
Chapter 19
Identifying Protein Complexes from Protein–Protein Interaction Networks
19.1 Introduction
Protein complexes are groups of proteins that interact with each other at the same time and place, forming a single multimolecular machine, such as the anaphase promoting complex, RNA splicing and polyadenylation machinery, and protein export and transport complexes. Protein complexes can be classified into obligate and nonobligate types according to whether a protein of a complex can form a stable crystal structure of its own. If it can, (without any other associated protein) in vivo, then a complex formed by such proteins is called a nonobligate protein complex. On the other hand, some proteins can't be found to create a crystal structure independently, but can be found as part of a protein complex that does create a stable crystal structure. Such a protein complex is called an obligate protein complex [1]. Protein complexes also can be divided into two classes: transient protein complexes and permanent complexes. Transient protein complexes form and break down transiently in vivo, whereas permanent complexes don't show such behavior but are typically dissociated by proteolysis [1]. Many protein complexes are well understood, particularly in the model organism Saccharomyces cerevisiae (a strain of yeast, e.g., baker's yeast). For this relatively simple organism, the study of protein complexes is now being performed genomewide, and the elucidation of most protein complexes of the yeast is continuing. Identification of protein complexes has significant meaning in mining and analyzing function of modules in a protein network, as it reveals protein function and explains the mechanism of a particular biological process. One method that is commonly used for identifying the members of protein complexes is immunoprecipitation. Computation methods are also used to predict protein complexes from prior knowledge of known complexes.
19.2 Density-Based and Local Search Methods
A protein–protein interaction network (PPI network) is represented as a undirected graph G(V,E), where the vertices represent proteins and edges represent interactions. A graph is unweighted if the relationship between two proteins can be the simple binary values 1 or 0, where 1 denotes that the two proteins interact and 0 denotes that they do not interact. Sometimes, a graph is weighted if the edges of graph G are weighted with a value between 0 and 1, and the weight represents the probability that this interaction is a true positive. Clustering in PPI networks consists in grouping the proteins into sets (clusters) that demonstrate greater similarity among proteins in the same cluster than in different clusters. In PPI networks, the clusters correspond to two types of module: protein complexes and functional modules; however, there is no clear distinction between them. The clusters predicted by the clustering methods discussed in this chapter are considered as protein complexes.
Assumpting that the members in the same protein complex strongly bind each other, a cluster can be referred to as a densely connected subgraph within a PPI network. The key idea in identifying a protein complex is to detect highly connected subgraphs (clusters) that have more interactions among themselves and fewer with the rest of the graph. A fully connected subgraph, or clique that is not a part of any other clique is an example of such a cluster. In general, clusters are not required to be fully connected; instead, the density of connections in the cluster is measured by the parameter d = 2m/(n(n − 1)), where n is the number of proteins in the cluster and m is the number of interactions between them. Many algorithms have been developed to identify clusters of sufficiently high d in a PIN.
Spirin and Mirny [2] present three methods for identifying clusters with high d.
Algorithms for finding clusters, or locally dense regions, of a graph are an ongoing research topic in computer science. To find locally dense regions of a graph, MCODE [3] used a vertex weighting scheme based on the core clustering coefficient. It consists of three stages: vertex weighting, complex prediction, and optionally postprocessing. The first stage of MCODE, vertex weighting, weights all the vertices on the basis of the core clustering coefficient. Different from the clustering coefficient, the core clustering coefficient of a vertex is defined as the density of the highest k core of the immediate neighborhood of (vertices connected directly to ) including . A k core is a graph of minimal degree k [graph G, for all in G, deg() > = k]. The second stage, molecular complex prediction, takes as input the vertex weighted graph, seeds a complex with the highest weighted vertex, and recursively moves outward from the seed vertex, including vertices in the complex whose weight is above a given threshold, which is a given percentage away from the weight of the seed vertex. In this way, the densest regions of the network are identified. The vertex weight threshold parameter defines the density of the resulting complex. A threshold that is closer to the weight of the seed vertex identifies a smaller, denser network region around the seed vertex. In the postprocessing step, MCODE filters or adds proteins according to connectivity criteria.
The aim of many density-based algorithms is to detect the densely connected subgraphs. However, ensuring density alone is not sufficient to achieve this objective. An algorithm DPClus [4] is based on the combination of density and periphery to mine dense subgraphs. Similar to MCODE, DPClus also weights all the vertices in its first step and starts at a highest weighted vertex. In DPClus, a vertex's weight is defined as the sum of the weights of the edges connected to the vertex and the weight of an edge (u,) is the number of the common neighbors of the vertices u and . DPClus takes the highest weighted vertex as an initial cluster and extends the cluster gradually by adding vertices from its neighbors. All neighbors are sorted according to their priorities. A neighbor's priority within a cluster is determined by the sum of the weights and the number of the edges between the neighbor and the vertices in the cluster. DPClus uses two parameters—din(a value of minimum density) and CPin (a minimum value for cluster property)—to determine whether a neighbor i should be added to the cluster n. For a given cluster n with density dn, the cluster property CPin of any vertex i is defined as CPin = |Ein|/(k × dn), where |Ein| is the total number of edges between the vertex i and the vertices of cluster n and k is the number of vertices in cluster n. Once a cluster is generated, DPClus removes it from the graph. Then, the weights of all the vertices in the remaining graph are recomputed and the next cluster is formed in the remaining graph. The process continues until no edge remains in the remaining graph. In such cases, DPClus can generate only nonoverlapping clusters. To generate overlapping clusters, DPClus extends the nonoverlapping clusters by adding their neighbors in the original graph (rather than in the remaining graph).
Li et al. [5] found out that most protein complexes have a very small diameter and a very small average vertex distance when investigating the structures of known protein complexes in MIPS, and proposed an algorithm abbreviated IPCA [5] to identify protein complex from PPI networks by combining vertex distance and subgraph density. IPCA consists of four stages: weighting vertex, selecting seed, extending cluster, and extending-judgment. IPCA uses diameter (or average vertex distance) and interaction probability IN to determine whether a neighbor should be added to a cluster k. For a cluster k, the interaction probability IN of a vertex to it is defined as IN = |E|/n. IPCA can generate overlapping clusters directly and does not need to consider the identified clusters' neighbors in the original graph.
Besides the typical methods discussed above, a number of other methods based on dense and local research have also been proposed to detect protein complex in PPI networks. Bu et al. [6] proposed a quasiclique algorithm to find clusters. Their key idea is that the proteins corresponding to absolutely larger components tend to form a quasiclique for each eigenvector with a positive eigenvalue. In addition to quasicliques, Bu et al. also detected the quasibipartites as clusters. Cui et al. [7] also developed an efficient algorithm for finding cliques and near-cliques in protein interaction networks and showed that a quasiclique as well as a clique often represented a biologically meaningful unit such as functional module or protein complex. Xiong et al. [8] applied an association pattern discovery method to find the hypercliques in the yeast protein interaction network as protein complexes.
19.3 Hierarchical Clustering Methods
Hierarchical clustering is one of the most common methods of classification used in PPI networks to detect protein complexes or functional modules. Generally, the hierarchical clustering algorithms can represent the hierarchy of a complex network as a tree. According to the difference between the processes of the tree's construction, hierarchical clustering algorithms can be divided into two classes: agglomerative algorithms and divisive algorithms. Agglomerative algorithms start at the bottom of the tree and iteratively merge vertices, whereas divisive algorithms begin at the top and recursively divide a graph into two or more subgraphs. For merging vertices or separating the graph, various heuristic rules have been used, such as betweenness centrality [9–11], clustering coefficient, and minimum cut.
Many studies have shown that PPI networks are composed of communities that are densely connected within themselves but sparsely connected with the rest of the network [9–13]. As the number of edges that connect communities is small and all shortest paths between different communities must traverse one of these edges, the edges that connect communities will have higher edge betweenness than those in communities. The edge betweenness of an edge e is defined as follows [10, 11]
19.1 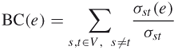
where σst(e) denotes the total number of shortest paths between s and t that pass through edge e.
On the basis of the analysis above, Girvan–Newman (GN) algorithm [10] was proposed to detect community structures in complex networks by removing edges with highest betweenness from the original graphs. As the original GN algorithm does not include a clear definition of module, it does not formally determine which parts of the tree are modules. To break through the limitation, Radicchi et al. proposed two new module definitions: strong module and weak module [14]. The community C in graph G is a strong module if
19.2 ![]()
where kin (i,C) is the number of edges that connect vertex i to other vertices in C and kout(i,C) is the number of edges that connect vertex i to other vertices in the rest of the graph G. The community C in graph G is a weak module if
19.3 ![]()
By combining the GN division process with the two new module definitions, Radicchi gave a new self-contained algorithm to identify modules from a network [14]. The main difference between Radicchi's algorithm and the GN algorithm is that when a graph or subgraph is split into two subgraphs, the two subgraphs are not split in Radicchi's algorithm if they are all modules.
Luo et al. modified the definition of a weak module by extending the concept of degree from a single vertex to a subgraph and developed an agglomerative algorithm MoNet [15]. MoNet initialed each vertex as a cluster and then assembled the clusters into modules by gradually adding edges to the clusters in the reverse order of deletion using the GN algorithm.
More definitions and modifications of modules and betweenness were proposed on the basis of Girvan, Newman's and Radicchi's work. The hierarchical algorithms were also extended in weighted PPI networks. More details are given by Wang et al. [16].
An alternative formulation of betweenness-based decomposition [17], which was based on vertex betweenness instead of edge betweenness, was proposed to allow vertices to be presented in multimodules, since most of the betweenness-based clustering algorithms grouped vertices into separated clusters. This algorithm guaranteed detection of overlapping modules by dividing the graph at the vertices with the highest betweenness and copying such vertices into the divided subgraphs.
Betweenness-based clustering approaches are computationally expensive because they require the repeated evaluation for each edge in the graph, and there have been many at tempts to speed up these algorithms. To develop a fast hierarchical clustering algorithm, Radicchi et al. [14] and Li et al. [18] used the local quantity (edge clustering coefficient) instead of the global quantity (betweenness). The clustering coefficient of an edge e(u,) is [14]
19.4 ![]()
where ![]() is the number of triangles built on that edge e(u,) and min [(ku − 1),(k − 1)] is the minimum possible degree of u and . The idea behind the definition is that many triangles exist in clusters and the edges between different clusters are seldom included in triangles. Thus, edges with small values of
is the number of triangles built on that edge e(u,) and min [(ku − 1),(k − 1)] is the minimum possible degree of u and . The idea behind the definition is that many triangles exist in clusters and the edges between different clusters are seldom included in triangles. Thus, edges with small values of ![]() tend to lie between different clusters.
tend to lie between different clusters.
Li et al. calculated the common neighbors instead of the triangles or high-order cycles and defined the clustering oefficient of an edge e(u,) as [18, 19]
19.5 ![]()
where Nu is the set of neighbors of vertex u and N is the set of neighbors of vertex , respectively. Nu ∩ N is the set of common neighbors of vertex u and vertex . Moreover, the edge clustering coefficient can be extended to a weighted PPI network [19, 20]
19.6 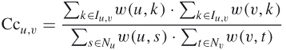
where (u,) denotes the weight of edge e(u,) and I denotes the set of common vertices in Nu and N (i.e., I = Nu ∩ N).
Li et al. also extended the weak module to the λ module [18, 19]. The community C in an unweighted graph G is a λ module if
19.7 ![]()
Similarly, the community C in a weighted graph G is a λ module if [23, 24]
19.8 ![]()
where kin(i,C) is the sum of weights of edges connecting vertex i to other vertices in C and kout(i,C) is the sum of weights of edges connecting vertex i to other vertices in the remainder of the graph G. Using the definitions of edge clustering coefficient and λ module, Wang et al proposed a fast agglomerative algorithm HC-PIN [19] that can be used in both weighted an unweighted PPI networks.
More recently, Wang et al. [21] combined the local metric (clustering coefficient, also termed commonality) and the global metric (betweenness) to generate clusters for balance and consistency.
Besides the two typical metrics discussed above, a number of other metrics have also been suggested for use in hierarchical clustering algorithms. Hartuv and Shamir [22] used the minimum cut to remove edges recursively and developed a divisive algorithm for the discovery of highly connected subgraphs (HCS). Arnau et al. [23] developed a hierarchical clustering algorithm, named UVCLUSTER, based on the shortest path between any two vertices in protein interaction networks. Lu et al. [24] suggested a simple graphical measure to depict the relationship between proteins and extracted the topological information of the network, such as quasicliques and spokelike modules, into a clustering tree. Several similarity measures, such as diffusion kernel similarity, shortest-path-based similarity, and adjacency matrix–based similarity, are evaluated by Wang et al. [25]. They proposed a nonnegative matrix factorization (NMF)-based method with the usage of diffusion kernel similarity for clustering complex networks and biological networks.
The definition of similarity metric or distance measure is a crucial step in hierarchical clustering. How to evaluate the metrics is another challenge in hierarchical clustering. Two evaluation schemes suggested by Lu et al. based on the depth of hierarchical tree and width of ordered adjacency matrix, may be useful. Moreover, Chen et al. [26] presented a formal definition of similarity metric and discussed the relationship between similarity metric and distance metric; they also presented general solutions to normalizing a given similarity metric or distance metric, which have provided a theory basis for constructing metrics.
The hierarchical clustering approach can present the hierarchical organization of protein interaction networks. Its drawback is that it cannot generate overlapping clusters, except that special preprocessing or other strategies are used. In addition, the hierarchical clustering approaches are known to be sensitive to the noisy data in protein interaction networks [27].
19.4 Finding Overlapping Clusters
More recently, much attention has been focused on the clustering algorithms for finding overlapping clusters. Most proteins have more than one biological function; thus each protein may be involved in multiple complexes or functional modules. In this section, we mainly discuss some representative algorithms that have been proposed for the purpose of finding overlapping clusters. Roughly, these algorithms can be classified into three groups:
Besides the three types of algorithms, described above, other methods, such as core attachment [33], MCL [34, 35], and fuzzy clustering [36], can also be used in PPI networks to find overlapping functional modules. Core attachment identifies the cores and attachments of functional modules in PPI networks separately. MCL (Markov cluster algorithm) has been proved to be a very successful clustering procedure. It constructs a stochastic Markovian matrix representing the transition probabilities between all pairs of vertices and simulates random walks on networks, by alternating two operations: expansion and inflation. As MCL is fast and scalable, it has been applied to find functional modules.
19.5 Identification of Protein Complexes by Integrating Multiple Biological Sources
The methods discussed above for identifying clusters are based mostly on graph-theoretic properties solely and require only protein interaction data. However, to some extent, a precise protein interaction network is more important than clustering methods, which not only contain less false-positive information but also can largely determine the accuracy of protein complex detection.
Other than the adoption of preprocessing, several researchers have suggested developing robust clustering algorithms by integrating data from multiple sources, such as genomic data, structure information, gene expression, and gene ontology (GO) annotations. The approaches differ in the way the sources are combined.
Zhang et al. [37] developed another multistep but easy-to-follow framework for the detection of protein complexes that estimated the affinity between each pair of proteins according to their copurification patterns derived from MS data. Jung et al. [38] presented a method for detecting protein complexes on the basis of integration of protein–protein interaction (PPI) data and mutually exclusive interaction information drawn from structural interface data of protein domains. By excluding interaction conflicts, Jung extracted cooperative sets of proteins such as the simultaneous protein interaction cluster (SPIC) from the protein interaction network.
Owing to the attribute that members in a cluster typically perform a specific biological function [39], several clustering algorithms have been proposed with a combination of PPI data and gene expression data. For example, Jansen et al. [40] related whole-genome expression data with PPIs and scored expression activity in complexes. Hanisch et al. [41] proposed a coclustering methodology by using a distance function based on the similarity of gene expression profiles with network topology. Ideker et al. [42] developed a clustering algorithm for the discovery of active subnetworks that showed significant changes in expression over a particular subset of the conditions. More recently, Ulitsky and Shamir [43] transformed the high-throughput expression data into similarity values, from which they found clusters, termed jointly active connected subnetworks (JACSs), that manifested high similarity. Ulitsky and Shamir [44] presented another novel confidence-based method for extracting functionally coherent coexpressed gene sets, named coexpression zone analysis using networks (CEZANNE), by using expression profiles and confidence-scored protein interactions.
With the availability of PPI data for most species (yeast, fly, worm, etc.), it has become feasible to use cross-species analysis to derive protein complex detection based on the evolution of the PPI networks. Sharan and coworkers proposed a series of methods for comparative analysis in two or more species. They used these methods for conserved pathway detection [45], protein function analysis [46], and conserved protein complex detection [47–49]. Basically, to detect the conserved protein complexes in two species, an orthology graph is constructed, whose nodes correspond to pairs of putative orthologous proteins (a protein may appear in multiple nodes with different orthologs), and whose edges correspond to protein interactions. The edges of the graph are assigned weights with probabilistic meaning, so that high-weight subgraphs correspond to conserved protein complexes. Then, a practical method is used to search the orthology graph for complexes of densely interacting proteins, which is based on forming high-weight seeds and extending them using local search.
Network alignment is also applied on conserved protein complex detection in two or more networks. Koyuturk et al. [50] proposed local alignment algorithm termed MaWISh, based on the duplication/divergence models that focus on understanding the evolution of protein interactions. The algorithm constructs a weighted global alignment graph and tries to find a maximum induced subgraph in it. The Graemlin algorithm developed by Flannick et al. [51] scored a possibly conserved module between different networks by computing the log ratio of the probability that the module is subject to evolutionary constraints and the probability that it is under no constraints, considering the phylogenetic relationships between species whose networks are being aligned. Dutkowski et al. [52] also proposed an evolution-based framework to detect conserved protein complexes across multiple species. First, all the proteins from different species are clustered by MCL with BLAST E scores as pairwise similarity. The proteins from each cluster are homologous and thus believed to have a common ancestral protein. Next, a gene tree is built for each protein group and reconciled with a common species tree. Then, the interactions between ancestral proteins are assigned weights based on the interactions observed in the input PPI networks and on the sequence of evolutionary events (duplications, speciations, and deletions) extracted from the reconciled gene trees. Finally, after removal of the edges with weights lower than an appropriate threshold in the conserved ancestral PPI network, the connected components are considered as conserved protein complexes.
With the rapidly expanding resource of microarray data and other biological information, such as structure profiles and phylogenetic profiles, combining multiple biological sources is believed to be an intriguing method for solving the problem of unreliable interaction data when clustering in protein interaction networks.
19.6 Identifying Protein Complexes From Dynamic PPI Network
Cellular systems are highly dynamic and responsive to cues from the environment. Their response to external stimuli is regulated by networks with diverse molecular interactions, such as protein–protein interactions and protein–DNA interactions. Despite the availability of large-scale biological network data, gaining a system-level understanding of the dynamic nature of cellular activity remains a difficult and frequently overlooked task. Since there rarely is any direct information available on the temporal dynamics of these network interactions, the majority of molecular interaction network modeling and analysis has focused solely on static properties. A static PPI network obtained from yeast two-hybrid experiments can be viewed as a comprehensive graph with edges (interactions) that eventually may occur under the set of tested conditions. Thus, it is not guaranteed that all interactions can occur simultaneously, and this may lead to inaccuratey protein complex identification. To address this issue, a number of researchers have attempted to model and analyze interactions and network dynamics [53–61]. A real protein interaction network in the cell changes over time, environments and different stages of the cell cycle [54]; this fluctuation is called dynamics. In the methods described [53–61], static protein interaction networks provide a scaffold by integrating other dynamic information, such as gene expression data and subcellular localization. Gene expression data under different conditions or different stages in cellular cycles can reflect the dynamics of the presence of protein. Some investigators [55] found out that most proteins interact with few partners, whereas a small but significant proportion of proteins, the hubs, interact with many partners. They further revealed that there are two types of hub: “party” hub, where proteins interact with most of their partners simultaneously, and “date” hub, in which proteins bind their different partners at different times or locations. Focused on the dynamic formation of protein complex, De Lichtenberg et al. [56] constructed time-dependent PPIN by integrating PPIN and gene expression. By analyzing the dynamics of protein complexes during the yeast cell cycle, they discovered that most complexes consist of both periodically and constitutively expressed proteins, suggesting a mechanism of just-in-time assembly. Other methods [57] combined gene expression data with PPI networks to classify dynamic proteins that are present (expressed) periodically and static proteins that are present (expressed) all the time, and further identified dynamic modules and static modules on static PPI networks. Tang et al. [60] split the original PPI network into timecourse subnetworks, by giving a potential threshold value in the gene expression data to determine whether a protein is expressed at this timepoint. The difference among these subnetworks is assumed to depict the dynamic changes of protein expression. Wang et al. [61] also tried to distinguish the key point of dynamics of protein interaction networks. They pointed out that the dynamic changes of protein activity are more meaningful than the dynamic changes of protein presence, and therefore proposed an active protein interaction network model. The active protein interaction network is extracted from PPI and gene expression data, by adopting a 3σ principle to identify active information of each protein. It is desirable that the dynamic protein interaction network combined with more biological information depict the real network more vividly, and make the analysis result more helpful for biologists. However, at the very beginning of dynamic network modeling, this method will be fraught with many problems. The first issue is whether a graph-based model is suitable for modeling a dynamic network. A new model might be needed to represent dynamic network.
19.7 Challenges and Future Research
In the postgenomic era, an important challenge is to analyze biological systems from the network level, in order to understand the topological organization of protein interaction networks, identify protein complexes and functional modules, discover functions of uncharacterized proteins, and obtain more exact networks. While some clustering approaches have been applied successfully in the discovery of protein complexes or functional modules, methods for clustering and analyzing protein interaction networks are less mature.
The main challenges for clustering protein interaction networks include: how to define the quality of a cluster and develop robust algorithm in the presence of noisy protein interaction networks; how heavily two clusters should overlap each other, and whether, it is computationally difficult for most current clustering algorithms to accurately identify protein complexes or functional modules from large-scale protein interaction networks, especially to discover mesoscale clusters. There is little a priori knowledge on clustering protein interaction networks, such as cluster number and cluster size. How to discriminate between complex and dynamic protein interaction networks is also a very difficult task.
The methods for distinguishing complex from dynamic protein interaction networks are in a nascent stage. Furthermore, techniques and methods for developing both robust and fast clustering algorithms are directions for further research. Moreover, it will be important to determine whether there is any relationship between the two properties of overlap and hierarchical organization among clusters, as these two issues were usually considered separately. Some research has been performed on complex networks, such as word association networks and scientific collaboration networks [31], to detect both the overlapping and hierarchical properties of a community structure. Do these properties also apply in protein interaction networks? Additionally, integration of multiple resources will help to detect clusters more accurately and will continue to attract interest.
References
1. Amoutzias G, Van de Peer Y, Single-gene and whole-genome duplications and the evolution of protein-protein interaction networks, Evolut. Genomics Syst. Biol. 413–429 (2010).
2. Spirin V, Mirny LA, Protein complexes and functional modules in molecular networks, Proc. Natl. Acad. Sci. USA 100:12123–12128 (2003).
3. Bader GD, Hogue CW, An Automated method for finding molecular complexes in large protein interaction networks, BMC Bioinformatics, 4:2 (2003).
4. Altaf-UI-Amin M, Shinbo Y, Mihara K, et al., Development and implementation of an algorithm for detection of protein complexes in large interaction networks, BMC Bioinformatics 7:207 (2006).
5. Li M, Chen J, Wang JX, et al., Modifying the DPClus algorithm for identifying protein complexes based on new topological structures, BMC Bioinformatics 9:398 (2008).
6. Bu D, Zhao Y, Cai L, et al., Topological structure analysis of the protein-protein interaction network in budding yeast, Nucleic Acids Res. 31(9):2443–2450 (2003).
7. Cui G, Chen Y, Huang DS, Han K, An algorithm for finding functional modules and protein complexes in protein-protein interaction networks, J. Biomed. Biotechnol. 1:10 (2008).
8. Xiong H, He X, Ding C, et al., Identification of functional modules in protein complexes via hyperclique pattern discovery, Proc. Pacific Symp. Biocomputing, 2005, vol. 10, pp. 221–232.
9. Narayanan S, The Betweenness Centrality of Biological Networks, MS in Computer Science, Virginia Polytechnic Inst. and State Univ., Sept. 16, 2005.
10. Girvan M, Newman M, Community structure in social and biological networks, Proc. Natl. Acad. Sci. USA 99:7821–7826 (2002).
11. Newman M, Girvan M, Finding and evaluating community structure in networks,. Phys Rev. E 69(2):1–16 (2004).
12. Rives AW, Galitski T, Modular organization of cellular networks, Proc. Natl. Acad. Sci. USA 100(3):1128–1133 (2003).
13. Barabasi A, Oltvai Z, Network biology: Understanding the cells's functional organization, Nat. Rev. Genet. 5(2):101–113 (2004).
14. Radicchi F, Castellano C, Cecconi F, Defining and identifying communities in networks, Proc Natl. Acad. Sci. USA 101(9):2658–2663 (2004).
15. Luo F, Yang Y, Chen CF, et al., Modular organization of protein interaction networks, Bioinformatics 23(2):207–214 (2007).
16. Wang J, Li M, Deng Y, Pan Yi, Recent advances in clustering methods for protein interaction networks, BMC Genomics 11(Suppl 3):S10 (2010) doi:10.1186/1471-2164-11-S3-S10.
17. Pinney J, Westhead D, Betweenness-based decomposition methods for social and biological networks, in Barber S, Baxter PD, Mardia KV, Walls RE, eds., Interdisciplinary Statistics and Bioinformatics, Leeds Univ. Press, 2006, pp. 87–90.
18. Li M, Wang JX, Chen J, et al., A fast agglomerate algorithm for mining functional modules in protein interaction networks, Proc. 2008 Int. Conf. Biomedical Engineering and Informatics, IEEE Press, 2008, pp. 3–7.
19. Wang J, Li M, Chen J, Pan Y, A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks, IEEE/ACM Trans. Comput. Biol. Bioinform. 8(3):607–620 (2011).
20. Li M, Wang JX, Chen J, Pan Y, Hierarchical organization of functional modules in weighted protein interaction networks using clustering coefficient, Proc. ISBRA2009, Lecture Notes in Bioinformatics (LNBI) Series, Vol. 5542, 2009, pp. 75–86.
21. Wang C, Ding C, Yang Q, Holbrook SR, Consistent dissection of the protein interaction network by combining global and local metrics, Genome Biol. 8:R271 (2007).
22. Hartuv E, Shamir R, A clustering algorithm based graph connectivity, Inform. Process. Lett. 76:175–181 (2000).
23. Arnau V, Mars S, Marín I: Iterative cluster analysis of protein interaction data, Bioinformatics 21:364–378 (2005).
24. Lu H, Zhu X, Liu H, Skogerbo G, Zhang J, Zhang Y, et al., The interactome as a tree: An attempt to visualize the protein-protein interaction network in yeast, Nucleic Acids Res. 32(16):4804–4811 (2004).
25. Wang RS, Zhang SH, Wang Y, et al., Clustering complex networks and biological networks by nonnegative matrix factorization with various similarity measures, Neurocomputing 72:134–141 (2008).
26. Chen S, Ma B, Zhang K, On the similarity metric and the distance metric, Theor. Comput. Sci. 410:2365–2376 (2009).
27. Cho YR, Hwang W, Ramanmathan M, Zhang AD, Semantic integration to identify overlapping functional modules in protein interaction networks, BMC Bioinformatics 8:265 (2007).
28. Palla G, Dernyi I, Farkas I, et al., Uncoverring the overlapping community structure of complex networks in nature and society, Nature 435(7043):814–818 (2005).
29. Li XL, Tan SH, Foo CS, et al., Interaction graph mining for protein complexes using local clique merging, Genome Informatics, 16:260–269 (2005).
30. Liu G, Wong L, Chua HN, Complex discovery from weighted PPI networks, Bioinformatics 25(15):1891–1897 (2009).
31. Shen H, Cheng X, Cai K, Hu MB, Detect overlapping and hierarchical community structure in networks, Physica A 388:1706–1712 (2009).
32. Lancichinetti A, Fortunato S, Kertesz J, Detecting the overlapping and hierarchical community structure in complex networks, New J. Phys. 11:1–17 (2009).
33. Leung HCM, Xiang Q, Yiu SM, Chin FYL, Predicting protein complexes from PPI data: A core-attachment approach, J. Comput. Biol. 16(2):133–144 (2009).
34. Dongen S, Graph Clustering by Flow Simulation, PhD dissertation, Centers for Mathematics and Computer. Science, Univ. Utrecht, 2000.
35. Enright AJ, Van Dongen S, Ouzounis CA, An efficient algorithm for large-scale detection of protein families, Nucleic Acids Res. 30(7):1575–1584 (2002).
36. Zhang S, Wang RS, Zhang XS, Identification of overlapping community structure in complex networks using fuzzy c-means clustering, Physica. 374(1):483–490 (2007).
37. Zhang B, Park BH, Karpinets T, Samatova NF, From pull-down data to protein interaction networks and complexes with biological relevance, Bioinformatics 24(7):979–986 (2008).
38. Jung SH, Jang W, Hur H, Hyun B, Han D, Protein complex prediction based on mutually exclusive interactions in protein interaction network, Genome Informatics 21:77–88 (2008).
39. Hartwell LH, Hopfield JJ, Leibler S, Murray AW, From molecular to modular cell biology, Nature 402(6761 Suppl) C47–C52 (1999).
40. Jansen R, Greenbaum D, Gerstein M, Relating whole-genome expression data with protein-protein interactions, Genome Res. 12:37–46 (2002).
41. Hanisch D, Zien A, Zimmer R, Lengauer T, Co-clustering of biological networks and gene expression data, Bioinformatics 18:S145–S154 (2002).
42. Ideker T, Ozier O, Schwikowski B, Siegel AF, Discovering regulatory and signalling circuits in molecular interaction networks, Bioinformatics 18:S233–S240 (2002).
43. Ulitsky I, Shamir R, Identification of functional modules using network topology and high-throughput data, BMC Syst. Biol. 1:8 (2007).
44. Ulitsky I, Shamir R, Identifying functional modules using expression profiles and confidence-scored protein interactions, Bioinformatics 25:1158–1164 (May 1, 2009).
45. Kelley RM, Sharan R, Karp RM, Sittler T, Root DE, Stockwell BR, Ideker T, Conserved pathways within bacteria and yeast as revealed by global protein network alignment, Proc. Natl. Acad. Sci. USA 100(20):11394–11399 (2003).
46. Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T, Conserved patterns of proteininteraction in multiple species, Proc Natl. Acad. Sci. USA 102(6):1974–1979 (2005).
47. Sharan R, Ideker T, Kelley BP, Shamir R, Karp RM, Identification of protein complexes by comparative analysis of yeast and bacterial protein interaction data, Proc RECOMB Conf., 2004, pp. 282–289.
48. Hirsh E, Sharan R, Identification of conserved protein complexes based on a model of protein network evolution, Bioinformatics 23(2):e170–e176 (2006).
49. Dost B, Shlomi T, Gupta N, Ruppin E, Bafna V, Sharan R, QNet: A tool for querying protein interaction networks, Proc. RECOMB Conf., 2007, pp. 1–15.
50. Koyuturk M et al., Pairwise alignment of protein interaction networks, J. Comput. Biol. 13:182–199 (2006).
51. Flannick J et al., Graemlin: General and robust alignment of multiple large interaction networks, Genome Res. 16:1169–1181 (2006).
52. Dutkowski J, Tiuryn J, Identification of functional modules from conserved ancestral protein-protein interactions, Intell. Syst. Mol. Biol. (Suppl. Bioinformatics) 23(13):149–158 (2007).
53. Bulashevska S, Bulashevska A, Eils R, Bayesian statistical modelling of human protein interaction network incorporating protein disorder information, BMC Bioinformatics 11:46 (2010).
54. Teresa MP, Mona S, Donna KS, Toward the dynamic interaction: It's about time, Brief. Bioinformatics 15–29 (Jan. 8, 2010).
55. Han JJ, Bertin N, Hao T, Evidence for dynamically organized modularity in the yeast protein-protein interaction network, Nature 430:88–93 (2004).
56. De Lichtenberg U, Jensen LJ, Brunak S, et al., Dynamic complex formation during the yeast cell cycle, Science 307:724–727 (2005).
57. Komurov K, White M, Revealing static and dynamic modular architecture of the eukaryotic protein interaction network, Mol. Syst. Biol. 3:110 (2007).
58. Jin R, McCallen S, Liu CC, Xiang Y, Almaas E, Zhou XJ, Identifying dynamic network modules with temporal and spatial constraints, Proc. Pacific Symp. Biocomputing 2009, pp. 203–214.
59. Bossi A, Lehner B, Tissue specificity and the human protein interaction network, Mol. Syst. Biol. 5:260 (2009).
60. Tang X, Wang J, Liu B, Li M, Chen G, Pan Y, A comparison of the functional modules identified from time course and static PPI network data, BMC Bioinformatics 12:339 (2011).
61. Wang J, Peng X, Li M, Luo Y, Pan Y, Active protein interaction network and its application in protein complex detection, Proc. IEEE Conf. Bioinformatics and Biomedicine (BIBM), 2011, pp. 37–42.