
Chapter 11
Progress in Prediction of Oxidation States of Cysteines via Computational Approaches
11.1 Introduction
Cysteine is one of the few amino acids that contain sulfur. The free thiol group allows it to bond with another cysteiene residue to form disulfide bond and help maintain the structure of proteins. The thiol group on cysteine residues is nucleophilic and easily oxidized. Because of this reactivity, cysteine residues serve numerous biological functions such as activation of certain biological activities [1], DNA binding [2], and reproductive systems [3] as well as the aging process of proteins [4]. As was shown in Figure 11.1, cysteine residues can be found in two chemical states in proteins: oxidized state and reduced state. The two states are interchangeable when proper conditions are met. When in reduced form, cysteine undergoes chemical reactions such as alkylation [5, 6], oxidation [7], or forming complex compounds with metal ions [8]. These chemical reactions play critical biological roles such as activation, deactivation of the active sites of enzymes, and altering the local environment of the proteins. In their oxidized form, two cysteine residues form disulfide bond and enable more complicated protein structures [9, 10] and functions [11, 12].
Figure 11.1 Reduced form (a) and oxidized form (b) of cysteine residues (twoStates.png).
The disulfide bond is the covalent bond formed between two cysteine residues on protein chains. The formation of disulfide bonds is a critical step for some of the membrane and secreted proteins in both eukaryotic and prokaryotic cells. Disulfide bonds are considered one of the elements of protein tertiary structure and directly contribute to the stability of the protein [13]. Figure 11.2 illustrates protein in 3D with the disulfide bonds denoted in light green.
Figure 11.2 Illustration of disulfide bonds (shown in light green) on protein (disulfideBond.png).
Disulfide bonds have been generally classified into three categories: structural disulfide bonds, catalytic disulfide bonds, and allosteric disulfide bonds. An important function of disulfide bonds is that they constrain the distal portions of protein chain and reduces the entropy of the unfolded molecule [14] and hence influence the thermodynamics of protein folding. Disulfide bonds also stabilize the folded state of a protein chain [14]. The stabilization of the protein structure helps in reducing protein damage in the presence of oxidants and proteolytic enzymes. Because of their biological importance, knowing the disulfide connectivity in the protein is essential for studying protein structure. Knowledge of the oxidation states of individual cysteines is the first step toward acquiring knowledge on disulfide bonds connectivity and hence inspired significant interest in developing effective and economical ways of finding the oxidation states of individual cysteines.
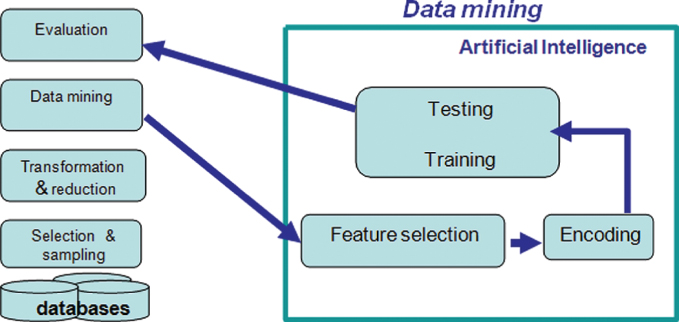
The bonding state of cysteines can be obtained both directly via lab methods or indirectly by information derived from sequence information. Experimentally, the determination of bonding states of cysteine and disulfide bonds often involves use of costly biomarker and thiol reagents and techniques such as electrophoresis [15], mass spectroscopy [16], and HPLC [17]. These experimental approaches are normally time-consuming and dependent on expensive equipment. Therefore, many researchers resort to computational approaches. Predicting the oxidation states of cysteines computationally usually involves first extracting commonalities from the available data obtained by observation or statistical analysis, or utilizing artificial intelligence. Then the acquired knowledge is applied to sequences with the bonding states unknown. Accuracy as calculation was based on the percentage of correct predictions among all test cases. To date, artificial intelligence achieves the highest accuracy among all computational methods attempted. Figure 11.3 is a flowchart of the prediction process via artificial intelligence. First, known data are extracted from database. Redundancy or over representation of certain protein families were avoided by careful selection and sampling. Then sequence information is transformed and reduced to fit the purpose of the data mining step, where relevant features are encoded and used to train the learning machine. After proper training, the resulted decision rules are used to evaluate to for the test data.
Figure 11.3 Information discovery via artificial intelligence (dataMining.png).
11.2 Survey of Previous Efforts to Predict Bonding State of Cysteine Residues on Protein Via Computational Approaches
In this chapter we surveyed ∼15 research efforts published since the 1980s in predicting cysteine oxidation states via computational methods, from the initial attempt using neural networks [18] to the present state of the art [19]. Most of these research efforts have reported prediction accuracy, although some of them used datasets that differed from the others. The essential information used for the predictions was first summarized all to determine the factors impacting the problem. In the following paragraphs we discuss prediction efforts in detail.
11.2.1 Major Factors Influencing Prediction of Cysteine Oxidation States
Figure 11.4 shows the information used in cysteine oxidation state prediction. Sequences local to the bonded cysteines provide a local chemical physical
Figure 11.4 Statistics on information used for predicting oxidation states of cysteines (featuresUsedStatistics.png).
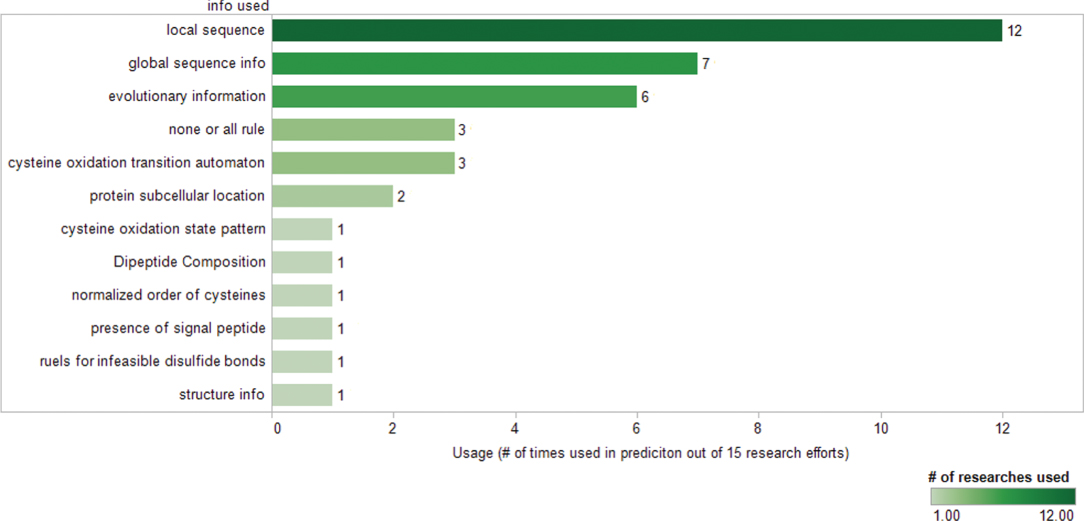
environment for the enzyme-mediated process of disulfide bond formation; therefore, it is critical for prediction. Out of the 15 research efforts surveyed, 12 (80%) included local sequence as input to their algorithms. Two of the early research efforts are based exclusively on the local flanking sequence [18, 20]. Local sequence information usually consists of amino acid composition, charge, and hydrophobicity of the neighboring amino acids. Except for the flanking amino acids on the protein sequence, spatially neighboring amino acids are also considered as part of the local sequence information in some more recent research efforts [21].
Global sequence information was perceived as a good indicator of the protein type and ranks as the second most popular input feature used in prediction algorithms. Features such as the amino acid composition of the whole chain, chain length, and total number of cysteine on the chain belong to the global sequence information category.
Because of their biological importance, disulfide bonds are considered highly conserved features in proteins. Thus evolution information derived from multiple-sequence alignment is utilized in much of the research on oxidation state prediction. Addition of evolution information has shown improved prediction accuracy by significant percentage [e.g., [22]].
Experiments have indicated that subcellular location of proteins is relevant to disulfide bond formation as well as stabilization. For example, disulfide bonds are hardly found in cytoplasm, and for eukaryotes, disulfide bond formation occurs mostly in the lumen of endoplasmic reticulum. By including the subcellular localization information as input, Savojardo et al. improved prediction accuracy was achieved for eukaryotes [19].
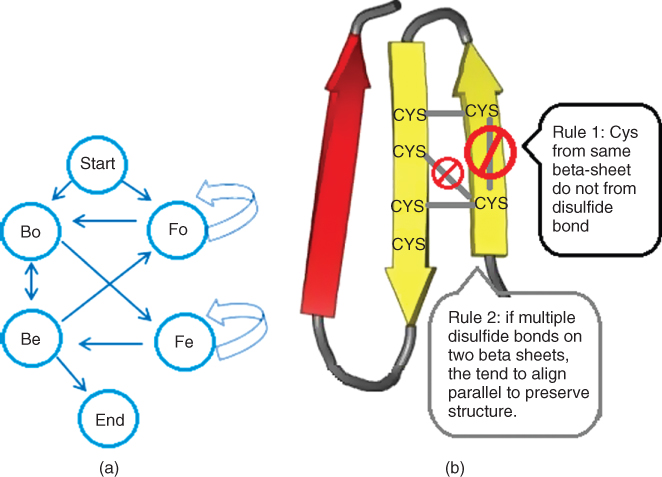
Other characteristics have also been explored such as the ‘none-or-all rule’ derived from direct observations of the test datasets, cysteine oxidation state transition automaton, dipeptide composition calculation, cysteine oxidation state pattern, normalized order of cysteines, the presence of signal peptide, and secondary structure information. Some of these features were used as an additional feature to train the learning machines. Some are used to fine-tune or correct the prediction results, including cysteine oxidation transition automaton [23–25] and the rules for infeasible disulfide bond [25] shown in Figure 11.5. Although not as popularly used as the local and global information and the evolution features mentioned above, each addition of these new features or new rules was reported to contribute to the overall prediction accuracy.
Figure 11.5 Fine-tuning steps reported in the literature: (a) cysteine oxidation transition automaton to limit the number of oxidized cysteines to even numbers; (b) rules for infeasible bonds (fineTunes.png).
11.2.2 Major Efforts in Prediction of Cysteine Oxidation States
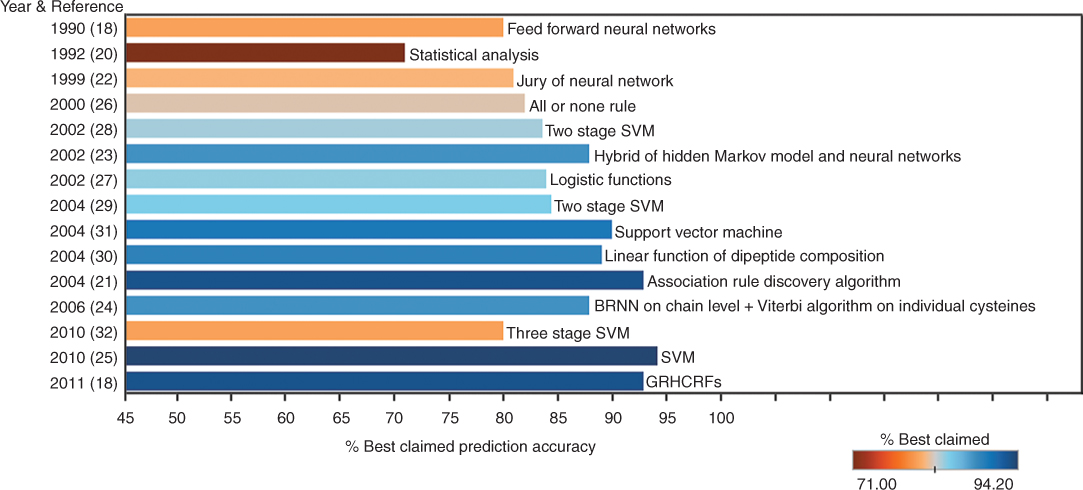
Figure 11.6 summarizes the efforts in predicting cysteine oxidation states. Since the 1980s, various computation techniques have been applied to the oxidation state prediction problem, ranging from single-layer neural networks to multistage support vector machines with increasingly relevant features added to the computation. As a result, prediction accuracy has increased from 71% for relatively small datasets to 93% for larger and more inclusive datasets. The details of these research are discussed in detail in the following paragraphs.
Figure 11.6 Research efforts on prediction of cysteine oxidation states since the 1980s (methodsSurvey.png).
11.2.2.1 Predicting Cysteine Bonding State from Local Amino Acid Sequence Only
Muskal et al. [18] trained simple feedforward neural networks with the flanking sequences around cysteine. This is the first effort reported in the literature to tackle the topic. The assumption was that local sequence is the determinant factor for the oxidation states of cysteines. Accuracy of 80% was achieved on 30 randomly selected proteins (15 sequences from sequences surrounding disulfide-bonded cysteines and 15 from non-disulfide-bonded) from a pool of 689. The window size of flanking sequences were also studied, and it was found that with increased window size, accuracy increases, but memorization becomes apparent after window size of 12 (6 before cysteine and 6 after).
Noticing the influence of flanking amino acids, Fiser et al. [20] took a statistical approach and analyzed a larger population of amino acids around cysteine residues (they studied 37,000 flanking residues vs. 9,000 residues studied in a previous effort). For each flanking sequence from −10 to +10 relative to the cysteine residue in consideration, the occurrence of the 20 amino acids were counted, and ratios of appearance as bonded and free cysteines were calculated on the basis of their appearance frequency. Finally, the disulfide forming potential
for a cysteine containing segment was calculated as the product of the corresponding values of ratio of appearance of all flanking amino acids. The accuracy for prediction is 71% with 1.0 as the threshold of the overall accuracy. This research effort further confirmed that the flanking sequences around bonded and free cysteines differ substantially and are applicable in prediction of bonding state of cysteines.
11.2.2.2 Predicting Cysteine Bonding State from Evolution Information and Local Sequence Information
Fariselli et al. [22] trained a neural network–based predicator to differentiate the bonding states of cysteine in protein chain in terms of both the flanking sequence and the evolution information via multiple-sequence alignment. Eight different encodings were used in this research. With only the local amino acid composition encoded, the prediction accuracy is ∼ 72%. Addition of evolution information such as multiple sequence plus charge, hydrophobicity, conservation weight, and relative entropy gave an improved accuracy of 80.1%. Finally, a jury of neural networks further improved prediction accuracy to 81%.
11.2.2.3 Predicting Cysteine Bonding State by Multiple Sequence Alignment Plus the “All or None” Rule
Fiser et al. [26] examined two datasets (81 protein chains from PDB and 233 protein chains, respectively, both from PDB) and found that only very few (2–4%) of the protein contain cysteines in both bonding states. In the majority of proteins containing cysteine residues, the cysteines are in either all-bonded state or in all-free state. This observation, combined with a conservation score calculated from physicochemical properties of the flanking amino acid, yielded a prediction approach with 82% in prediction accuracy. The dataset used here relatively small (81 protein chains included).
11.2.2.4 Predicting Cysteine Bonding State by Global Sequence Information
Mucchielli-Giorgi et al. [27] used information on either the residues flanking the cysteines or the amino acid content of the whole protein chain to train single logistic function. Two descriptors were used to train the function: (1) the cysteine environment descriptor corresponding to the local flanking sequence and (2) the protein descriptor. The latter was calculated as ![]() , where
, where ![]() is the occurrence frequency of amino acid l in this protein chain and
is the occurrence frequency of amino acid l in this protein chain and ![]() is the total number of occurrences of this amino acid in all protein chains in the DB. The prediction performance rates of the descriptors were compared and the weights associated with the logistic function analyzed. Final results showed that the local sequence environment of the cysteines contains less information than the global amino acid composition of the protein chain. The prediction accuracy in a dataset of 559 protein chains was ∼84%.
is the total number of occurrences of this amino acid in all protein chains in the DB. The prediction performance rates of the descriptors were compared and the weights associated with the logistic function analyzed. Final results showed that the local sequence environment of the cysteines contains less information than the global amino acid composition of the protein chain. The prediction accuracy in a dataset of 559 protein chains was ∼84%.
11.2.2.5 Cysteine Bonding State Prediction via Combined Global and Local Sequence Information
Martelli et al. [23] designed a hybrid system based on the hidden Markov model (HMM) and neural networks (NN). A standard feedforward neural network was first trained with the local sequence of 27 residue-long input window around the cysteine. Then a Markov model with N states connected by means of transition probabilities is applied. The transition between each possible state is restrained only to those that make sense. (If the chain contains disulfide bonds, then the number of bonded cysteine is bounded to be even. Transitions ending up with odd number of boned cysteines are not possible with the transition chart.) The prediction accuracy was up to 88% with this hybrid system.
11.2.2.6 Cysteine Bonding State Prediction Using a Two-Stage SVM Architecture
Because cysteines and half-cysteines rarely occur simultaneously in the same protein chain, Frasconi et al. [28] designed a system based on SVM architecture with two cascaded classifiers. The first classifier predicts the type of protein (all, none, or mix, indicating all, none, or a mixed number of oxidized cysteines on the protein chain) based on the whole sequence. In this stage the inputs include amino acid composition of the protein chain, the length percentage of the protein chain compared with that of the training set (calculated as length of protein under consideration divided by the average protein length in the training set). Also, a flag is encoded in input features indicating whether the number of cysteine count is odd. According to the output of the first classifier, the second stage classifier further classifies cysteines from those protein chains that were marked as mix. The stage 2 classifier uses the multiple-sequence alignment profile of the local input window to predict the bonding states of each individual cysteine on the chain. The prediction accuracy reported was 83.6% for a test set of 716 chains from 2001 PDB database.
11.2.2.7 Cysteine Bonding State Prediction Using a Combination of Kernel Machines
Ceroni et al. [29] extended the two-stage SVM architecture described above with a combination of kernel machines. First, a kernel machine based on the spectrum kernel was adopted and whole-protein sequence characteristics were used as input to this stage. In second stage, evolution information in the form of multiple-sequence alignments around cysteines were used. The accuracy was improved to 84.5% for the same protein dataset described in Section 11.2.2.7.
11.2.2.8 Cysteine Bonding State Prediction Based on Dipeptide Composition
Song et al. [30] implemented a two-class predicator based on dipeptide composition information to improve the bonding state prediction accuracy to 89.1% for 8114 cysteine-containing protein chains selected from PISCES culled PDB. The linear function of the probability of dipeptides Pab (which can be the same or a different amino acid) is used to calculate characteristic index ![]() , specifically, Qk = ∑ ab VabPab (k), where Vab is constant for all proteins.
, specifically, Qk = ∑ ab VabPab (k), where Vab is constant for all proteins.
11.2.2.9 Signal Peptide as a Strong Indicator for Cysteine Bonding States in Extracellular Proteins
To gain full understanding of the biological factors during the process of disulfide bonds formation in cells, Tessier et al. [21] collected sequential and spatial neighborhood information, structural information (such as secondary structure information and the calculated hydrophobic regions on the chain), and evolutionary information as well as other important information about the protein such as the presence of a signal peptide, parity of the cysteines, and the subcellular location of the protein. By applying the associating rule discovery algorithm a priori, it was observed that for extracellular proteins, the presence of a signal peptide is a strong descriptor of the bonding state of cysteines. This rule can be further reinforced when there are an even number of cysteines on the chain. Such an association was only found in extracellular proteins and not valid in membrane proteins and proteins from other compartments.
11.2.2.10 Cysteine Bonding State Prediction Using SVM Based on Local and Global Sequences Combined with Cysteine State Sequence
Chen et al. [31] integrated all the ideas described in earlier research efforts as inputs to a two-stage approach based on SVM. The decision value from the first SVM is normalized by the arctan transfer function before being used to predict the bonding state prediction. The second stage used the branch-and-bound algorithm to optimize the probability of cysteine state sequences (CSSs) while constrain the number of oxidized cysteines to an even number. With the local sequence information and the global amino acid composition alone, with SVM alone, an accuracy of 86% was obtained. Further coupling with the CSS yielded up to 90% in the overall prediction accuracy.
11.2.2.11 Cysteine Bonding State Prediction Using SVM and Bidirectional Recurrent Neural Networks
Ceroni et al. [24] employed SVM binary classifier to predict the overall cysteine bonding states for the whole chain (not on an individual cysteine basis), followed by a refinement step with bidirectional recurrent neural network (BRNN). For each cysteine, the BRNN output was computed using the logistic function. Then the number of the bonded cysteine was enforced to be even using a finite-state automaton. This approach achieved an accuracy of 88%.
11.2.2.12 Cysteine Bonding State Prediction with SVM Based on Protein Types
Lin et al. [25] used a SVM-based two-stage system to bring the prediction accuracy to 94.2%. In the first stage, an iterative protein level “type” (none, mix, or all) classification was performed. Each iteration uses the output probabilities of SVM of the previous iteration as the new feature until the result converges. The second stage is the mix classification using SVM for those chains that were classified as mix chain. Inputs to the SVM in this stage include position-specific scoring matrix (PSSM), normalized order of the cysteine in the protein chain, normalized length of protein length. In addition, a procedure called simple tune (Fig. 11.5b) is applied on the output from SVM. The simple tune step is based on two assumptions: (1) disulfide bonds in one chain do not locate in the same β sheet and (2) in case there are multiple disulfide bonds on two peptide chains of a β sheet, the disulfide bonds that are parallel to each other would be biologically favored so as to preserve the protein structure. The simple tune step consists of four tuning steps: boundary adjustment, oxidized inversion, reduced inversion, and odd–even revision. These fine-tune steps was reported to noticeably improve the prediction accuracy.
11.2.2.13 Prediction of Cysteine Oxidation States Using Three-Stage SVM
Guang et al. [32] proposed a three-stage SVM to predict the oxidation of cysteines: (1) protein chains are classified into ‘none’ or ‘have’ categories to indicate whether there are disulfide bonds on the protein chain; (2) the have sequences are further classified to mix and all, indicating whether all cysteines on the chain are in the oxidized form; and (3) cysteiens in the ‘mix’ sequences are analyzed with respect to a sliding window centered by cysteine. Features used as input to SVM include amino acid composition, local sequence around cysteines, evolution information, and secondary structure information as well as the existence of signal peptide from protein annotation. Accuracies of 90.05%, 96.36%, and 80% were achieved for the three stages of prediction, respectively.
11.2.2.14 Cysteine Oxidation State Prediction with Grammatically Restrained Hidden Conditional Random Fields (GRHCRFs) Based on Protein Subcellular Localization and Position-Specific Scoring Matrix (PSSM)
Motivated by the fact that subcellular localization provides a suitable ambient environment for redox reaction of cysteines to occur, Savojardo et al. [19] included predicted protein subcellular localization in addition to other sequence information such as PSSM. It was found that inclusion of protein subcellular localization improves the prediction accuracy by two or three percentage points. The machine learning technique used here is GRHCRFs, which was found to perform better than other machine learning methods [33]. The prediction accuracy reached 93%. At the protein level, the prediction accuracy is reported to be 86%. It's worth noting that the dataset PDBCYS used in this study has excluded the trivial cases and that protein chains containing a single cysteine are not present.
11.3 Summary
Since the 1980s, tremendous progresses have been made in the area of predicting oxidation states of cysteines. From simply utilizing the local sequences and single-layer neural networks to a broad spectrum of protein characteristics combined with multistage machine learning algorithms, researchers have improved the prediction accuracy from 71% for a relatively small dataset containing ≤93% accuracy for a larger and more selective dataset excluded proteins with single cysteine (in which case, the oxidation states for those cysteines would have been obvious without performing complicated calculations). Research on the prediction of oxidation states of cysteines is surveyed, and the details of each study are discussed in this chapter. With the rapid advancement in biological research and better understanding of the mechanism of protein oxidative folding [34, 35], prediction of oxidation states of cysteines on protein chains is more selective in terms of substrate (eukaryotes vs. prokaryotes or, e.g., whether catalyzed by a certain enzyme family) and focuses more attend on the mechanism of disulfide bond formation. This chapter is intended to serve as a reference source for future developments in the most recent efforts.
References
1. Chamberlain LH, Burgoyne RD, Activation of the ATPase activity of heat-shock proteins Hsc70/Hsp70 by cysteine-string protein, Biochem. J. 322(3):853–858 (1997).
2. McBride AA, Klausner RD, Howley PM, Conserved cysteine residue in the DNA-binding domain of the bovine papillomavirus type 1 E2 protein confers redox regulation of the DNA- binding activity in vitro, Proc. Natl. Aca. Sci, USA 89(16):7531–7535 (1992).
3. Hatch TP, Allan I, Pearce JH, Structural and polypeptide differences between envelopes of infective and reproductive life cycle forms of Chlamydia spp., J. Bacteriol. 157(1):13–20 (1984).
4. Berlett BS, Stadtman ER, Protein oxidation in aging, disease, and oxidative stress, J. Biol. Chem. 272(33):20313–20316 (1997).
5. Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S, Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region, Proc Natl. Acad. Sci. 96(16):9112–9117 (1999).
6. Zhang Z-Y, Dixon JE, Active site labeling of the Yersinia protein tyrosine phosphatase: The determination of the pKa of the active site cysteine and the function of the conserved histidine 402, Biochemistry 32(8):9340–9345 (1993).
7. Arne ES and Holmgren A, Physiological functions of thioredoxin and thioredoxin reductase, Eur J. Biochem. 267(2):6102–6109 (2000).
8. Vallee BL, Auld DS, Zinc coordination, function, and structure of zinc enzymes and other proteins, Biochemistry 29(24):5647–5659 (1990).
9. Lehrer SS, Effects of an interchain disulfide bond on tropomyosin structure: intrinsic fluorescence and circular dichroism studies,. J Mol. Biol. 118(2):209–226 (1978).
10. Wagner DD, Lawrence SO, Ohlsson-Wilhelm BM, Fay PJ, Marder VJ, Topology and order of formation of interchain disulfide bonds in von Willebrand factor, Blood 69(1):27–32 (1987).
11. Reiter Y, Brinkmann U, Webber KO, Jung S-H, Lee B, Pastan I, Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv, Protein Eng. 7(5):697–704 (1994).
12. Reiter Y, Brinkmann U, Jung SH, Lee B, Kasprzyk PG, King CR, Pastan I, Improved binding and antitumor activity of a recombinant anti-erbB2 immunotoxin by disulfide stabilization of the Fv fragment, J. Biol. Chem. 269(28):18327–18331 (1994).
13. Horton HR, Moran LA, Ochs RS, Ravn JD, Scrimgeour KG, Principles of Biochemistry, 2nd ed., Prentice-Hall, Upper Saddle River, NJ, pp. 102
14. Wedemeyer WJ, Welkler E, Narayan M, et al, Disulfide bonds and protein folding, Biochemistry 39:4207–4216 (2000).
15. Huck CW, Bakry R, Bonn GK, Progress in capillary electrophoresis of biomarkers and metabolites between 2002 and 2005, Electrophoresis 27(1):111–125 (2005).
16. Kim SO, Merchant K, et al, OxyR a molecular code for redox-related signaling, Cell 109(3):383–396 (2002).
17. Toyo'oka T et al, Amino acid composition analysis of minute amounts of cysteinecontaining proteins using 4-(aminosulfonyl)-7-fluoro-2,1,3- benzoxadiazole and 4-fluoro-7-nitro-2,1,3-benzoxadiazole in combination with HPLC, Biomed Chromatogr. 1(1):5–20 (1986).
18. Muskal S, Holbrook S, Kim S, Prediction of the disulfide-bonding state of cysteine in proteins, Protein Eng. 3(8):667–672 (1990).
19. Savojardo C, Fariselli P, Alhamdoosh M, Martelli PL, Pierleoni A, and Casadio R, Improving the prediction of disulfide bonds in eukaryotes with machine learning methods and protein subcellular localization, Bioinformatics: 27(16):2224–2230 (2011).
20. Fiser AM, Cserzo ET, Simon I, Different sequence environments of cysteines and half cystines in proteins, application to predict disulfide forming residues, FEBS Lett. 302:117 (1992).
21. Tessier D, Bardiaux B, Larré C, Popineau Y, Data mining techniques to study the disulfide-bonding state in proteins: signal peptide is a strong descriptor, Bioinformatics 20(16):2509–2512 (2004).
22. Fariselli P, Riccobelli P, Casadio R, Role of evolutionary information in predicting the disulfide-bonding state of cysteine in proteins, Proteins 36:340 (1999).
23. Martelli PL, Fariselli P, Malaguti L, Casadio R, Prediction of the disulfide bonding state of cysteines in proteins at 88% accuracy, Protein Sci. 11:2735–2739 (2002).
24. Ceroni A, Passerini A, Vullo A, Frasconi P, DISULFIND: A disulfide bonding state and cysteine connectivity prediction server, Nucleic Acids Res. 34:W177–W181 (2006).
25. Lin CY, Yang CB, Hor CY, and Huang KS, Disulfide bonding state prediction with SVM based on protein types, Proc. 5th Int. IEEE Conf., Bio-Inspired Computing: Theories and Applications (BIC-TA), Changsha, 2010, pp. 1436–1442.
26. Fiser A, Simon I, Predicting the oxidation state of cysteines by multiple sequence alignment, Bioinformatics 16:251 (2000).
27. Muccielli-Giorgi MH, Hazout S, Tuffery P, Predicting the disulfide bonding state of cysteines using protein descriptors, Proteins 46:243–249 (2002).
28. Frasconi P, Passerini A, Vullo A, A two-stage SVM architecture for predicting the disulfide bonding state of cysteines, Proc. 12th IEEE Workshop on Neural Networks for Signal Processing, 2002, pp. 25–34.
29. Ceroni A, Frasconi P, Passerini A, Vullo A, Predicting the disulfide bonding state of cysteines with combinations of kernel machines, J. VLSI Signal Process. 35:287–295 (2003).
30. Song JN, Wang ML, Li WJ, Xu WB, Prediction of the disulfide-bonding state of cysteines in proteins based on dipeptide composition, Biochem. Biophys. Res. Commun. 318:142–147 (2004).
31. Chen YC, Lin YS, Lin CJ, Hwang JK, Prediction of the bonding states of cysteines using the support vector machines based on multiple feature vectors and cysteine state sequences, Proteins: Struct. Funct. Bioinformatics 55(4):1036–1042 (2004).
32. Guang X, Guo Y, Xiao J, Wang X, Sun J, Xiong W, Li M, Predicting the state of cysteines based on sequence information, J. Theor. Biol. 267:312–318 (2010).
33. Savojardo C, Fariselli P, Martelli PL, Shukla P, Casadio R, Prediction of the bonding state of cysteine residues in proteins with machine-learning methods, Computational Intelligence Methods for Bioinformatics and Biostatistics, Lecture Notes in Computer Science, 6685, pp 98–111 (2011).
34. Mamathambika BS, Bardwell JC, Disulfide-linked protein folding pathways, Annu. Rev. Cell Devel. Biol. 24:211–235 (2008).
35. Sevier CS, New insights into oxidative folding, J. Cell Biol. 188:757–758 (2010).