid sequence, BMC Bioinformatics 6:248 (2005).
49. Zhang H, Zhang T, Chen K, Kedarisetti KD, Mizianty MJ, Bao Q, Stach W, Kurgan L, Critical assessment of high-throughput standalone methods for secondary structure prediction, Brief. Bioinformatics 12:672–688 (2011).