
Chapter 8
Prediction of RNA Binding Sites in Proteins
8.1 Introduction
Protein–RNA interactions play a key role in a number of biological processes in DNA packaging and replication, mRNA processing, protein synthesis, assembly, and function of ribosomes and eukaryotic spliceosomes. A reliable identification of RNA binding sites in proteins is important for functional annotation and site-directed mutagenesis. However, it is time-consuming and labor-intensive to detect the interaction sites in proteins by traditional experimental methods. There are some computational methods that have been proposed to address this challenge. Generally, prediction of RNA binding sites is based on the sequence and structure features identified from protein and its RNA partner. The residue properties as well as various element features are detected and combined together into description vectors to represent the interacting events. For the encoding scheme, numerous methods have been proposed to describe the interacting preferences of protein residue and its RNA partners. In this chapter, we provide an introduction for the prediction of RNA binding sites in proteins by machine learning algorithms, such as neural network, naive Bayes, support vector machines, and random forest. On the basis of these classification methods, we can identify the RNA binding sites of proteins by various features underlying the interaction.
8.2 Background
It is crucial to decipher the mechanism of how proteins interact with other molecules in the understanding of cellular processes [2, 12, 38]. RNA undergoes diverse posttranscriptional regulation of gene expression, including regulation of its transportation, localization, and decay [10]. In many cases, this process occurs through elements on the mRNA molecule that interact with the hundreds of RNA binding proteins existing in the cell [12, 24]. Interactions between proteins and RNA molecules play an essential role in a variety of biological activities, such as posttranscriptional gene regulation, alternative splicing, translation, and infections by RNA viruses [31]. Therefore, it is important to understand the principle of protein–RNA interactions and identify their interaction sites when selectingactivators and inhibitors in rational drug design. RNA recognition by proteins is mediated primarily by certain classes of RNA binding domains and motifs [25, 28]. The correlated patterns of sequence and structure in RNA binding proteins can then be recognized to bind to specific RNA sequences and folds. In recognition of RNA functional importance in living molecules and close association with protein in its activities, experimental and computational studies of protein–RNA complexes have been substantially increased [8, 15]. Various approaches have been proposed to study protein–RNA interactions [24], but precise mechanisms of interaction are far from for fully understood. Therefore, to clearly elucidate protein–RNA interacting patterns, it is necessary to develop a reliable method for predicting protein–RNA interacting sites, in particular by exploiting the increasingly accumulated data of protein–RNA complexes.
Many studies indicate that there is a strong relationship between interaction residues and their compositions in protein–RNA complexes [7, 8, 18]. In building a machine learning method for predicting protein–RNA binding sites, two factors play crucial roles in the predictor:
In this chapter, we mainly describe the protocols of predicting RNA binding sites in proteins by feature based machine learning methods. The available protein–RNA complexes from the Protein Data Bank (PDB) [4] are selected to build the data source and define the binding sites in proteins. Various features of sequence and structure are derived comprehensively to represent protein residues for characterizing the protein–RNA binding events. The features are collected and combined together by encoding feature vectors. Machine learning methods (e.g., NN, NB, SVM, RF) are implemented to ascertain the features underlying the RNA binding residues as well as those of non-RNA-binding residues. To check the prediction performance of these predictors, cross-validation processes are used to show the efficiency and effectiveness of these pipelines. Furthermore, the trained predictors are used to predict novel RNA binding sites in proteins. In this chapter, we also compare these features and those of the existing methods. In particular, we identify the importance of each feature in determining the specificity of protein–RNA interaction, as well as the contribution of various hybrid features in the prediction.
8.3 Framework of Prediction
Figure 8.1 is a flowchart showing steps involved in the prediction of RNA binding sites in proteins. The framework of these steps can be summarized as follows. Given a protein [sequence or structure shown in diagram (a)], the task involved in this prediction is to annotate where the RNA binding events take place [shown in structure (c)] by a predictor [shown in diagram (b)]. The predictor is often built by a classifier trained on available protein–RNA binding information. Given protein–RNA complexes, we first define the RNA binding residues from their three-dimensional (3D) structures [diagram (d)]. In these feature-based machine learning approaches, a crucial step is to identify the features of these binding residues, such as sequence-based features of physicochemical atoms and structure-based features of solvent accessible surface areas. Often, we also identify some defined features that are considered to be closely related to the interaction between protein and RNA. Each protein residue is then represented by a derived feature vector [diagram (e)]. In the training dataset, we implement a machine learning algorithm to learn the distinctive features underlying these RNA binding residues as well as non-RNA-binding residues. The predictor is trained to distinguish the features of RNA binding residues and those of non-RNA-binding residues simultaneously [graph (f)]. The prediction performance of the classifier is often evaluated by a cross-validation process [graph (g)]. After we achieve acceptable training results, we can use the predictor to predict
Figure 8.1 Framework for predicting RNA binding sites in proteins.
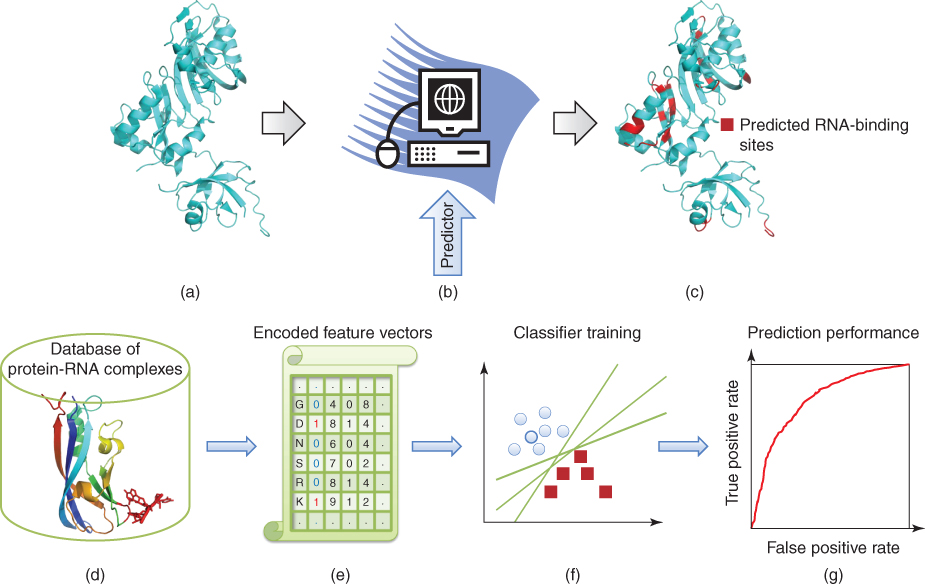
RNA binding sites in a novel protein after the target protein is encoded with the same feature vectors as described in the training data.
The overall prediction performance for a predictor is first evaluated by the statistical measurements in the cross-validation, such as fivefold cross-validation; that is, the whole dataset is randomly partitioned into five groups of equal size. To ensure that the training process is completely independent from the test data, the classifier is trained on the four groups and tested on the control group, and each of them is chosen for the assessment one by one. The predictive results are evaluated by different measures, including sensitivity (SN), specificity (SP), accuracy (ACC), ![]() measure, and Matthews correlation coefficient (MCC). Mathematically, they are defined by the following equations:
measure, and Matthews correlation coefficient (MCC). Mathematically, they are defined by the following equations:
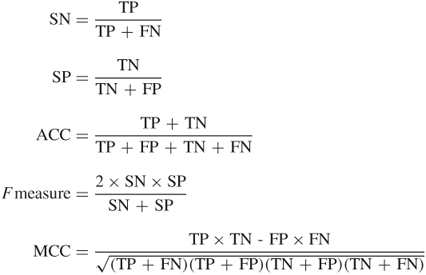
where TP, FN, FP, and TN are the numbers of true positive, false negative, false positive, and true negative residues in the prediction, respectively. To present the interrelationship of specificity and sensitivity of the identification, the receiver operating characteristic (ROC) curve is often used, and the area under the ROC curve (AUC) is also calculated. The ![]() measure is the harmonic mean of sensitivity and specificity. Usually the maximum
measure is the harmonic mean of sensitivity and specificity. Usually the maximum ![]() measure point is chosen as the cutoff for sensitivity and specificity in ROC curves of the prediction performance. The MCC value ranges between 1 (all predictions are correct) and
measure point is chosen as the cutoff for sensitivity and specificity in ROC curves of the prediction performance. The MCC value ranges between 1 (all predictions are correct) and ![]() 1 (none are correct).
1 (none are correct).
The purpose of cross-validation is to evaluate the efficiency and effectiveness of the proposed prediction method on available protein–RNA complexes. When we intend to identify the RNA binding sites in a novel protein, the former built classifier can be trained in the whole dataset. After the corresponding features of the predicting protein are encoded, the RNA binding residues can be predicted by the trained predictor. The generalization and extension ability of prediction plays a crucial role in the applications. The flexibility and scale of application are highly related to the required features in the proposed predictor. For instance, some structure-based features in a protein are not available when there is no 3D structure. In these cases, we can train the predictor by sequence-based features, and then the RNA binding sites can be predicted only by sequence information.
8.4 Description Features of Protein RNA Binding Sites
For predicting RNA binding sites in proteins by machine learning methods, we need to encode the residues by various descriptors mined from available protein sequences and structures. Compared to some energy-based methods for defining features of the protein–RNA interaction sites, sequence-derived and structure-derived features focus mainly on the physicochemical patterns, evolutionary information, solution accessible surfaces, and sidechain environment of residues. Roughly, these features can be categorized into sequence-derived and structure-derived classes. Some of them, such as interaction propensity, are also defined by this available information.
8.4.1 Definition of RNA Binding Sites
RNA binding events often take place at the pockets or cavities on protein surfaces [1, 22, 26]. When we train the predictor, we need to define positive samples as well as negative samples, that is, true RNA binding residues and true non-RNA-binding residues in the proteins. When the 3D structure of protein–RNA complex is available, the closest distance between the atoms of protein residue and that of its partner RNA nucleotide residue can be easily calculated. When the distance is shorter than that of a given threshold, the amino acid residue is defined as RNA binding residue [32]. Furthermore, some energy-based methods are proposed to define the RNA binding residues in proteins. For instance, a residue is defined as an RNA binding residue if any of its nonhydrogen atoms is within van der Waals contact or hydrogen binding distance to any RNA nonhydrogen atom directly or indirectly by a bridging water molecule [1, 39]. Several methods, such as ENTANGLE, can be used to detect the interacting residues by searching appropriate hydrogen bonding and stacking, electrostatic, hydrophobic, and van der Waals interactions between protein and RNA residues [1]. The structure information can also be implemented to define RNA binding sites. For instance, on comparison of solvent accessible surface area of the protein structure with and without RNA, the degree of difference of residues can be used to distinguish RNA binding residues with non-RNA-binding residues [18].
8.4.2 Sequence-Based Features
It is relatively easy to obtain the sequence information of interacting protein and RNA. Hence, the sequence-derived features are wildly implemented in building the feature vectors. Strong biases of different types of amino acid residues in RNA binding sites have been reported, such as the abundancy of arginine-rich motifs [1]. This motivates the inclusion of residue information in the prediction. Often, 20 types of amino acids and their physicochemical characteristics are identified andrepresented as the feature descriptors. The physicochemical characteristics of an amino acid residue can be described by three values: number of atoms, number of electrostatic charge, and number of potential hydrogen bonds [21, 23, 29]. The hydrophobic effect is also shown to be important in protein–RNA binding. The hydrophobicity of an amino acid residue can be described by the hydrophobic index designed by Sweet and Eisenberg [30].
Evolutionary information is often used to locate the functional sites of RNA binding. The position-specific scoring matrix (PSSM) is a commonly used representation of evolutionary patterns in biological sequences. For a PSSM profile, a residue ![]() at position
at position ![]() in a protein sequence is presented by an evolutionary information vector consisting of loglikelihoods for 20 different amino acids [6]. The sequence conservation status is given by the weighted positions of RNA binding events. The values of sequence conservation for amino acids are often obtained by PSI-BLAST [3] search of the protein chain sequence in a nonredundant sequence database [34]. The round of iteration can be set to be 3, and the result of the PSI-BLAST search well be a PSSM. Specifically, the conservation score of each residue is referred to the corresponding diagonal value of the matrix [23].
in a protein sequence is presented by an evolutionary information vector consisting of loglikelihoods for 20 different amino acids [6]. The sequence conservation status is given by the weighted positions of RNA binding events. The values of sequence conservation for amino acids are often obtained by PSI-BLAST [3] search of the protein chain sequence in a nonredundant sequence database [34]. The round of iteration can be set to be 3, and the result of the PSI-BLAST search well be a PSSM. Specifically, the conservation score of each residue is referred to the corresponding diagonal value of the matrix [23].
8.4.3 Structure-Based Features
These former values are related only to protein sequence and do not contain any structural information. When the 3D structure of the protein–RNA complex is available, some important structure features can be derived to characterize these amino acid residues. Secondary structure information has been shown to be correlated with protein–RNA interaction [1, 31]. Two types of secondary structures are RNA binding type. (1) binding between ![]() -helix or loop and a groove of the RNA pockets and (2) binding between
-helix or loop and a groove of the RNA pockets and (2) binding between ![]() -sheet surface and unpaired RNA bases [11, 39]. It is expected to improve the prediction accuracy of RNA binding residues after inclusion of such features. The secondary structure of an amino acid residue can be calculated by DSSP algorithm [16]. It would be divided into three states: helix, sheet, and coil. DSSP secondary structure types I, G, and H are considered as helix; types E and B are considered as sheet; types T, S, and blank are considered as coil. We can use (1,0,0), (0,1,0), and (0,0,1) to represent the three types, respectively [23, 37].
-sheet surface and unpaired RNA bases [11, 39]. It is expected to improve the prediction accuracy of RNA binding residues after inclusion of such features. The secondary structure of an amino acid residue can be calculated by DSSP algorithm [16]. It would be divided into three states: helix, sheet, and coil. DSSP secondary structure types I, G, and H are considered as helix; types E and B are considered as sheet; types T, S, and blank are considered as coil. We can use (1,0,0), (0,1,0), and (0,0,1) to represent the three types, respectively [23, 37].
The area accessible to a solvent on a protein surface is important in the protein function of binding RNA. The relative accessible surface area delineates the local solvent environment of protein–RNA interaction [23]. This should improve the prediction of RNA binding residues in proteins. The accessible surface area of an amino acid can generally be calculated by DSSP [16]. Then we calculate the relative property by dividing the accessible surface area by the accessible surface area of fully exposed amino acids. The accessible surface areas ofthe fully exposed amino acids are available according to Rost and Sander [27].
The p![]() value of an amino acid sidechain defines the pH-dependent characteristic of a protein. The descriptor represents the pH-dependence of activity and protein stability. This is an important factor in determining environmental characteristics of a protein [9]. It is widely used in the prediction of protein–RNA binding sites for its important effect on the protein environment [23, 35]. The normal p
value of an amino acid sidechain defines the pH-dependent characteristic of a protein. The descriptor represents the pH-dependence of activity and protein stability. This is an important factor in determining environmental characteristics of a protein [9]. It is widely used in the prediction of protein–RNA binding sites for its important effect on the protein environment [23, 35]. The normal p![]() values of amino acids in calculating protein sidechain environment properties [9] can be implemented in the descriptors of characterizing the protein residues.
values of amino acids in calculating protein sidechain environment properties [9] can be implemented in the descriptors of characterizing the protein residues.
To distinguish the RNA binding sites of protein, various descriptors are derived from available information. They are usually combined together as feature vectors to encode the residues. We often define some new descriptors to represent the residue specificity to distinguish the RNA binding events. For instance, some statistical information of binding or interaction propensity between protein and RNA residues can be derived [23, 31].
8.4.4 Derived Features of Interaction Propensity
To improve the state of the art in machine learning methods for predicting protein–RNA binding residues, some methods have been developed to define various measurements of binding propensity between protein and its RNA partners [31]. We also introduced a new interaction propensity for binding residues that highlights residue pairs on protein–RNA interface [23]. Protein–RNA interactions are reported to involve a number of nonpolar weak interactions. Also, the interactions often occur in a patch on the surface [15, 18]. Therefore, a statistical measure of binding residue pairs in the protein–RNA interface definitely sheds new light on the binding characteristics and features. These derived features are expected to improve the accuracy of predicting RNA binding residues in proteins.
In several previous studies, strong biases have been reported in the types of amino acids, presenting in protein–RNA interfaces such as the abundant occurrence of arginine-rich motifs [15, 17, 18, 31]. Terribilini et al. [31], defined the interface propensity for each amino acid type ![]() as
as
![]()
Apparently, an interface propensity value greater than 0 indicates that an amino acid is overrepresented in RNA–protein interfaces relative to the protein sequence as a whole.
Kim et al. [17] defined an interaction propensity for each of the 20 amino acids binding each of the four nucleotides, respectively. Amino acids on the protein surface were determined if the relative accessibility was ![]() % [20]. The interaction propensity
% [20]. The interaction propensity ![]() between amino acid
between amino acid ![]() and nucleotide
and nucleotide ![]() was then defined by [17]
was then defined by [17]
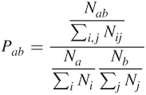
where
![]() = number of amino acid residues
= number of amino acid residues ![]() interacting with nucleotide residue
interacting with nucleotide residue ![]()
![]() = total number of interacting pairs of any amino acid and nucleotide
= total number of interacting pairs of any amino acid and nucleotide
![]() = number of amino acids
= number of amino acids ![]()
![]() = total number of amino acids
= total number of amino acids
![]() = number of nucleotides
= number of nucleotides ![]()
![]() = total number of nucleotides
= total number of nucleotides
![]() = ratio of occurrence of amino acid
= ratio of occurrence of amino acid ![]() andnucleotide
andnucleotide ![]() to total number of all amino acids binding to any nucleotide on protein surface.
to total number of all amino acids binding to any nucleotide on protein surface.
![]() = ratio of frequency of nucleotide
= ratio of frequency of nucleotide ![]() to that of all nucleotides on surface.
to that of all nucleotides on surface.
In 2006, another interaction propensity was defined by amino acid residue singlet interface propensity and residue doublet propensity in protein–RNA interfaces [18]. The residue singlet interface propensity (![]() ) was calculated for each amino acid type
) was calculated for each amino acid type ![]() as a fraction of the frequency that
as a fraction of the frequency that ![]() contributes to a protein–RNA interface compared to the frequency that
contributes to a protein–RNA interface compared to the frequency that ![]() contributes to a protein surface:
contributes to a protein surface:

Here, ![]() is the number of amino acid type
is the number of amino acid type ![]() on the protein surface and
on the protein surface and ![]() is that in the RNA interface. The number
is that in the RNA interface. The number ![]() was obtained from the population of nonhomologous proteins in the PDB and
was obtained from the population of nonhomologous proteins in the PDB and ![]() was determined from the data for protein–RNA complexes [18]. Similarly, the residue doublet interface propensity
was determined from the data for protein–RNA complexes [18]. Similarly, the residue doublet interface propensity ![]() was calculated as follows. The frequency
was calculated as follows. The frequency ![]() of doublet amino acid type
of doublet amino acid type ![]() on the protein surface and that in the protein–RNA interface
on the protein surface and that in the protein–RNA interface ![]() were calculated from the number of residue doublets as
were calculated from the number of residue doublets as
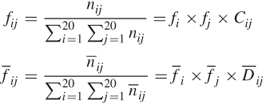
where ![]() is the number of doublet type
is the number of doublet type ![]() on the protein surface and
on the protein surface and ![]() is that in the RNA interface. Terms
is that in the RNA interface. Terms ![]() ,
, ![]() ,
, ![]() ,
, ![]() are given in the single interface propensity, and
are given in the single interface propensity, and ![]() and
and ![]() are the surface and interface residue doublet coefficient, respectively. If amino acid types
are the surface and interface residue doublet coefficient, respectively. If amino acid types ![]() and
and ![]() have no correlation on the protein surface, then
have no correlation on the protein surface, then ![]() and in the RNA interface,
and in the RNA interface, ![]() . Then, the residue doublet preference in the RNA interface was determined to be
. Then, the residue doublet preference in the RNA interface was determined to be
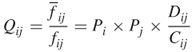
where ![]() and
and ![]() are the residue singlet interface propensities. The residue doublet interface propensity was defined as
are the residue singlet interface propensities. The residue doublet interface propensity was defined as
![]()
The mutual interaction propensity between protein residues and its binding RNA partners will discriminatively characterize the RNA binding residues in protein sequences, and the derived features from the structure information will benefit the prediction accuracy of a classifier that considers the mutual relationship between interacting residues and nucleotides. With this in mind, we identified and quantified the mutual dependence between protein residues and RNA nucleotide by calculating a new measure: mutual interaction propensity [23]. We highlighted the important role of the neighbor residues in determining the specificity of biochemical features and the preference for interaction with nucleotides for an amino acid residue [31, 36]. Hence, we defined the mutual interaction propensity of a residue triplet and a nucleotide. A triplet is regarded as interacting with a nucleotide when its central residue interacts with the nucleotide. The mutual interaction propensity was defined as
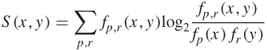
where ![]() represents a residue triplet,
represents a residue triplet, ![]() represents a nucleotide (i.e.,
represents a nucleotide (i.e., ![]()
![]() );
); ![]() represents the frequency of
represents the frequency of ![]() interacting
interacting ![]() in the protein–RNA pair
in the protein–RNA pair ![]() , where
, where ![]() is the number of residue triplet
is the number of residue triplet ![]() binding to nucleotide
binding to nucleotide ![]() and
and ![]() is the total number of residue triplet and nucleotide pairs in the protein–RNA pair
is the total number of residue triplet and nucleotide pairs in the protein–RNA pair ![]() ; and
; and ![]() represents the frequency of the residue triplet
represents the frequency of the residue triplet ![]() in protein
in protein ![]() , where
, where ![]() is the number of residue triplet
is the number of residue triplet ![]() and
and ![]() is the total number of all residue triplets in the protein
is the total number of all residue triplets in the protein ![]() . Similarly,
. Similarly, ![]() represents the frequency of a nucleotide
represents the frequency of a nucleotide ![]() , where
, where ![]() is the number of nucleotide
is the number of nucleotide ![]() and
and ![]() is the total number of nucleotides in the RNA
is the total number of nucleotides in the RNA ![]() . The interaction propensity of a triplet
. The interaction propensity of a triplet ![]() and a nucleotide
and a nucleotide ![]() is calculated on all interacting protein–RNA pairs in the dataset. We identify the binding specificity of mutual interaction propensity between the existing triplets and four RNA nucleotides. A protein sequence of length
is calculated on all interacting protein–RNA pairs in the dataset. We identify the binding specificity of mutual interaction propensity between the existing triplets and four RNA nucleotides. A protein sequence of length ![]() residues corresponds to
residues corresponds to ![]() triplets, and every triplet will get its corresponding values of mutual propensity with four types of nucleotides individually. The derived feature of mutual interaction propensity of each residue (
triplets, and every triplet will get its corresponding values of mutual propensity with four types of nucleotides individually. The derived feature of mutual interaction propensity of each residue (![]() centers in the triplets) was described by the four values [23].
centers in the triplets) was described by the four values [23].
8.4.5 Encoding Scheme
We determine the properties and various features of binding residue from sequence and structure profiles. The combination of information from different sources would enable us to encode protein residues into feature vectors after deriving the features of RNA binding residues as well as those of non-RNA-binding residues. They are encoded into a vector to represent the elements of residue characterization. The information of neighbor residues is usually contained when we formulate the feature vectors. A sliding-window technique is often implemented to encode the amino acid residues. From instance, we can use the windows of odd number ![]() size of residues in the encoding scheme. Whether a residue binds RNA is determined by the middle residue (itself) and its neighbor
size of residues in the encoding scheme. Whether a residue binds RNA is determined by the middle residue (itself) and its neighbor ![]() residue profile. The feature vectors representing the residue in the window were then encoded by properties of the
residue profile. The feature vectors representing the residue in the window were then encoded by properties of the ![]() residues. Individual residues were then represented in feature vectors with the same-length elements. The number of elements in the feature vector would be determined by the chosen feature descriptors.
residues. Individual residues were then represented in feature vectors with the same-length elements. The number of elements in the feature vector would be determined by the chosen feature descriptors.
8.5 Existing Methods
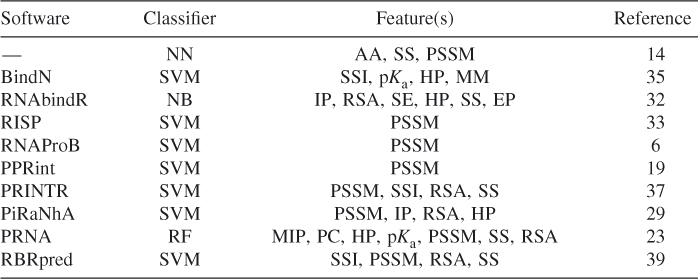
Methods NN, NB, SVM, and RF are the classic machine learning methods. So far, some methods based on these classifiers have respectively been proposed to predict RNA binding sites in proteins by various features. Table 8.1lists some methods and their corresponding features used to train the predictors. Jeong et al. [13] proposed a NN-based method for predicting RNA binding sites by using amino acid types (AA) and secondary structure elements (SS). Then, Jeong and Miyano [14] improved the prediction performance of NN classifier by using the weighted profiles of PSSM. Wang and Brown [35] proposed a SVM-based classifier to predict RNA binding residues in proteins by using single-sequence information (SSI) plus three biochemical sequence features, including side chain p![]() value (p
value (p![]() ), hydrophobicity index (HP), and molecular mass of amino acid (MM). RNABindR [32] implemented a NB classifier to predict RNA binding amino acid residues in proteins by features of interface propensity (IP), relative accessible surface area (RSA), sequence entropy (SE), hydrophobicity (HP), secondary structure (SS), and electrostatic potential (EP). PPRint [19] implemented a SVM-based method in the identification using PSSM. RNAProB [6] is also a SVM-based method by using the evolutionary information of smoothed PSSM. Tong et al. [33] also proposed a RNA interaction site prediction (RISP) method using SVM in conjunction with evolutionary information of amino acid sequences in terms of their PSSMs. PRINTR [37] developed a method for the prediction of protein residues that interact with RNA using SVM by single-sequence information (SSI), PSSM, predicted secondary structure (SS), relative solvent-exposed surface area (RSA). Spriggs et al. [29] improved the prediction performance by a SVM-based classifier using four properties of PSSM, interface propensity (IP), relative solvent accessibility (RSA), and hydrophobicity (HP). We proposed a RF method for combining various features of interacting protein and RNA [23]. Mutual interaction propensity (MIP) and sequence-derived features, such as physicochemical characteristics (PC), hydrophobicity (HP) and sidechain p
), hydrophobicity index (HP), and molecular mass of amino acid (MM). RNABindR [32] implemented a NB classifier to predict RNA binding amino acid residues in proteins by features of interface propensity (IP), relative accessible surface area (RSA), sequence entropy (SE), hydrophobicity (HP), secondary structure (SS), and electrostatic potential (EP). PPRint [19] implemented a SVM-based method in the identification using PSSM. RNAProB [6] is also a SVM-based method by using the evolutionary information of smoothed PSSM. Tong et al. [33] also proposed a RNA interaction site prediction (RISP) method using SVM in conjunction with evolutionary information of amino acid sequences in terms of their PSSMs. PRINTR [37] developed a method for the prediction of protein residues that interact with RNA using SVM by single-sequence information (SSI), PSSM, predicted secondary structure (SS), relative solvent-exposed surface area (RSA). Spriggs et al. [29] improved the prediction performance by a SVM-based classifier using four properties of PSSM, interface propensity (IP), relative solvent accessibility (RSA), and hydrophobicity (HP). We proposed a RF method for combining various features of interacting protein and RNA [23]. Mutual interaction propensity (MIP) and sequence-derived features, such as physicochemical characteristics (PC), hydrophobicity (HP) and sidechain p![]() value (p
value (p![]() ), as well as structure-derived features, including PSSM conservation value, relatively accessible surface (RAS), and secondary structure (SS), are integrated together to train the predictor. Zhang et al. [39] proposed a sequence-based model for the prediction of RNA binding residues. They implemented five feature sets (12 features) based on the single-sequence information (SSI), evolutionary conservation (PSSM), the predicted secondary structure (SS), and the predicted relative solvent accessibility (RSA). A SVM classifier was built on these features after processing by feature selection.
), as well as structure-derived features, including PSSM conservation value, relatively accessible surface (RAS), and secondary structure (SS), are integrated together to train the predictor. Zhang et al. [39] proposed a sequence-based model for the prediction of RNA binding residues. They implemented five feature sets (12 features) based on the single-sequence information (SSI), evolutionary conservation (PSSM), the predicted secondary structure (SS), and the predicted relative solvent accessibility (RSA). A SVM classifier was built on these features after processing by feature selection.
Table 8.1 Some Available Methods for Predicting RNA Binding Sites in Proteins
8.6 Feature Analysis and Comparison Study
We represented protein residues as feature vectors individually by identifying sequence and structure information potentially contributing to the RNA binding events. These features of binding residues and nonbinding residues are implemented to train a classifier to learn the underlying residue patterns of protein–RNA interaction. Some of the descriptors in the feature vector, such as relative accessible surface and secondary structure, can be calculated only after 3D protein structure is available. The defined feature of interaction propensity can also be implemented when structure information is known. Apparently, it is valuable to determine and compare the importance of these different descriptors about their contributions to the prediction. Moreover, the comparisons of machine learning methods will provide valuable information on the discriminative classifiers for predicting RNA binding sites in proteins.
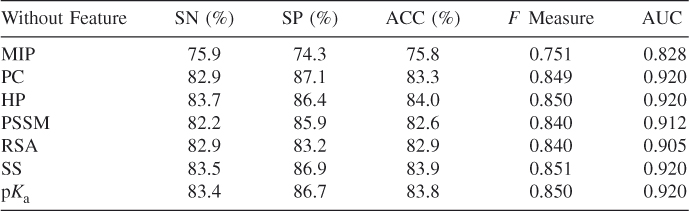
In our published paper on predicting RNA binding residues in proteins [23], we investigated comparisons between various methods and numerous features. The effectiveness and efficiency provided evidence for substantial feasibility of the prediction for protein–RNA binding residues by feature-based machine learning methods. We generally categorized our residue features into sequence-based and structure-based descriptors. We composed a dataset of 205 protein–RNA interaction pairs from RsiteDB [28]. As shown in Table 8.1, we identified MIP, PC, HP, SS, and PSSM conservation values and p![]() values of these residues to encode the feature vectors. We trained and tested our RF-based predictors and achieved high performance of prediction by combining various features [23]. To evaluate the contribution of each feature for the prediction accuracy, we tested the performance of some selected features in these descriptors. Table 8.2 presents the results of prediction performance of the fivefold cross validation by subtracting one of the descriptors individually in the scoring scheme. After subtracting each descriptor individually while describing these residues, we found decreased accuracy of prediction in comparison to the accuracy obtained when we used all the descriptors. For instance, when we omitted the defined MIP, ACC, and
values of these residues to encode the feature vectors. We trained and tested our RF-based predictors and achieved high performance of prediction by combining various features [23]. To evaluate the contribution of each feature for the prediction accuracy, we tested the performance of some selected features in these descriptors. Table 8.2 presents the results of prediction performance of the fivefold cross validation by subtracting one of the descriptors individually in the scoring scheme. After subtracting each descriptor individually while describing these residues, we found decreased accuracy of prediction in comparison to the accuracy obtained when we used all the descriptors. For instance, when we omitted the defined MIP, ACC, and ![]() measure, the AUC became 75.8%, 0.751, and 0.828, respectively, in comparison to 84.5%, 0.859, and 0.923 that we obtained individually using all descriptors.
measure, the AUC became 75.8%, 0.751, and 0.828, respectively, in comparison to 84.5%, 0.859, and 0.923 that we obtained individually using all descriptors.
Table 8.2 Prediction Results Obtained by Subtracting Descriptor
We also investigated the prediction performance by combining different sequence-derived features and structure-derived features [23]. The details of prediction results are shown in Table 8.3. Table 8.3 also shows the results of the prediction based on the combined sequence features and those of structure features, respectively. When we employed the scheme of combining these features based only on sequence (without Structure features), our RF-based method achieved a prediction accuracy of 81.4%, 0.832 ![]() measure and 0.905 AUC value. Our method can obtain 82.6% ACC, 0.847
measure and 0.905 AUC value. Our method can obtain 82.6% ACC, 0.847 ![]() measure, and 0.917 AUC without sequence features. The results indicate that with the combination of all these descriptors, the predictor can identify more information for better classifying protein–RNA binding residues from nonbinding ones. Here, MIP represents mutual interaction propensity, PC represents physicochemical characteristics, HP represents hydrophobicity, PSSM represents PSSM conversation value, ACC represents accessible surface, SS represents secondary structure, and p
measure, and 0.917 AUC without sequence features. The results indicate that with the combination of all these descriptors, the predictor can identify more information for better classifying protein–RNA binding residues from nonbinding ones. Here, MIP represents mutual interaction propensity, PC represents physicochemical characteristics, HP represents hydrophobicity, PSSM represents PSSM conversation value, ACC represents accessible surface, SS represents secondary structure, and p![]() represents sidechain p
represents sidechain p![]() value, respectively. The comparison study results have shown the importance of residue properties in the identification of binding specificity. These features can be used to construct the structural markers and motifs for discriminating RNA binding sites as well as nonbinding ones [22, 23].
value, respectively. The comparison study results have shown the importance of residue properties in the identification of binding specificity. These features can be used to construct the structural markers and motifs for discriminating RNA binding sites as well as nonbinding ones [22, 23].
Table 8.3 Predictive Results Obtained by Combinations of Different Descriptors

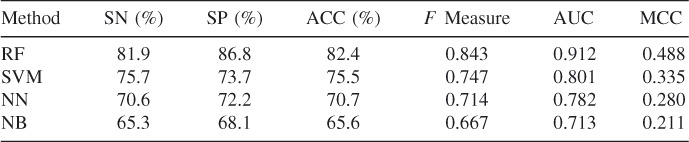
For available machine learning methods of NN, NB, SVM, and RF, we also compared the prediction results of these predictors on the benchmark dataset. We explored these algorithms using the same process of performing training and test steps [23]. In the 205 protein chains, we randomly conducted a training set of 105 protein chains as well as a test set of the remaining 100 chains. We trained these classifiers and then implemented the prediction and validation in the test set to evaluate the performance of the machine learning methods. Figure 8.2 shows the ROC curves of prediction by different classifiers in the test set. The performance details are given in Table 8.4. In Figure 8.2, AUC values of RF-based, SVM-based, NN-based, and NB-based predictors are 0.912, 0.801, 0.782, and 0.713, respectively. The RF-based method performs better than other classifiers in our dataset. As to the prediction results of the other known methods listed in Table 8.1, such as RNABindR, BindN, and PPRint, they are also available in our published paper [23].
Figure 8.2 ROC performance of several classifiers.
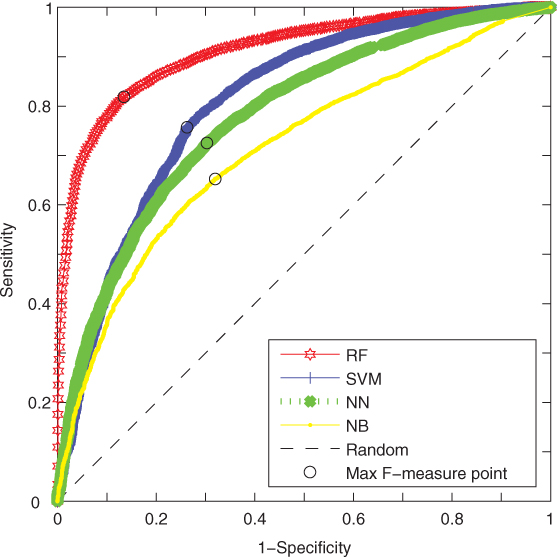
Table 8.4 Comparison of Results for Different Machine Learning Methods
8.7 Conclusion
In this chapter, we provided an overview for predicting RNA binding sites in proteins on the basis of various features by machine learning methods. The physicochemical features, sequence-derived features, and structure-derived features were identified in various protein–RNA complexes. The interacting sites of protein and RNA were encoded into features, and the interaction events between these residues were signed with labels to distinguish the binding residues and nonbinding residues. Various features representing the interaction of an amino acid and its partner nucleotides were encoded into feature vectors. The features were then used to train a machine learning classifier. To assess the prediction performance of these predictors, currently the cross-validation technique is widely implemented. The statistical assessments were analyzed and compared by various strategies of encoding features and selecting machine learning algorithms. The comparisons in these existing methods show their advantages and characteristics. In the training set, the residues were encoded into feature vectors representing the RNA binding specificities in proteins. The learning and validation processes in protein–RNA complexes will provide evidence for the effectiveness of predictors. After training of those computational methods, the predictors can directly be used to predict the RNA binding residues in a protein. The sequence information and structure information, if available, of the targeted protein will be identified as the input features. The methods based only on sequence would provide broader coverage of prediction. Clearly, the accuracy and coverage of prediction are closely related to the available information, encoded mechanisms, and the classifiers.
Acknowledgments
We thank Professor Xiang-Sun Zhang, Dr. Ling-Yun Wu, and Dr. Yong Wang of the Chinese Academy of Sciences (CAS) for helpful discussions and comments. This work was supported by the National Natural Science Foundation of China (NSFC) under Grants 31100949, 61134013, 91029301, and 61072119; by Shanghai NSF under Grant 11ZR1443100; and by the Knowledge Innovation Program of Shanghai Institutes for Biological Sciences (SIBS) of CAS with Grant 2011KIP203; by the Chief Scientist Program of SIBS, CAS with Grant 2009CSP002; and by the SA-SIBS Scholarship Program; This research was also partially supported by the National Center for Mathematics and Interdisciplinary Sciences, CAS.
References
1. Allers J, Shamoo Y, Structure-based analysis of protein-RNA interactions using the program ENTANGLE, J. Mol. Biol. 311:75–86 (2001).
2. Chen L, Wang RS, Zhang XS, Biomolecular Network: Methods and Applications in Systems Biology, John Wiley & Sons. 2009.
3. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ, Gapped BLAST and PSI-BLAST: A new generation of protein database search programs, Nucleic Acids Res. 25:3389–3402 (1997).
4. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P, The Protein Data Bank, Nucleic Acids Res. 28:235–242 (2000).
5. Breiman L, Random forests, Machine Learn. 45:5–32 (2001).
6. Cheng CW, Su EC, Hwang JK, Sung TY, Hsu WL, Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinformatics 9(Suppl 12):S6 (2008).
7. Doherty EA, Batey RT, Masquida B, Doudna JA, A universal mode of helix packing in RNA, Nat. Struct. Biol. 8(4):339–343 (2001).
8. Ellis JJ, Broom M, Jones S, Protein-RNA interactions: Structural analysis and functional classes, Proteins 66:903–911 (2007).
9. Gibas CJ, Subramaniam S, Explicit solvent models in protein pKa calculations, Biophys. J. 71:138–147 (1996).
10. Glisovic T, Bachorik JL, Yong J, Dreyfuss G, RNA-binding proteins and post-transcriptional gene regulation, FEBS Lett. 582:1977–1986 (2008).
11. Hall KB, RNA-protein interactions, Curr. Opin. Struct. Biol. 12:283–288 (2002).
12. Han LY, Cai CZ, Lo SL, Chung MC, Chen YZ, Prediction of RNA-binding proteins from primary sequence by a support vector machine approach, RNA 10:355–368 (2004).
13. Jeong E, Chung IF, Miyano S, A neural network method for identification of RNA-interacting residues in protein, Genome Inform. 15:105–116 (2004).
14. Jeong E, Miyano S, A Weighted Profile Based Method for Protein-RNA Interacting Residue Prediction, LNCS Series, vol. 3939, 2006, pp. 123–139.
15. Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM, Protein-RNA interactions: A structural analysis, Nucleic Acids Res. 29:943–954 (2001).
16. Kabsch W, Sander C, Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features, Biopolymers 22:2577–2637 (1983).
17. Kim H, Jeong E, Lee SW, Han K, Computational analysis of hydrogen bonds in protein-RNA complexes for interaction patterns, FEBS Lett. 552:231–239 (2003).
18. Kim OT, Yura K, Go N, Amino acid residue doublet propensity in the protein-RNA interface and its application to RNA interface prediction, Nucleic Acids Res. 34:6450–6460 (2006).
19. Kumar M, Gromiha MM, Raghava GP, Prediction of RNA binding sites in a protein using SVM and PSSM profile, Proteins 71:189–194 (2008).
20. Lee B, Richards FM, The interpretation of protein structures: Estimation of static accessibility, J. Mol. Biol. 55:379–400 (1971).
21. Li N, Sun Z, Jiang F, Prediction of protein-protein binding site by using core interface residue and support vector machine, BMC Bioinformatics 9:553 (2008).
22. Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L, Bridging protein local structures and protein functions, Amino Acids 35:627–650 (2008).
23. Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L, Prediction of protein-RNA binding sites by a random forest method with combined features, Bioinformatics 26:1616–1622 (2010).
24. Lunde BM, Moore C, Varani G, RNA-binding proteins: Modular design for efficient function, Nat. Rev. Mol. Cell. Biol. 8:479–490 (2007).
25. Morozova N, Allers J, Myers J, Shamoo Y, Protein-RNA interactions: Exploring binding patterns with a three-dimensional superposition analysis of high resolution structures, Bioinformatics 22:2746–2752 (2006).
26. Perez-Cano L, Solernou A, Pons C, Fernandez-Recio J, Structural prediction of protein-RNA interaction by computational docking with propensity-based statistical potentials, Proc. Pacific Symp. Biocomputing, Kamuela, Hawaii, 2010, vol. 15, pp. 293–301.
27. Rost B, Sander C, Conservation and prediction of solvent accessibility in protein families, Proteins 20:216–226 (1994).
28. Shulman-Peleg A, Shatsky M, Nussinov R, Wolfson HJ, Prediction of interacting single-stranded RNA bases by protein-binding patterns, J. Mol. Biol. 379:299–316 (2008).
29. Spriggs RV, Murakami Y, Nakamura H, Jones S, Protein function annotation from sequence: Prediction of residues interacting with RNA, Bioinformatics 25:1492–1497 (2009).
30. Sweet RM, Eisenberg D, Correlation of sequence hydrophobicities measures similarity in three dimensional protein structure, J. Mol. Biol. 171:479–488 (1983).
31. Terribilini M, Lee JH, Yan C, Jernigan RL, Honavar V, Dobbs D, Prediction of RNA binding sites in proteins from amino acid sequence, RNA 12:1450–1462 (2006).
32. Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, Dobbs D, RNABindR: A server for analyzing and predicting RNA-binding sites in proteins, Nucleic Acids Res. 35:W578–W584 (2007).
33. Tong J, Jiang P, Lu Z, RISP: A web-based server for prediction of RNA-binding sites in proteins, Comput. Methods Programs Biomed. 90:148–153 (2008).
34. The UniProt Consortium: The Universal Protein Resource (Uniprot), Nucleic Acids Res. 36:D190–D195 (2008).
35. Wang L, Brown SJ, BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences, Nucleic Acids Res. 34:W243–W248 (2006).
36. Wang L, Eghbalnia HR, Markley JL, Nearest-neighbor effects on backbone alpha and beta carbon chemical shifts in proteins, J. Biomol. NMR 39(3): 247–257 (2007).
37. Wang Y, Xue Z, Shen G, Xu J, PRINTR: Prediction of RNA binding sites in proteins using SVM and profiles, Amino Acids 35:295–302 (2008).
38. Weigt M, White RA, Szurmant H, Hoch JA, Hwa T, Identification of direct residue contacts in protein-protein interaction by message passing, Proc. Natl. Acad. Sci. USA 106:67–72 (2009).
39. Zhang T, Zhang H, Chen K, Ruan J, Shen S, Kurgan L, Analysis and prediction of RNA-binding residues using sequence, evolutionary conservation, and predicted secondary structure and solvent accessibility, Curr. Protein Pept. Sci. 11(7): 609–628 (2010).