
Chapter 23
Protein-Related Drug Activity Comparison Using Support Vector Machines
23.1 Introduction
At present, combinatorial chemistry can produce millions of new molecules at a time. However, this high level of production cannot exhaust the trillions of potential combinations within a few thousand years. The quantitative structure–activity relationship (QSAR) analysis is required to restrict the search space to avoid produce and test every possible molecular combination. QSAR analysis is very important for understanding the correlation between the molecule's activities and structure. Intelligent machine learning techniques are important tools for QSAR analysis. As a result, these techniques are integrated into the drug production process. The effective intelligent computational model can reduce the cost of drug design significantly by producing the sublibrary of molecular combination derived from a much larger library. This survey compares the performance of several popular machine learning technique for drug activity. The machine learning techniques introduced in this chapter are used to predict activity of pyrimidines and triazines based on the structure–activity relationship of these compounds. Pyrimidines and triazines are two important inhibitors of Escherichia coli dihydrofolate reductase (DHFR). Analysis and prediction of activities of these two inhibitors is very important for finding potential treatment agents for malaria, bacterial infection, and other serious disease. This chapter focuses especially on granular kernel trees (GKTs). GKTs are designed to include previous domain knowledge and voting schemes in order to optimize the performance of the SVM kernels and reduce the training time substantially.
23.2 Related Studies for Pyrimidines Drug Activity Comparison
Combinatorial chemistry has helped produce hundreds, thousands, and even millions of new molecular compounds at a time [3]. This high-throughout production cannot exhaust the trillions of potential combinations within a few thousand years. Consequently, much faster searching process is required to produce and test thousands of molecular combinations in a short amount of time. quantitative structure–activity relationship (QSAR) analysis has been increasingly applied to the drug production process [3]. QSAR is one of the most important techniques in reducing the search space for new drugs. An effective intelligent system based on QSAR analysis for drug screening can produce significant economic benefit. The assumption of QSAR is that the variation of biological activity among a group of molecular compounds is closely related to the variation of their respective structural and chemical features. QSAR tries to search a set of rules or functions that can predict a molecule's activity using its physicochemical descriptors. QSAR focuses on chemical reactivity, biological activity, and toxicity [3]. This kind of analysis is needed to study the relationship between attributes of a finite number of compounds and a known target activity. The dataset in the QSAR analysis usually contains few compounds with many attributes, which are difficult for standard statistical approaches. The solution of the QSAR analysis can be used to design the combinatorial library in the initial stage of the production process. Because the number of potential molecular combinations from existing databases is huge, the search space for potentially useful combinations must be reduced greatly. During the virtual screening, QSAR analysis can distinguish molecules that potentially produce a desired effect on synthesis. Consequently, QSAR analysis can help the drug designer construct a small sub-library containing suitable molecules selected from a large and more diverse library [3]. This combinatorial sublibrary is important to identify an interesting molecule, which might potentially lead to drug discovery.
The intelligent computational system has become an important tool for QSAR analysis. After the compounds having diverse biological and chemical activities and functionality are discovered using this system, detailed QSAR analysis is conducted to analyze their relationship. Finally, the predictive rules are used to predict activity of the molecules according to their chemical and physical properties and to discern the relationships among biological activities of compounds. During the analysis process, the biological and chemical activities are evaluated by the log function defined by log(1/C), where C is the constant value for the inhibitory growth concentration [5].
Many machine learning techniques, including the genetic algorithm (GA) [1], inductive logic programming (ILP) [2], and support vector machines (SVMs) [3], have been proposed for QSAR analysis and drug activity comparison. In these work, one type of E. coli dihydrofolate reductase (DHFR) called pyrimidines is closely studied [5]. These inhibitors are potential therapeutic agents for treating malaria, bacterial infection, toxoplasma, and cancer.
Hirst and his colleagues compared neural networks (NNs), ILP, and linear regression for modeling activities of pyrimidines using QSAR analysis [2]. They use cross-validation methods in order to conduct a statistically rigorous evaluation of the prediction performance of various methods. The training set and testing set are chosen randomly, and the performances of different methods are developed on the basis of the same training dataset. Molecules in the ILP analysis are represented by features instead of Hansch parameters [2]. Hirst's study used 74 pyrimidines. Biological activities are evaluated by the association constant defined by log (Ki). The dataset used in this study has been widely studied by other QSAR methods. Furthermore, the results generated by the QSAR model are compared with the X-ray stereochemistry of interaction [2]. This particular study used the fivefold cross-validation test. Each of the 55 pyrimidines in the cross-validation study shows up only once in one of the test sets [2]. This study applies neural networks for linear regression. The neural network includes a large number of basic computational units that are connected to one another. Each computational unit performs a weighted sum of incoming signals. The neural networks are organized into several layers including an input layer, hidden layers, and an output layer. In these layers, the signal is propagated forward from the input layers to the output layer through any number of hidden layers. The neural network is modeled to study the mapping function between input and output signals. By updating the weights in well-defined manners based on the learning rules, the sum of the squared error between the target signal and training signals is minimized. Besides the neural network, this work also useds inductive logic programming (ILP) for drug design problems since ILP can be specially designed to discern the relationship between different molecule structures. The logical relationship between different objects can be expressed by a subset of predicate calculus [2]. The ILP program needs three types of facts: (1) positive, (2) negative, and (3) background. The positive facts are the paired example of greater activity [2]. The negative facts are the paired example of lower activity. The ILP program needs both positive and negative facts to generate the balanced results. The background facts consist of the chemical structures of the drugs and the properties of the substituent. The fivefold cross-validations include 2198 background facts, 1394 negative facts, and 1394 positive facts.
Their studies revealed that neural networks and ILP have better performance than linear regression using the attribute representation. ILP analysis can also generate rules for explaining the relationship between the inhibitors' activities and their chemical structures. Rule generation provides better interpretation of the biological activity of the inhibitors and their chemical structure. The study also shows that neural networks tend to overfit on the basis of attribute representation. Hidden units cannot improve the performance of neural networks for QSAR analysis. Experiential results generated from the linear regression and the neural network indicate that the 5-substituent should have a low metabolic rate (MR) value. In contrast, the 3-substituent has a high MR value. The neural network often produced different sets of important attributes as compared to regression analysis. For small datasets, full capability of neural networks may not be fully utilized [2].
Burbride et al. [3] compared several learning algorithms for structure–activity relationship (SAR) analysis, which can be used to reduce the search for new drugs since the different molecular combinations can be studied using the intelligent system. The effective solution for drug search can provide significant economic benefit by accelerating the search process substantially. The goal of SAR analysis is to find a rule that predicts a molecule's biological activities from its physicochemical attributes. In this particular study, they are interested in predicting qualitative biological activity for drug design, using a publicly downloadable dataset [3]. Machine learning techniques are used to study the relationships between different known target activities with respect to the attributes of a finite number of compounds. SAR analysis is used to study many areas of modern drug design. For example, the system developed by this study can be applied to design combinatorial libraries in the early stage of the production process. SAR analysis can be used to search for appropriate molecules for the sublibrary selected from a larger and more diverse library [3]. The datasets used in this study are obtained from the UCI Data Repository. The machine learning techniques are applied to predict the inhibition ability of dihydrofolate reductase using pyrimidines. The QSAR system generally tries to solve the regression problem by learning the posterior probability of the target, given some predictive features. There are three positions of possible substitution in each drug. Each substitution position has several important descriptors, including polarity, size, flexibility, and hydrogen bond donor and acceptor. The system can be used to generate the classification rule. Each rule can be used to predict which of two unseen compounds can potentially perform greater activities. In this study, several machine learning algorithms, including neural networks, the decision tree, and the support vector machine are compared [2]. In the experiment, the prediction performance of SVM for the inhibition of dihydrofolate reductase by pyrimidines is compared with that of several other popular machine learning algorithms. The experimental results show that the prediction performance of SVM is significantly better than that of the artificial neural networks, a radial basis function (RBF) network, and a C5.0 decision tree.
23.3 Feature Granules and Hierarchical Kernel Design
Support vector machines (SVMs) have been extensively applied to many data mining applications with strong generalization capabilities. The training complexity of SVM is highly related to the size of the training sample. For many drug-related studies, the size of the molecule combination is usually potentially very large. How to shorten the SVM training time for a large dataset remains one of the most challenging problems in SVM research. In order to explore the SVM's capacity for drug design and reduce the training time significantly, we focus on the hierarchical kernel design proposed by Jin [5] for drug activity comparison. The principle of the hierarchical kernel design is applied to produce powerful and flexible structures called granular kernel trees (GKTs).
The construction of the GKT is divided into four phases:
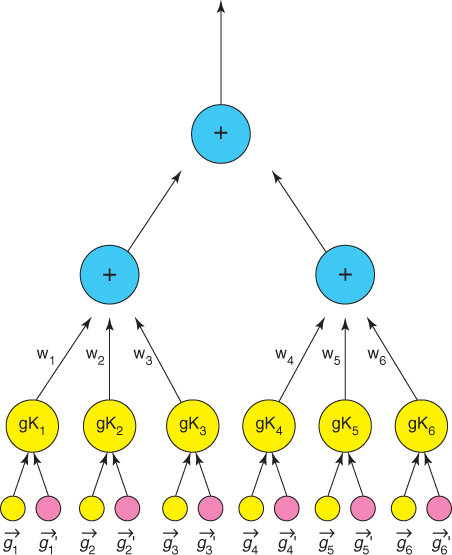
All GKTs take advantage of biological and chemical properties of molecules to optimize kernels effectively. The GKT model can be easily parallelized to reduce the training time significantly. For the GKT model, the input vectors are decomposed into several feature granules according to the possible substituent locations. As a result, each pyrimidine drug pair has six feature granules, each of which has nine features. Two GKT variants, GKT1 and GKT2, are used to model drug activity for pyrimidines [5]. The structures for GKT1 and GKT2 are shown in Figures 23.1, 23.2 and 23.3. GKT1 is a two-layer kernel tree with all granules fused by a sum operation. Each granule pair of GKT2 is represented by a two-layer subtree.
Figure 23.1 Feature granules in the pyrimidine drug pair [5].
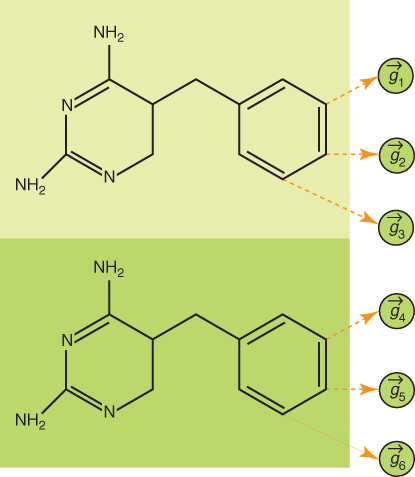
Figure 23.2 GKTs-1 [5].
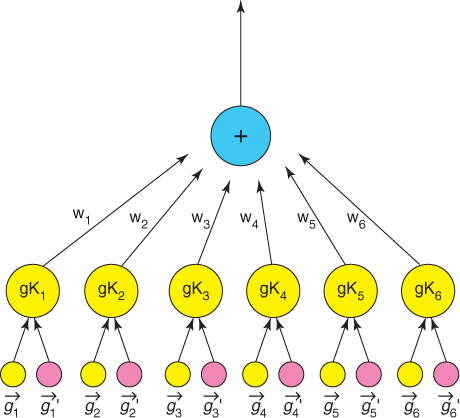
Figure 23.3 GKTs-2 [5].
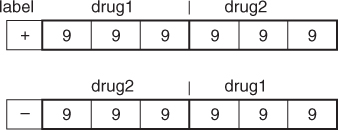
Both GKT1 and GKT2 models use 55 drugs in the pyrimidines dataset [5]. Each drug has three possible substitution positions (R3, R4, and R5). Each substitution position includes nine chemical and biological features. These nine features include polarity, size, flexibility, hydrogen bond donor, and acceptor, π donor and π acceptor, polarizability, and σ effect [5]. Figure 23.4 shows the structure of pyrimidines. Each substituent is represented by nine chemical properties features including polarity, size, flexibility, hydrogen-bond donor, hydrogen-bond acceptor, π donor, π acceptor, polarizability and σ effect. The substituent is used to identify drug activities. Each input vector represents features of two drugs in a specific order. The vector is labeled as positive if the activity of the first drug is higher than that of the second one. Otherwise, it is labeled as negative. The features of input vectors are shown in Figure 23.5. The total number of features for each vector is 54. The pyrimidine–dataset is randomly divided into the training set and testing set in the proportion of 4:1. The training set has 44 compounds, and the testing set has 11 compounds.
Figure 23.4 Structure of pyrimidines [5].
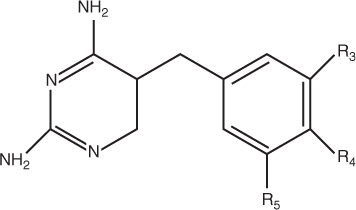
Figure 23.5 Sample input vector [5].
23.4 Experimental Results for Different Machine Learning Models
Burbidge compared the performance of SVMs, neural networks, RBF networks, and decision trees on this dataset [3]. Table 23.1 indicates that the prediction accuracy of SVMs is much better than that of other learning algorithms.
Table 23.1 Performance Comparison of SVMs, MLP, RBF Networks, and C5.0 on the Pyrimidine Dataset
| Algorithm | Testing Accuracy (%) |
| SVMs + RBF | 87 |
| MLP | 86 |
| Pruned neural network | 84 |
| Dynamic neural network | 85 |
| RBF network | 77 |
| C5.0 | 81 |
Source: Jin [5].
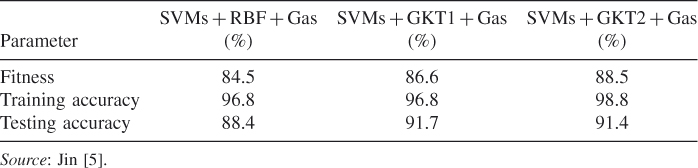
Bo discusses performance comparison for SVM, granular kernel trees (GKTs), and evolutionary granular kernel trees (EGKTs) [5]. The RBF kernel functions are selected for SVM training. Fivefold cross-validation is adopted for the pyrimidine training set. For the GA optimization, the probability of crossover is 0.7 and the mutation rate is 0.5 [5]. Performance rates of SVM and three other types of kernel machines are compared in Table 23.2 [5]. This table shows that SVMs combined with GKTs outperform SVMs by 3% and 3.3% on the testing dataset. Furthermore, the fitness values of SVMs using GKTs-1 and GKTs-2 have also improved compared with SVMs. The testing accuracy of SVMs with GKTs-1 is higher than that of SVMs with GKTs-2 [5].
Table 23.2 Performance Comparison for SVMs, GKT1, and GKT2 on the Pyrimidine Dataset
23.5 Summary
In this chapter, we have surveyed several machine learning models for drug activity comparison. In particular, we have discussed SVMs and the more advanced model GKT in detail. Since the SVM is a robust and highly accurate intelligent classification model, the kernel of SVM can be customized for QSAR analysis. The comparative study for the sample dataset demonstrates that the GKT1 and GKT2 models outperform several of the most frequently used learning algorithms [5]. The experimental results also demonstrate that the MLP model can achieve similar prediction accuracy but that the training period required for this model is an order of magnitude longer [2]. This becomes a serious issue when using QSAR on a large number of molecular compounds. The performance of other machine techniques such as an RBF network or decision tree is far inferior to that of the SVM [3]. However, since the SVM is a deterministic learning algorithm, it can produce reproducible and verifiable results. During the optimization process, SVM converges to the global, rather than local, optimum, which is a significant advantage as compared to other machine learning techniques. Our survey shows that the SVM has a great potential for application to QSAR analysis in the future.
References
1. Devillers J, Neural Networks and Drug Design, Academic Press, 1999.
2. Hirst JD, King RD, Sternberg MJE, Quantitative structure-activity relationships by neural networks and inductive logic programming. I. The inhibition of dihydrofolate reductase by pyrimidines, J. Comput. Aided Mol. Design 8(4):405–432 (1994).
3. Burbidge R, Trotter M, Buxton B, Holden S, Drug design by machine learning: Support vector machines for pharmaceutical data analysis, Comput. Chem. 26(1):4–15 (2001).
4. Newman DJ, Hettich S, Blake CL, Merz CJ, UCI Repository of Machine Learning Databases, Dept. Information and Computer Science, Univ. California, Irvine, 1998.
5. Bo J, Evolutionary Granular Kernel Machines, PhD dissertation, Georgia State University, 2007.