
29
SOLID FORM DEVELOPMENT FOR POORLY SOLUBLE COMPOUNDS
Alessandra Mattei, Shuang Chen, Jie Chen, and Ahmad Y. Sheikh
Solid State Chemistry, AbbVie Inc., North Chicago, IL, USA
29.1 INTRODUCTION
In recent years the number and complexity of targets being studied for developing drugs have increased dramatically across multiple therapeutic areas. Additionally, modalities for developing therapeutics for these targets have increased well beyond typical small molecules and humanized monoclonal antibodies to include many new approaches, such as various constructs of antibody drug conjugates and cell‐based therapies. Even many of the recently approved chemically synthesized small molecules are not small, with molecular masses well in excess of 500 Da. Consequently, the percentage of drug candidates with poor aqueous solubility emerging from drug discovery has steadily increased. As observed by Loftsson and Brewster in 2010 [1], “while 40% of currently marketed drugs are poorly soluble based on the definition of the biopharmaceutical classification system (BCS), nearly 90% of drug molecules in the development pipeline can be characterized as poorly soluble compounds.” Figure 29.1 illustrates how the majority of the portfolio of developmental drugs falls into the two low aqueous solubility categories of the BCS (i.e. classes II and IV) [2]. An increasing trend toward poorly soluble drug candidate molecules represents a major challenge for drug development as the formulation of poorly soluble compounds can be very complex.

FIGURE 29.1 Trend of pipeline drugs toward low aqueous solubility.
Source: Data from Lipp [2].
Many small molecule drug candidates are delivered to patients in a crystalline state, due to its more desirable properties, including stability, purity, reproducibility, and ease of manufacture. The crystalline state of a molecule is highly consequential to two important elements of the target product profile in drug development: bioavailability and shelf life. Bioavailability and shelf life are in turn related to solubility and stability, respectively. These two attributes can be in conflict, and finding an optimum between the two is an essential element of preclinical development. Higher solubility and faster dissolution rate can lead to measurable increases in bioavailability and, likely, therapeutic efficacy. A number of pharmaceutical strategies can be applied to enhance solubility, dissolution rate, and ultimately bioavailability of poorly water‐soluble drug candidates. Crystal structure modifications, particle size reduction, and transformation of a crystalline active pharmaceutical ingredient (API) into a stabilized amorphous state are some of the main approaches that can be utilized to overcome drugability challenges. The crystal‐structure‐modification‐based approach includes, but is not limited to, the use of higher solubility polymorphs, salts, and cocrystals. Micronization and nano‐milling can be used for dissolution rate enhancement due to increased surface area; and amorphous solid dispersions (ASDs) offer a viable path to increase solubility while ensuring the stability of the high‐energy amorphous state.
Enhancement of solubility and dissolution rate can be achieved by molecular modifications. Different crystal forms from the same organic molecule, known as polymorphs [3], exhibit unique X‐ray diffraction patterns and have distinct melting points and solubility properties in addition to other well‐defined physicochemical properties. Up to fivefold solubility differences between different crystal forms have been reported in the literature [4, 5]. As such, higher‐energy crystal forms that offer acceptable solid‐state properties and are also kinetically stable can be considered for development with well‐defined and robust control strategies.
At a fundamental level, the solubility enhancement via salt and cocrystal formation is a consequence of the pH‐solubility profile of acidic/basic drugs and the solubility profile of a cocrystal as a function of the coformer concentration, as illustrated in Figure 29.2. Sound understanding of concepts, including intrinsic solubility, Ka (ionization constant), Ksp (solubility product), pHmax (pH of maximum solubility), and critical coformer activity for cocrystals, is essential to fully leverage the opportunities offered by salts and cocrystals. As such, salts and cocrystals are routinely explored in the search of commercially viable solid forms. Pharmaceutical cocrystals can in fact be considered a natural extension of the opportunity to modify molecular properties of drugs that cannot form salts either due to the lack of ionizable moieties or insufficient pKa differences to make a stable salt. Indeed, the stoichiometric nature of cocrystals predisposes them to large yet predictable changes in solubility and thermodynamic stability as solution conditions vary (i.e. pH and drug solubilizing agents) [7–9]. Physical and chemical stability of salts or cocrystals is also a key consideration in their development. It is important to understand the dissociation of a salt to its parent species or of a cocrystal to its individual components during processing conditions, storage, or in the presence of formulation excipients as it can lead to poor performance.

FIGURE 29.2 (a) pH‐solubility profile for a weak base. (b) Solubility curve as a function of coformer concentration.
Source: Adapted from Serajuddin [6] and Kuminek et al. [7] with permission.
The dissolution rate and solubility of crystalline drugs can be affected by the particle size. In fact, particle size reduction is a fairly common and at times effective approach for dissolution rate enhancement of poorly soluble compounds [10, 11]. Conventional mechanical milling can reduce particle size to the 2–5 μm range, resulting in a moderate enhancement of surface area and, hence, dissolution rate. Nano‐milling can further reduce the size to the 100 nm range. Particle size reduction into the nanometer range can generate a 100‐fold increase in surface‐area‐to‐volume ratio. This increase in surface area is often accompanied by an increase in surface energy due to the generation of structural disorder or exposure of high‐energy crystal facets. The net result can be not only a profound improvement in dissolution rate but also an increase in apparent solubility. However, nano‐milling is not possible if molecular organic crystals are soft and undergo elastic deformation. A careful understanding of intrinsic mechanical properties is thus a requisite for materials undergoing nano‐milling as well as conventional high‐impact mechanical milling. Even when material properties are conducive to nano‐milling, nanometer‐sized drug particles can show a tendency for agglomeration due to attractive interparticle forces, which can decrease the effective surface area, thereby negating the benefit of size reduction on dissolution enhancement. In such cases, wetting agents, such as surfactants, can be used to retain the effective surface area. Implications of high‐energy surfaces for physical and chemical stability also need to be thoroughly assessed in the context of shelf life determination for the drug substance.
When crystal structure modifications or particle size reduction are insufficient to improve solubility and dissolution rate, enabling formulations that do not incorporate the drug substance in the crystalline state can be evaluated. Chief among such options is ASDs, which can provide superior bioavailability when compared with drug products containing a crystalline drug. At the most basic level, ASDs favor enhanced dissolution or increased solubility of poorly soluble crystalline compounds due to the formation of a high‐energy amorphous state [12–14]. The increased solubility can also be attributed to solubilization effects of the polymer, often used in ASDs to stabilize the amorphous state during manufacturing and storage [15]. Polymers are also selected to help maintain supersaturation during in vivo dissolution by inhibiting recrystallization within the time scale relevant to absorption. It has been reported that even the flux across a membrane can be enhanced by the polymer in supersaturated solutions [16]. At a fundamental level, in vitro and in vivo stabilization of ASDs depends on both thermodynamic and kinetic factors. Maximum physical stability is achieved when the drug and excipients remain intimately mixed. Assurance of thermodynamic stability requires quantitative understanding of the solubility/miscibility of the drug in the polymer‐rich excipient matrix. Recently, relatively simple but reliable experimental approaches have been developed and implemented to obtain the phase diagram of API/polymer systems and determine the maximum drug load for a fully miscible ASD [17–19]. Mathematical modeling solutions to estimate solubility/miscibility of drugs in polymers have been also reported, with the perturbed‐chain statistical associating fluid theory (PC‐SAFT) [20, 21] being especially noteworthy due to its origins in polymer chemistry. Designing a thermodynamically stable and molecularly dispersed ASD, wherein the drug load does not exceed the solubility/miscibility limit in the polymer matrix, offers excellent stability. However, this ASD system often translates in relatively low drug load, which can lead to pill burden challenges for high dose drug candidates. In such cases, kinetically frozen ASDs, where drug molecules are not molecularly mixed, can be considered with appropriate process controls and storage conditions.
In vivo stability of an ASD is an equally important consideration in achieving target bioperformance. For an enhanced absorption to take place, supersaturation must be obtained and maintained in the gastrointestinal environment. This can be achieved by designing ASD formulations that upon dissolution form nanosized drug‐rich droplets. Implicit in the drug‐rich domain formation is the benefit offered by rapid dissolution rates and equilibration with the aqueous phase, saturated with the drug at the amorphous solubility, to replenish the molecularly dissolved drug, as the drug diffuses into systemic circulation. Therefore, drug‐rich droplets can serve as reservoirs to sustain the thermodynamic activity of the drug at its maximum value [22, 23]. The underlying phenomenon causing the formation of drug‐rich droplets upon dissolution of an ASD is the liquid–liquid phase separation (LLPS) [24]. A schematic representation of the drug uptake of an ASD undergoing LLPS is displayed in Figure 29.3. Whether an ASD dissolves to form a solution in which LLPS occurs is likely to depend on a number of factors, including inherent drug properties (e.g. crystallization tendency), drug loading, and polymer type in the formulation [25]. The role of LLPS is the foundation for a well‐designed ASD formulation; as long as LLPS can be maintained within the gastrointestinal absorption window, the formulation is close to optimal. Multiple processing options including drum drying, spray drying, and hot melt extrusion exist to manufacture ASDs. Advances in the hot melt extrusion technology have in particular accelerated industrial applications of ASDs for the delivery of poorly soluble drugs.

FIGURE 29.3 Schematic representation illustrating the drug uptake of an ASD formulation that undergoes LLPS.
Source: Adapted from Raina et al. [22] with permission of Elsevier.
As part of the process of identifying and efficiently selecting the most appropriate solubility‐enhancing strategy, solid form development plays an integral role in the development of new chemical entities. Solid form screening experiments can be tailored not only to the physicochemical properties of the drug molecule but also the downstream formulation options. As a consequence, solid form selection criteria can be adapted to the specific objectives for the drug substance molecule. In this chapter, we provide a general current perspective on solid form development in the pharmaceutical industry and two case studies on solid form development of poorly soluble compounds. Through the case studies we illustrate the impact of solid form on drug substance as well as on drug product manufacturing processes.
29.2 PERSPECTIVE ON SOLID FORM SCREENING IN DRUG DEVELOPMENT
The solid form development process starts with a discussion on strategic objectives related to dosage forms to achieve a target product profile. Determinations are made on whether chemical (salts/cocrystals) and/or physical (polymorphs) modifications are needed based on the physicochemical properties of the molecule and early predictions of the projected dose by physiologically based pharmacokinetics models. The aim is to generally identify a crystalline form that shows optimal solid‐state properties and a relatively simple solid form landscape. In addition, a developable solid form is able to reject impurities during crystallization, is amenable to downstream processing, and ensures consistency in the safety and efficacy profile of the drug product throughout its shelf life. Overall, among the potentially many solid forms of an API, a crystal form that is stable, helps achieve bioperformance, and can be manufactured into the dosage form is generally progressed in pharmaceutical development. A thermodynamically most stable form is highly desirable, because a significant drop in solubility due to the discovery of a more stable form can profoundly affect bioperformance and dosage form. This is especially true for soluble compounds where solubility modification techniques to maintain supersaturation through in vivo dissolution and absorption are not built into the dosage form.
When salts and/or cocrystals are anticipated to address significant solubility or stability issues, experimentation begins with screening appropriate counterions and/or coformers. Some of the recently matured in silico tools can be used to augment experimental efforts to improve outcomes and save resources [26]. Generally, salt or cocrystal screening does not add value for molecules that are deemed to be developable only in the amorphous state via, for instance, the ASD route. Once the species (salt, cocrystal, or parent molecule) has been selected, extensive polymorph screens are conducted in stages during preclinical development to eventually identify a developable commercially viable crystal form generally well before registration supporting clinical studies. Although the solid form screen is a routine activity for any given small molecule drug candidate, the actual recipe for searching the crystal forms of a compound for which the crystal form landscape has never been explored is uniquely tied to the structure of the molecule itself [27]. In other words, polymorph screening is by no means routine [28], and the design of each solid form screen has to be customized based on (i) physicochemical properties of the molecule and (ii) the intended formulation.
Figure 29.4a illustrates some of the key considerations that drive customization of form screening experiments for commercially viable crystal form selection. The interconnectivity between crystal form complexity, formulation, manufacturing processes, and bioperformance, along with the associated need for customization of screens, becomes even more important for poorly soluble compounds. By way of example, Figures 29.4b and c illustrate an approach to customization based on solid form complexity and manufacturing process choices. The illustrations capture a scenario where wet milling is intended to be used either to induce nucleation under high‐shear conditions or control particle size and hot melt extrusion for the preparation of an ASD‐based dosage form. For the latter, since hydrates or low‐melting but sufficiently stable solid forms are preferable over high‐melting polymorphs, a strong emphasis is placed on searching for hydrates and crystallizing under high‐supersaturation conditions. The use of shear can reduce the induction time and facilitate nucleation and even isolation of metastable forms. Relatively large molecules that do not nucleate under mild supersaturation conditions afforded by the classic solution‐mediated phase transformation‐based polymorph screening or that have the tendency of oiling out can especially benefit from high‐shear/high‐supersaturation conditions. Miniaturized Taylor–Couette [29] flow system can be used for screening purposes to mimic high‐shear conditions and achieve Reynold’s numbers not too dissimilar to those generated in commonly used rotor–stator‐based wet mills. Complexity of a crystal form landscape would necessitate focusing more experiments in the process‐relevant space. In the end, the number of experiments performed is somewhat commensurate with the solid form complexity.

FIGURE 29.4 (a) Schematic representation of the strategy for a customized solid form screen, (b) solvent selection guidance based on the solid form complexity and formulation platform, and (c) experimental condition guidance based on the ease of crystallization and the crystallization platform.
A comprehensive perspective on a solid form screen can provide a route map toward a fully integrated, holistic product design process and enable the journey from molecule to crystal to product performance. The framework needs to be customized for each drug molecule due to the distinct nature of crystal chemistry and different crystallization tendencies, either of which can limit the diverse experimental conditions that can be applied in the search for crystal forms. For these reasons, solid form control is, in the end, primarily a risk assessment and mitigation exercise for the selected, commercially viable crystalline form, from drug substance isolation through drug product manufacture and storage. Rapidly evolving in silico tools can also effectively augment the well‐established crystal form risk assessment tools typically used in pharmaceutical development and help de‐risk the detrimental implications of late discovery of a new crystal form.
29.2.1 In Silico Modeling in Pharmaceutical Solid‐State Chemistry
Advances in computational solid‐state chemistry are increasingly becoming important for de‐risking crystal form complexity in pharmaceutical development [30]. In silico modeling, for example, provides unique insights into the fundamentals of crystal chemistry that often are inaccessible through traditional experimental methods. Within the broad spectrum of computational solid‐state chemistry, multiple approaches exist that help de‐risk pharmaceutical solid forms. Herein, a brief overview of two such approaches – structural informatics and crystal structure prediction (CSP) – is provided.
29.2.1.1 Structural Database Mining
The identification of unusual structural features, such as an unusual molecular conformation, a geometrically unusual hydrogen bonding interaction, and/or an unusual donor–acceptor pair, can be very powerful in assessing risk and form complexity in a new API molecule. The role of hydrogen bonding in particular toward the formation and stabilization of molecular crystals is a foundational element of structural diversity seen in pharmaceutical materials [31–33]. Hydrogen bonding interactions, which are strong and directional, often contribute significantly to lattice energies, even though van der Waals interactions also play a significant role in the formation of molecular organic crystals [34–37]. Systematic assessment of inter‐ and intramolecular hydrogen bond‐forming propensity, as well as its comparison to the known packing motifs of molecular organic crystals, can help estimate the form complexity of a new API molecule even in the absence of a solved single‐crystal structure. To this end, Cambridge Structural Database (CSD) [38, 39] organized by the Cambridge Crystallographic Data Centre (CCDC) and internal proprietary structure databases can be leveraged. At the time of this writing, more than 900 000 crystal structures of organic and metal‐organic small molecules have been collected.
The CSD contains information about intramolecular geometries and molecular conformations as well as intermolecular interactions. Once a crystal structure of an API molecule has been solved, additional insights can be obtained through the analysis of full interaction maps within the crystal lattice and with the comparison of the known structures. A stage‐wise combination of hydrogen bond propensity analysis and full interaction maps can even provide insights into the relative stability of polymorphs and aid polymorph risk assessment/mitigation. The CSD offers a suite of software that provides a wide range of tools for both of these structural informatics‐based approaches to readily execute the assessment at a quite modest computational cost.
Consistent with the important correlation between diversity of hydrogen bonding motifs and polymorphism, CSD analysis shows that when molecules have multiple hydrogen bond donors and acceptors, a variety of hydrogen bond pairings can be observed among polymorphs. Statistical models of experimental observations in the crystallographic database can be applied to compute the likelihood that a given hydrogen bond pairing will occur. The hydrogen bond propensity model offers a prediction for the presence or absence of a hydrogen bond between a specified donor and acceptor atom in a crystal structure, based on related known crystal structures, their chemical functionality, and their molecular environment [40, 41]. The key assumption is that actual hydrogen bonds directing the formation of a crystal structure will be those with the highest likelihood of forming among all possible donor–acceptor pairs. Based on this approach, crystal forms that satisfy donor–acceptor pairs with the highest predicted propensity are likely the most stable crystal forms. In contrast, the lack of strongest donor and acceptor pairs indicates a high likelihood that a more stable form is possible and/or other competitive crystal forms might exist. Assessment of the famous case of ritonavir polymorphs via hydrogen bond propensity analysis is indeed very revealing [5]. The stable Form II of ritonavir contains a hydrogen bonding interaction with a higher propensity to occur that is absent in the metastable Form I [42], as depicted in Figure 29.5a. In contrast, two hydrogen bonds in the initially marketed Form I are unusual and show low propensity values. Proactive application of the knowledge‐based methodology to the structure of Form I at early stage of drug development would have warranted further experimental solid form screening.

FIGURE 29.5 In silico tools for solid form de‐risking: (a) donors and acceptors in ritonavir molecule with the highest propensity of the hydrogen bonding observed in Form II predicted between the amide donor and the hydroxyl acceptor, (b) full interaction map for ritonavir Form II, and (c) crystal energy landscape of the model pharmaceutical crizotinib
Source: Adapted with permission from Abramov [43]. Copyright © 2013 American Chemical Society.
The Full Interaction Map tool within the CSD helps visualize molecular interactions in three dimensions and also helps evaluate whether the molecular packing of a crystal structure satisfies the interactions of functional groups [44]. The tool computes maps around a molecule where hydrogen bonds are likely to be found based on IsoStar [45] interaction data from the CSD. The IsoStar program is a library of graphical and numerical information about intermolecular interactions in the form of scatter plots that relate a pair of functional groups. First, the Full Interaction Map tool breaks down the molecule into a set of central groups and then assembles the group‐based interaction data for selected donor/acceptor/hydrophobic probes around each central group. Environmental effects, including steric factors, are taken into account to create a full three‐dimensional picture of molecular interactions. A comparison of the fit of the calculated maps with the observed interactions allows the crystal structure to be assessed in terms of how well intermolecular interactions are satisfied by the existing lattice. For a polymorphic compound, the different interactions observed in each polymorph can be examined and assessed. Revisiting the case of ritonavir with full interaction maps shows that Form II has a crystal structure with hydrogen bonding consistent with the most favorable interactions, as shown in Figure 29.5b. A complementary method of comparing known crystal forms is represented by Hirshfeld fingerprint plots, which represent a two‐dimensional visualization of the space (surface) occupied by a molecule in a crystal. The fingerprint plots provide a quantitative analysis of various intermolecular interactions, including close contacts in the crystal [46, 47].
29.2.1.2 Crystal Structure Prediction
As stated by Cruz‐Cabeza et al. [27], targeted polymorphism remains out of reach for molecular organic crystals, such as pharmaceuticals. In fact, given the unique features of every compound, we have no means of knowing the number of crystal forms that can exist, nor the number or type of experiments that need to be carried out. Structural informatics and risk experimental methodologies described above can help reduce risk within the limitations of the approaches but not eliminate it. Ab initio CSP is in principle the most comprehensive approach to explore the complete form landscape and fully understand risks. The field has made significant strides since the gauntlet was thrown down by Dunitz in the early 1990s [48].
CSP attempts to predict all the possible crystal structures of a molecule, given only the two‐dimensional chemical structure of the compound in question. The most general and commonly applied method that has been developed for CSP is global lattice energy minimization [49]. The approach involves locating and assessing the relative stabilities of all local energy minima on the lattice energy landscape. The result is an assembly of plausible crystal structures (i.e. a crystal energy landscape) that are ranked in order of their lattice energy, computed at absolute zero temperature, and separated by their density, as shown in Figure 29.5c. In this example, the lowest‐energy predicted structure of the pharmaceutical molecule crizotinib corresponds to the experimental crystal structure and is approximately 7.5 kJ/mol below the next lowest‐energy structure [43]. A key assumption of CSP is that the structure corresponding to the global minimum is the most likely observed structure [50]. Because the lattice energies of possible crystal structures in a given molecule are often found to be within a few kilojoules per mole [51], ranking crystal structures can be a challenge. However, CSP can be applied to identify and rank potential polymorphs of a given crystal form by lattice energy.
Many theoretical methods of lattice energy calculations have been developed. The performance of different approaches has been assessed in six blind tests hosted by the CCDC [50, 52–56], where predictions are performed in advance of the real crystal structures being made available. Genuine successes in predicting crystal structures have been achieved during these blind tests. The latest blind test [56], held in 2016, shows the continuing development and increased maturity of the computational methods with more challenging target systems included in the test, such as two large conformational flexible molecules, a salt hydrate, and a cocrystal as multicomponent systems. All experimental crystal structures of the target systems but one were predicted by at least one submission. Thus, tasks that were thought to be unmanageable in the early days of CSP have become possible.
Despite tremendous progress, computational CSP still faces two important challenges: (i) a ranking problem and (ii) a sampling problem. A main limitation is related to the lattice energy model – the atom–atom force field – currently used [57]. Predictive relative energies can be within the expected errors, due to the model potential and inability to accurately account for thermal effects in lattice energy calculations. This creates a physical challenge in accurately describing and thus ranking the relative stabilities of all possible crystal packing alternatives. In order to address this challenge, the field has evolved toward higher quality and more sophisticated energy models, including those based on density functional theory (DFT) augmented by the empirical dispersion correction (DFT‐d). A significant advancement in the DFT‐based approach has been made by Neumann and coworkers [58, 59]. It involves using the DFT‐d theory to generate reference data from which force field parameters, tailored to the molecule under investigation, are derived for candidate crystal structure generation. The final energy minimization of low‐energy minima among the computed structures is then performed by DFT‐d calculations. While refining the relative lattice energies of the computed structures by periodic electronic structure calculations improves the accuracy of reliable ranking, the faithfulness of predictions can be compounded for enantiotropic polymorphic systems where the most stable crystal phase changes with temperature. Simulations of free energy tackle the finite temperature effect directly. Enhanced molecular dynamics algorithms, based on multiscale modeling, have been developed with the aim of addressing theoretically challenging questions on computed crystal energy landscape at ambient temperature with high accuracy [60, 61]. Benchmark free energy calculations of small rigid molecules, such as benzene and naphthalene, have proven to be successful for generating and thermodynamically ranking their crystal structures [62]. It is worth emphasizing that, at present, free‐energy‐based approaches can only handle molecules with significantly low structural complexity compared with lattice‐energy‐based methods.
A second challenge relies on finding all low minima locations on the multidimensional lattice energy surface, which is defined by the degrees of freedom or structural variables of a system. Navigating and exploring the complex energy surface is not a trivial task. Most of the plausible crystal structures for a molecule are generated based on the input molecular connectivity and stoichiometry and the user‐specified range of space groups and number of independent molecules in the unit cell. In order to determine the relevant local minima, the vast majority of studies have utilized the random sampling approach, which has proven to be fairly effective with structures up to 20 degrees of freedom [63]. This number of degrees of freedom covers structure generation for a rigid molecule with up to two independent molecules in any space group. Nevertheless, it is known that sampling an energy landscape well enough to locate all local minima becomes more difficult as the flexibility of the target molecule, the number of symmetrically independent molecules in the crystal unit cell, and the number of components increase. This means that by scaling the sampling with molecular flexibility and the computational cost with molecular size, current ab initio methods cannot be scaled to a molecule like ritonavir. To date, the computational expense of successful blind test submissions for more challenging target systems corresponds to a few months of dedicated use of high performance computing clusters. Even if the field continues to advance, CSP does not aim to replace experimental solid form studies. Rather, it can be applied as part of a suite of in silico tools to assess the likelihood that other crystal forms could exist for a given molecule and manage the risk of the selected solid form if a more stable one has yet to be identified.
29.3 SOLID FORM CONTROL OF LINIFANIB
29.3.1 Introduction
Linifanib is a multi‐targeted receptor tyrosine kinase inhibitor [64]. Linifanib is lipophilic at neutral pH with a distribution coefficient, Log D, between n‐octanol and a pH 7.4 buffer of 4.2. The compound has high permeability and is practically insoluble, with a solubility of 27 ng/ml in aqueous media at pH 5; thus, it is classified as a BCS class II compound. Linifanib exhibits an exceedingly slow dissolution rate, suggesting that oral absorption through the membrane is dictated completely by dissolution (i.e. the absorption is dissolution rate limited). Simply increasing the dissolution rate via salt formation or particle size reduction may not be sufficient to achieve the desired bioavailability. An ASD‐based formulation will be required for overcoming problems associated with dissolution rate absorption, as it ensures that the drug is present in a reservoir with a low barrier to permeate through the membrane. Linifanib is formulated as ASD by hot melt extrusion technology for enhancing solubility, thereby improving dissolution rate and oral bioavailability.
The molecular structure of linifanib, shown in Figure 29.6, contains hydrophobic aromatic rings as well as polar substituents (i.e. urea and indazole groups), which provide hydrogen bonding donating and accepting capacity. The presence of rotatable bonds permits the molecule to exist in a number of different plausible conformations. In principle, these features of the molecular structure should create the opportunity for linifanib to form a variety of potential molecular packing motifs, giving rise to different polymorphic forms. This case study illustrates the application of systematic yet comprehensive solid form screening experiments coupled with in silico modeling tools for solid form risk assessment. This assessment includes crystallographic knowledge and structural informatics toward selecting a solid form that possesses desirable solid state and physicochemical properties and also enables both the drug substance quality and an optimal drug product dosage form.

FIGURE 29.6 Molecular structure of linifanib, highlighting donor–acceptor functional groups.
29.3.2 Structural Analysis of Linifanib Ethanol Solvate
29.3.2.1 Crystal Structure
During the early stage of development, various isomorphic solvated crystalline forms, including ethanol and toluene solvates, were discovered as part of the initial solid form screening. These solid forms were characterized by single‐crystal X‐ray structure determination. The crystal structure of the ethanol solvate, named as Form I, is shown in Figure 29.7a. Form I crystallizes in the triclinic lattice system and with the space group P‐1, wherein linifanib molecules form a layered structure. Solvent molecules are arranged in “pockets” between drug chains. The spacing between drug chains can increase in order to accommodate solvent molecules larger than ethanol, thus accounting for the ability of linifanib to form structurally similar solvates with various organic solvents.

FIGURE 29.7 (a) Molecular packing and (b) intermolecular hydrogen bonding network, highlighted as dashed lines, of linifanib ethanol solvate Form I.
The analysis of the strength and directionality of intermolecular interactions in a crystal structure usually allows the classification of molecular organic crystals depending upon the type of their basic structural motif or synthon. Supramolecular synthons express the core features of a crystal structure; as such, they are considered a reasonable approximation of the entire crystal [65]. Linifanib contains a N‐N′‐diaryl urea moiety. Even though the urea functional group has only one acceptor atom, the carbonyl oxygen, it can form N─H···O interactions with two neighboring molecules. Crystal structures characterized by the urea functional group can be principally classified into two categories depending upon the common hydrogen bond pattern or supramolecular synthon: the urea tape α‐network (synthon I) and the non‐urea tape structure (synthon II), as displayed in Scheme 29.1. Typically, synthon I (i.e. urea···urea hydrogen bond motif) is known to be the strongest and most dominant motif of the urea functional group [66]. In the urea tape α‐network, the acceptor oxygen atom receives hydrogen bonds from two equivalent donor groups, thus forming a cyclic eight‐member ring. The isostructural solvated forms of linifanib are, however, characterized by synthon II (non‐urea tape motif) as the dominant hydrogen bond motif, wherein N─H···N interactions disrupt the robust tape α‐network of the urea functionality. A close inspection of the Form I crystal lattice indicates that linifanib molecules adopt hydrogen‐bonded chains between the carbonyl oxygen of the urea functionality and the NH of the indazole ring and between NH donors of the urea group and the nitrogen of the indazole ring, as displayed in Figure 29.7b. In Form I, donor and acceptor groups of the indazole ring are not engaged in intermolecular interactions with solvent molecules; as such, the acceptor nitrogen atom of the indazole ring accepts hydrogen bonds from the urea NH donors, while the donor group of the indazole moiety is involved in a hydrogen bonding with the carbonyl oxygen of the urea functional group. These interactions alter the structural arrangement of linifanib molecules and result in a synthon that prevents the geometry and topology of the strong hydrogen bonding urea tape motif.

SCHEME 29.1 Common supramolecular synthons formed from urea functional groups.
The hydrogen bond motif present in the crystal structure of Form I is thought to be related to the molecular conformation. The urea moiety in the Form I crystal structure shows the characteristic feature of adopting a near‐planar conformation with N–Ph torsional angles of 176.1° and 158.3°. This conformation is further stabilized by an intermolecular interaction with the indazole ring of a neighboring drug molecule. Thus, in the Form I crystal structure, drug molecules reside in a stable conformation with approximate coplanarity between the urea group and the aryl rings, but do not generate the strong hydrogen bonding of the urea tape α‐network. To go beyond the functional group‐based viewpoint, intermolecular interactions of the whole molecule in the crystal structure of linifanib Form I were then evaluated using the Full Interaction Map tool.
29.3.2.2 Full Interaction Map
Using the Full Interaction Map tool in the CCDC Mercury software package [67], the molecular conformation in the Form I crystal structure was analyzed and compared with fragments exhibiting similar chemical features to the many published structures in the CSD. A map describing the molecular environment was then prepared; changes in both the chemical nature of the fragments and molecular conformations are reflected in the resulting full interaction map. The full interaction map for linifanib Form I is shown in Figure 29.8. The clouds indicate regions where hydrogen bond acceptors and hydrogen bond donors are most likely to be found, based on interaction data mined from the CSD. The most intense contour maps indicate a greater likelihood that an interacting group will be found in that region.

FIGURE 29.8 Full interaction map around molecular conformations of linifanib ethanol solvate Form I.
The donor and acceptor atoms of the indazole moiety together with the donors of the urea group form hydrogen bonds in the predicted locations. Careful investigation reveals that the carbonyl oxygen of the urea in Form I shows a hydrogen bond with suboptimal geometry, which results in a donor outside the closest interaction map cloud. The presence of an unsatisfied acceptor in the crystal structure of Form I indicates a sign of metastability. The metastability is attributed to the formation of a rare hydrogen bond synthon stabilized by proximal weak interactions. Thus, the in silico Full Interaction Map tool helps confirm a weakness in the crystal structure of linifanib Form I.
Further, the ethanol solvate Form I was found to represent a challenge, as the solid form for pharmaceutical development for two main reasons: (i) the high residual solvent level in the drug substance, due to the “pocket‐like” crystal structure, and (ii) the high melting point (204 °C) that results in a high temperature for the drug substance dissolution in the extrudate matrix with potential high risk of processability for the hot melt extrusion. Therefore, based on structural informatics principles, the statistical treatment of intermolecular interactions in the Form I crystal structure, together with its solid‐state properties and drug product manufacturability, warranted the search for alternative solid forms, including low‐melting anhydrates or hydrates with low dehydration temperatures. In silico methods for the hydrate formation prediction have proven to be of high value in guiding a more systematic experimental screening. In the following section, additional details on the approach and its application to linifanib are summarized.
29.3.3 Linifanib Hydrate Formation: Computational and Experimental Approaches
The solid form screening conducted early in drug development did not yield crystalline hydrates, despite including crystallization conditions favoring their formation. As such, computational approaches were undertaken to predict the hydration propensity of linifanib. Computational and statistical models have been used to rationalize hydrate formation of molecular organic crystals [68–70]. The free energy of mixing of the molecule of interest with water in a supercooled liquid phase has been shown to be an effective descriptor of the propensity of small molecule drug candidates to form hydrates [71]. Hydrogen bond propensity analysis has been also utilized to understand hydration in molecular organic crystals [40]. However, two main considerations are (i) the probability of low‐energy geometrical distortions of a molecule from its ideal geometry and (ii) the existence of multiple conformers on the intramolecular energy surface. Building upon these concepts, a method based on molecular conformation estimation and hydrogen bond propensity has been developed for predicting hydrate formation [72]. Gauging molecular conformations representative of the solid state can guide a more accurate description of intermolecular hydrogen bonding interactions in hydrates and anhydrous crystals, because intramolecular interactions and sterically inaccessible polar regions can significantly affect whether intermolecular interactions can form in the solid state.
A combined quantum mechanics and data‐driven approach, according to the schematic illustrated in Figure 29.9, has been developed and utilized to predict the hydrate formation of linifanib. Conformers were generated using quantum mechanical calculations by the conductor‐like screening model for realistic solvation (COSMO‐RS). This method adopts a quantum approach in combination with statistical thermodynamics [73, 74]. Donor and acceptor groups available for intermolecular interactions were analyzed by evaluating representative molecular conformations. API–water and API–API propensities for each donor/acceptor group were determined to provide insights on potential hydrate formation. Hydrogen bond propensities, modeled by a probability function using data mined from relevant CSD crystal structures, were predicted for linifanib fragments. API–water and API–API propensities were evaluated in a hierarchical manner by pairing the best donor with the best acceptor, the next best donor with the next best acceptor, and so on. The results of the analysis are presented in Table 29.1. The strongest donor–acceptor pair is between the primary amine and water, followed by water and the urea group, with propensity values of 0.71 and 0.70, respectively. The strongest API–API propensity is between the primary amine and the urea groups with a value of 0.76. Since each functional group is allowed to donate and/or accept once, another strong API–API interaction is between urea and indazole groups. Using hydrogen bond propensities, a multi‐differential hydrogen bond propensity score can be calculated, according to the equation
The overall hydrogen bond propensity score of the model is −0.08. A negative score indicates that hydrate formation is favorable for linifanib. This prompted the initiation of a more systematic solid form screen with the intent to find developable crystalline hydrates.

FIGURE 29.9 Schematic of the combined quantum mechanics and data‐driven approach for the hydrate prediction of linifanib.
TABLE 29.1 Hydrogen Bond Propensities Predicted for the Hydrate Formation of Linifanib Molecule. Observed Donor–Acceptor Combinations in the Monohydrate Form V Are Indicated
| Donor | Acceptor | Propensity | Form V |
| NH amine | O urea | 0.76 | X |
| NH amine | O water | 0.71 | √ |
| OH water | O urea | 0.70 | X |
| NH amine | N indazole | 0.67 | X |
| OH water | O water | 0.65 | X |
| OH water | N indazole | 0.60 | √ |
| NH urea | O urea | 0.58 | √ |
| NH indazole | O urea | 0.55 | X |
| NH urea | O water | 0.51 | X |
| NH indazole | O water | 0.48 | √ |
| NH urea | N indazole | 0.46 | X |
| NH indazole | N indazole | 0.44 | √ |
Note: √, observed; X, not observed.
The solid form screening of linifanib required overcoming challenges presented by its poor aqueous solubility properties and the relative ease with which the molecule seems to crystallize as solvates. The screen comprised mainly solution‐based recrystallizations and included both thermodynamic and kinetic approaches. The thermodynamic screening was carried out in organic solvents with a wide range of properties and various combinations of aqueous organic solvent mixtures. The screen was designed by varying the water content of binary and ternary solvent compositions as a means of regulating the water activity, so as to favor the formation of potential hydrates. For the kinetic screening, stressed crystallization experimental conditions were employed (e.g. elevated supersaturation ratio, high antisolvent addition rate, forward/reverse antisolvent addition, high shear rate, and/or fast cooling rate) with the aim of identifying metastable forms. These targeted experiments resulted in the isolation of a monohydrate crystal form, designated as Form V.
29.3.4 Structural Analysis of Linifanib Monohydrate
29.3.4.1 Crystal Structure
In the crystal structure of the monohydrate Form V, linifanib molecules are arranged in a linear array via the urea tape α‐network, as shown in Figure 29.10. Intermolecular interactions between the indazole rings contribute to hydrogen‐bonded chains, while water molecules interact within the indazole hydrogen bonding network.

FIGURE 29.10 Molecular packing and intermolecular hydrogen bonding network, highlighted as dashed lines, of linifanib monohydrate Form V.
Molecules in the Form V crystal structure adopt a twisted conformation with N–Ph torsional angles of −132.6° and 147.6°. An overlay of Form I and Form V conformers is displayed in Figure 29.11. The rotation of the N–Ph moiety in the molecule, from a near‐planar conformation (as observed in Form I) to a twisted conformation (as observed in Form V), is required for the urea tape synthon I self‐assembly. In order to form the urea tape α‐network, the molecule has to take an energy penalty by rotating around the N–Ph bond that is energetically compensated by forming a strong intermolecular hydrogen bond. This implies that the molecular conformation is related to the urea tape α‐network and non‐urea hydrogen bonding synthon. This leads to the idea of exploiting the hard and soft acids and bases (HSAB) principle [75] to evaluate intermolecular interactions and understand molecular assembly in molecular organic crystals. It has been shown that the twisting of the aryl groups in flexible diaryl urea structures makes the carbonyl oxygen of the urea functional group a better hydrogen bond acceptor [76], able to interact with the strong urea NH donors and form the urea tape array. Thus, there is a key link between the intrinsic molecular structure and the crystal packing, allowing a profile of important interactions to be built up within families of compounds. To provide additional support for intermolecular packing effects, two CSD‐based structural informatics approaches – hydrogen bond propensity and full interaction map – have been applied.

FIGURE 29.11 Overlay of Form I and Form V conformers. The three pairs of atoms used for the overlay are highlighted.
29.3.4.2 Hydrogen Bond Propensity
As discussed previously, a predictive indicator of whether a hydrogen bonding interaction is unusual is vital in assessing solid form stability during pharmaceutical product development. Table 29.1 shows hydrogen bonding interactions in the known monohydrate Form V compared with the predictions of a set of potential donor–acceptor pairs, as an assessment of solid form stability. In the known hydrate crystal structure, the observed hydrogen bonding between the amine group of linifanib and the oxygen atom of the water molecule is ranked the second most likely donor–acceptor pair (0.71) and shows a hydrogen bond propensity higher than that predicted between water molecules. The results suggest that intermolecular interactions between linifanib and water molecules are favored compared with those between water molecules. This may also imply that the direct formation of linifanib hydrate is possible when water is used as the crystallization solvent.
29.3.4.3 Full Interaction Map
A full interaction map was computed for linifanib Form V using data from fragments found in the CSD. The results are shown in Figure 29.12. The urea and indazole groups in Form V have distinct regions of interaction density located near them. By overlaying the intermolecular interactions observed in the crystal structure of Form V, donor and acceptor groups of the indazole group and the urea functional group form hydrogen bonds in the exactly predicted locations. The donor and acceptor groups of the water molecule are also satisfied and fit the map well. Overall, the crystal structure of linifanib Form V matches the predicted interaction geometry. Thus, the known monohydrate crystal form exhibits a satisfactory network of intermolecular interactions. This supports the hypothesis that a different molecular packing would not likely result in a more stable hydrate form.

FIGURE 29.12 Full interaction map around the molecular conformation of linifanib monohydrate Form V.
29.3.5 Solid Form Selection and Impact on the Downstream Processing
Linifanib monohydrate Form V was selected as the solid form for continued development based on its favorable solid state and physicochemical properties. Obtaining an API in the right solid form and with consistent physical properties is critical not only from the drug substance manufacture standpoint but also from the perspective of drug product processing, performance, and stability.
The high‐energy amorphous state of the hot melt extrusion process features the dissolution of the drug substance into the polymer matrix, which is facilitated by applying shear stress and thermal energy to a powder blend. During processing, drug substances are thereby exposed to elevated temperatures for prolonged periods of time. High processing temperatures together with shear stresses and melt viscosities can induce decomposition of thermally unstable drugs. Linifanib drug substance degrades under high temperatures. Linifanib Form V melts at a lower temperature compared with the ethanol solvate Form I; as such, the manufacture of linifanib Form V drug substance allows the hot melt extrusion technology to operate at a reduced temperature in order to molecularly dissolve the drug substance into the polymer matrix and achieve a homogeneous ASD. The ability to operate at a lower temperature helps control the risk of chemical degradation and increase the processing window for the hot melt extrusion.
The process of reaching the thermodynamic end state at which the drug can form a molecular‐level single‐phase system is accelerated as the temperature is increased. Nevertheless, other material properties determine the kinetics of reaching the thermodynamic endpoint of the process. It is expected that surface area increase or particle size reduction enhances the dissolution kinetics by reducing the diffusive mixing required for dissolution. Mathematically, this can be described by the traditional Noyes–Whitney equation [77], wherein the dissolution rate or rate of mass transport is a function of the difference in solubility of the drug in the polymer, Cs, and the concentration of the drug dissolved in the polymer, C(t); the diffusion coefficient, D; the surface area, A; and the boundary layer thickness, h, as follows:
It becomes apparent that the dissolution rate can be enhanced by increasing the surface area of the components through effective dispersion or decreasing the boundary layer thickness through shearing of the particles. Linifanib Form V crystallizes as small and thin needlelike particles, resulting in high surface area. This is beneficial for the complete dissolution of linifanib drug substance in the hot melt extrusion process. The resulting extrudates, manufactured using Form V, were crystal‐free, while those manufactured using Form I were found to contain small amounts of residual crystallinity, as illustrated in Figure 29.13. The incomplete suppression of the drug crystallinity, as observed by polarized light microscopy, is attributed to the specific surface area and particle morphology of the drug substance.

FIGURE 29.13 Scanning electron microscopy images of linifanib (a) Form I and (b) Form V and their corresponding extrudates.
29.3.6 Manufacturing Process of Linifanib Monohydrate
Based on slurry competition studies, linifanib Form V was established to be thermodynamically stable only at high water activities (>0.9). This represents a significant challenge for a poorly soluble molecule in terms of solvent selection and conceptual design of the crystallization process. Based on experiments conducted in various solvent compositions to evaluate solubility and purification potential, a crystallization solvent system comprising water, ethanol, and ethyl acetate was selected. The relative stability of process‐relevant solid forms in the ternary solvent system was assessed based on the equilibrium solubility, which was determined by high performance liquid chromatography, and the solid phase, which was characterized by X‐ray powder diffraction. The phase diagram built in the ternary solvent system, based on the mole fraction of solvent mixtures, shows that either an anhydrate crystal form (Form VII) or a mixture of polymorphs was obtained in most of the solvent region, as displayed in Figure 29.14. This implies that most of the solvent domain is not appropriate for the consistent isolation of Form V under thermodynamic control. Despite challenges related to the very tight region involved in Form V stability, the low solubility, and chemical instability in many organic solvents, a crystallization process was designed to selectively yield the monohydrate Form V by direct precipitation without interferences from Form I and/or Form VII. The developed crystallization process consisted of charging a solution of linifanib in ethanol/water to water for seed generation. The seed slurry was added to a concentrated solution of linifanib with an appropriate ethyl acetate/water composition. The product slurry was then charged to water while distilling to remove organic solvents and maintain a water‐rich solvent composition. The product was isolated by filtration, washed with water, and then dried. During the drying process, the integrity of the hydrate state of the desired solid form was carefully studied, and appropriate controls were established. Appropriate storage conditions were also instituted to preserve the monohydrate Form V, as it is stable at a relative humidity equal to or greater than 30% at 25 °C.
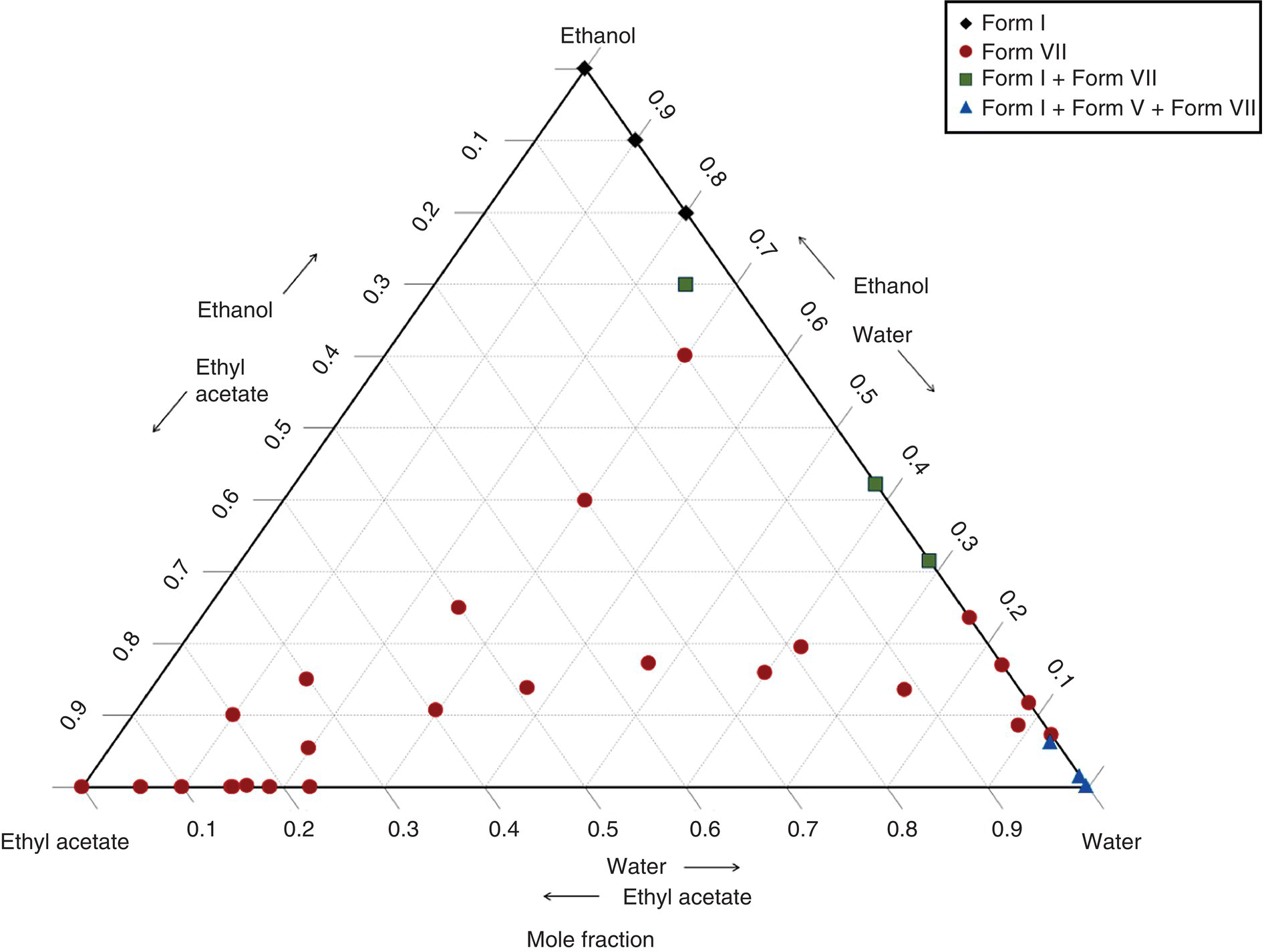
FIGURE 29.14 Phase diagram of linifanib in the crystallization solvent system at room temperature.
29.3.7 Summary
A multidisciplinary approach, involving solid form screening experiments and in silico modeling tools as well as the correlation with information extracted from a given single‐crystal structure, is of paramount importance to better understand the structure and function of small pharmaceutical molecules. Through tailored designed experiments and in silico modeling tools, a fundamental understanding of the structure, thermodynamics, and kinetics of process‐relevant solid forms has been gained for the poorly soluble compound linifanib. When multiple crystalline forms are identified from solid form screens, it is the connection between crystal structure, solid‐state properties, drug substance, and drug product processing and performance that ultimately determine which form advances the development of the drug product. A metastable form of linifanib was selected for development to meet the nature or requirements of the drug product, thus facilitating the enabling formulation technology. This case study thus emphasizes the importance of applying integrated evaluation of drug substance properties and drug product processing technology and their interplay to enable rational design and development of crystalline drugs with poor aqueous solubility.
29.4 SOLID FORM CONTROL OF DASABUVIR
29.4.1 Introduction
Dasabuvir is a non‐nucleoside inhibitor of the hepatitis C virus (HCV) NS5B polymerase. The parent compound of dasabuvir, shown in Scheme 29.2, is lipophilic with the distribution coefficient, Log D, between octanol and a pH 7.4 buffer of 4.5 and exhibits low aqueous solubility (0.15 μg/ml at pH 7.4, 25 °C). Due to its low aqueous solubility, good permeability by Caco‐2 assay (~40 × 10−6 cm/s), and projected human dose (400 mg per day), the free form of dasabuvir is classified as a BCS class II compound. It is a highly crystalline solid exhibiting a high melting point (>200 °C), which limits the feasibility of the hot melt extrusion technology as an enabling formulation.

SCHEME 29.2 Preparation of dasabuvir monosodium monohydrate.
Based on in vitro tests, the dissolution of the parent form of dasabuvir in aqueous media is extremely slow. Correspondingly the compound exhibits low bioavailability (9.3%), as determined by in vivo dog PK studies. It was, therefore, desirable to conduct salt screening to identify a faster dissolving solid form that enables formulating the compound into a conventional oral dosage form. The free form of dasabuvir is a weak acid drug with pKa values of 8.2 and 9.2. The weak acidic functional groups (i.e. uracil and sulfonamide moieties) of the compound limit its salt formation to only a few pharmaceutically acceptable strong bases, including sodium hydroxide, potassium hydroxide, and choline hydroxide. As a result of the salt screening, a monohydrate form of the monosodium salt was discovered and selected as the lead solid form for development based on its favorable solid‐state properties, physicochemical properties, and manufacturability. The monosodium salt shows a higher apparent water solubility (0.47 mg/ml) and a faster dissolution rate compared with the parent compound. The dissolution rate enhancement was confirmed in a dog PK study, where the aqueous suspension of the salt achieved a sevenfold increase (72.6%) in bioavailability.
29.4.2 Solid Form Screening and Salt Formation Process
The main purpose of the solid form screening was to verify that the dasabuvir monosodium salt monohydrate selected for development was thermodynamically stable under process conditions. The polymorph screen consisted of thermodynamic and kinetic approaches. Emphasis was placed on identifying anhydrates and hydrates of the sodium salt and its free acid, as well as process‐relevant solvates.
In the thermodynamic solid form screening, the starting material dasabuvir monosodium salt monohydrate was suspended in various organic solvents at ambient temperature. Wet cakes were sampled at different time points up to three months. The design of the thermodynamic polymorph screening was based on solution‐mediated phase transformation to facilitate the conversion from metastable solids to stable phases (more stable solid species, polymorphs, hydrates, and solvates). The guiding principles for selecting non‐process‐relevant solvents for the thermodynamic polymorph screen included (i) the selection of a diverse range of solvent properties within a practical number of experiments, (ii) the search for potential anhydrate and/or hydrate forms, and (iii) the consideration of the water‐soluble formulation platform.
As the parent compound of dasabuvir is a weak acid, the manufacturing of its sodium salt hydrate poses a few challenges, such as the disproportionation and solid form control of the monosodium salt. In order to design the process and control salt formation to ensure consistent isolation of the desired monosodium salt monohydrate, a series of experiments were conducted to, first, select a solvent system for the conceptual design of the crystallization process and, second, identify process‐relevant solid forms. As a result of comprehensive solvent screening studies, a solvent system composed of dimethyl sulfoxide (DMSO), water (H2O), and isopropanol (IPA) was selected for the crystallization of the monosodium salt. DMSO provides most of the solubility for the API, while IPA and H2O act as antisolvents, with the latter providing the required water activity for yielding the monohydrate. The general scheme for manufacturing dasabuvir monosodium salt monohydrate from its free acid is represented in Scheme 29.2.
Detailed analysis of solvent compositions at various steps of the process – salt formation, nucleation and growth, and isolation of the monosodium salt monohydrate – was crucial to define the solvent compositions used in the thermodynamic screening. Various solvent combinations of DMSO, IPA, and H2O were used in the thermodynamic screen as process‐relevant solvents. In the kinetic solid form screening, crystallization experiments of dasabuvir monosodium salt were executed under stressed conditions, including (i) spiking with structurally similar impurities, (ii) low seed amounts and holding temperatures, (iii) fast and slow antisolvent addition rates, (iv) fast and slow cooling rates, and (v) a process‐relevant high‐shearing milling environment. The main purpose of performing crystallization experiments under these stressed conditions was to look for any potential metastable solid forms that could be present at the various stages of the manufacturing process (i.e. nucleation, crystal growth, and isolation of the monosodium salt monohydrate).
As a result of thermodynamic and kinetic screens, the following process‐relevant dasabuvir species and solid forms thereof were discovered: monosodium salt monohydrate, monosodium salt monoDMSO solvate, free acid triDMSO solvate, free acid anhydrate, and disodium salt DMSO/H2O mixed solvate. Specifically, during the polymorph screening and crystallization development, it was observed that the disodium salt DMSO/H2O mixed solvate could appear as a kinetic form during self‐nucleation and/or addition of a low seed amount of monosodium salt monohydrate. The disodium salt DMSO/H2O mixed solvate converted to the desired monosodium salt monohydrate during the holding period post‐seeding addition or IPA/H2O antisolvent addition. The monosodium salt monoDMSO could be also observed as a kinetic form, being promoted by high‐shear milling, and it converted to the monosodium salt monohydrate during IPA/H2O antisolvent addition. Finally, an uncontrolled crystallization of the monosodium salt could yield a hydrate crystalline form, solvates, or anhydrate of the free acid in organic/water solvent mixtures.
29.4.3 Solid Form Phase Diagram and Interconversion Pathways
The polymorph screening results indicate that multiple species and solid forms could be present in the crystallization process of dasabuvir. To assess the risk of these species and solid forms on dasabuvir drug substance manufacture, it is vital to (i) determine the phase diagram in DMSO/IPA/H2O of the relevant solid forms and relationships thereof; (ii) understand the basis for the formation of various species, including the free acid and disodium salt solid forms, in the monosodium salt reaction crystallization; and (iii) evaluate how well the crystallization process controls the desired monosodium salt monohydrate.
The dasabuvir solid form phase diagram in the DMSO/IPA/H2O ternary solvent system at ambient conditions was constructed based on the thermodynamic screening results, as shown in Figure 29.15. A schematic representation of the chemical species interconversion pathways in DMSO/IPA/H2O solvent system is depicted in Scheme 29.3. Given the complexity of the solid form and species landscape and their interconversions, a detailed assessment is provided.

FIGURE 29.15 Phase diagram of dasabuvir in the crystallization solvent system at room temperature.

SCHEME 29.3 Interconversion pathways of dasabuvir species and solid forms.
As shown in both Figure 29.15 and Scheme 29.3, dasabuvir monosodium salt undergoes disproportionation to yield the corresponding free acid and disodium salt in DMSO–H2O‐rich regions. The disproportionation of monosodium salt in a DMSO–H2O‐rich region is likely due to two factors. First, the monosodium salt and its disproportionation products (i.e. free acid and disodium salt) differ in solubility; second, water (pKa 14) acts as proton donor and acceptor, thus facilitating the disproportionation process. When the DMSO/H2O volume ratio was high enough, the free acid is converted to triDMSO solvate. Similar behavior occurred in DMSO/IPA/H2O systems in which DMSO was the dominant component. When the DMSO content was at its extreme (i.e. 100% DMSO), the free acid triDMSO solvate became the least soluble solid form due to very high DMSO activity, and it precipitated out with or without trace amounts of water. In addition, the disodium salt, the other portion of the disproportionation product, could exist as a DMSO/H2O mixed solvate or remained in solution in the aforementioned DMSO–H2O‐rich solvent regions. When the monosodium salt monohydrate was reslurried in DMSO–IPA‐rich regions, the monosodium salt was stable as a monoDMSO solvate and did not undergo disproportionation. Besides solubility reasons, the lack of disproportionation may be due to the fact that IPA (pKa 16.5) is a less efficient protic solvent than water and thus not able to facilitate the process.
When the monosodium salt monohydrate was reslurried in IPA–H2O‐rich regions, the monosodium salt was stable and remained as the monohydrate crystalline form. This is most likely due to the very limited solubility of the compound in such solvent compositions and to the water activity being not high enough to effectively promote disproportionation of the monosodium salt. However, when the water content was significantly high (e.g. 30% or above), the disproportionation of the monosodium salt to its free form occurred, and thus the free acid anhydrate became more stable. It is noteworthy that in neat water the monosodium salt monohydrate was found to be stable over four weeks. This further confirms that both a good solubility differential between the monosodium salt and free acid/disodium salt and an effective protic solvent are necessary to effectively promote the disproportionation of the monosodium salt. This also holds true in neat IPA where the monosodium salt monohydrate was found to be stable over 12 weeks. Yet, from a theoretical point of view, the monosodium salt should eventually undergo disproportionation in both H2O and IPA to yield the corresponding hydrate or anhydrate/solvate of the free acid and disodium salt. Practically though, the time frame for this to happen is much longer relative to the process time needed for manufacturing and, therefore, is not a risk to the process.
The detailed analysis of the phase diagram built at ambient temperature helps understand the complexity of the solid form landscape associated with the crystallization process. It also helps visualize the conceptual design of the process. The commercial crystallization process consisted of slurrying dasabuvir free acid in DMSO and charging an aqueous solution of sodium hydroxide to form monosodium salt. This salt formation step corresponds to the path from star point o to a on the phase diagram in Figure 29.15. The IPA/H2O solvent mixture was added to the salt solution to adjust DMSO/IPA/H2O volume ratio and create supersaturation for seeding/self‐nucleation of the monosodium salt monohydrate. This is represented by the path from star point a to b on the phase diagram. The addition of the bulk antisolvent IPA/H2O (represented as star point d on the phase diagram) drives the crystallization from star point b to c before isolation. The crystallization slurry was then filtered, and its wet cake was washed with IPA/H2O solvent mixture (star point e). Thus, dasabuvir monosodium salt monoDMSO solvate, free acid anhydrate, free acid triDMSO solvate, and disodium salt DMSO/H2O mixed solvate were observed in DMSO/IPA/H2O mixtures outside of the operational range, suggesting a minimal impact of these forms on the crystallization of the desired monosodium salt monohydrate. This further ensures that the desired monosodium salt monohydrate can be consistently isolated from the process and provides assurance that no form conversion would occur.
It is important to emphasize that the phase diagram was constructed for process‐relevant solid forms at ambient temperature. This phase diagram may not necessarily provide an accurate view of the solid phase stability at elevated temperatures, particularly during the initial crystallization steps. Constructing a complete and accurate phase diagram at high temperatures is experimentally challenging given the potential solvent evaporation and the compound stability over a prolonged period of time. Nevertheless, the ambient temperature phase diagram provides highly valuable information regarding potential solid forms present at the beginning of the crystallization step, as well as guidance on how to conduct process control justification work with appropriate solid form focus.
29.4.4 Summary
The salt formation strategy was utilized to improve the bioavailability of the poorly soluble HCV polymerase inhibitor dasabuvir. In order to ensure a robust solid form control in the manufacturing process of dasabuvir monosodium salt monohydrate, comprehensive thermodynamic and kinetic solid form screens were carefully designed and executed. The results from the solid form screening led to the construction of the phase diagram and to the fundamental understanding of interconversion pathways between different chemical species and solid forms of dasabuvir. The solid form development undertaken to ensure that every stage of the manufacturing process (i.e. seeding, crystal growth, and isolation) consistently produced the desired monosodium monohydrate allowed a robust commercial manufacturing process and a sound control strategy to be developed.
ACKNOWLEDGMENTS
The authors wish to thank Ann Czyzewski, James Marek, and John Gaertner of Process Engineering; Michael Rozema, Travis Dunn, Lawrence Kolaczkowski, and David Barnes of Process Chemistry; and Weifeng Wang, and Lewis Meads of Process Analytical. The authors would also like to thank Rodger Henry of Structural Chemistry, Geoff G.Z. Zhang and Xiaochun Lou of Drug Product Development, and Gao Yi of Science and Technology.
All authors are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
REFERENCES
- 1. Loftsson, T. and Brewster, M.E. (2010). Pharmaceutical applications of cyclodextrins: basic science and product development. Journal of Pharmacy and Pharmacology 62 (11): 1607–1621.
- 2. Lipp, R. (2013). The innovator pipeline: bioavailability challenges and advanced oral drug delivery opportunities. American Pharmaceutical Review 16 (3): 10–16.
- 3. Bernstein, J. (2002). Polymorphism in Molecular Crystals. New York: Oxford University Press.
- 4. Chemburkar, S.R., Bauer, J., Deming, K. et al. (2000). Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Organic Process Research & Development 4 (5): 413–417.
- 5. Bauer, J., Spanton, S., Henry, R. et al. (2001). Ritonavir: an extraordinary example of conformational polymorphism. Pharmaceutical Research 18 (6): 859–866.
- 6. Serajuddin, A.T.M. (2007). Salt formation to improve drug solubility. Advanced Drug Delivery Reviews 59 (7): 603–616.
- 7. Kuminek, G., Cao, F., Bahia de Oliveira da Rocha, A. et al. (2016). Cocrystals to facilitate delivery of poorly soluble compounds beyond‐rule‐of‐5. Advanced Drug Delivery Reviews 101: 143–166.
- 8. Good, D.J. and Rodríguez‐Hornedo, N. (2009). Solubility advantage of pharmaceutical cocrystals. Crystal Growth & Design 9 (5): 2252–2264.
- 9. Blagden, N., Coles, S.J., and Berry, D.J. (2014). Pharmaceutical co‐crystals – are we there yet? CrystEngComm 16 (26): 5753–5761.
- 10. Kawabata, Y., Wada, K., Nakatani, M. et al. (2011). Formulation design for poorly water‐soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. International Journal of Pharmaceutics 420 (1): 1–10.
- 11. Merisko‐Liversidge, E. and Liversidge, G.G. (2011). Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly‐water soluble compounds using wet media milling technology. Advanced Drug Delivery Reviews 63 (6): 427–440.
- 12. Newman, A. (2015). Pharmaceutical Amorphous Solid Dispersions. Hoboken, NJ: Wiley.
- 13. Baghel, S., Cathcart, H., and O’Reilly, N.J. (2016). Polymeric amorphous solid dispersion: a review of amorphization, crystallization, stabilization, solid‐state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. Journal of Pharmaceutical Sciences 105 (9): 2527–2544.
- 14. DeBoyace, K. and Wildfong, P.L.D. (2018). The application of modeling and prediction to the formation and stability of amorphous solid dispersions. Journal of Pharmaceutical Sciences 107 (1): 57–74.
- 15. Law, D., Krill, S.L., Schmitt, E.A. et al. (2001). Physicochemical considerations in the preparation of amorphous ritonavir–poly(ethylene glycol) 8000 solid dispersions. Journal of Pharmaceutical Sciences 90 (8): 1015–1025.
- 16. Miller, J.M., Beig, A., Carr, R.A. et al. (2012). A win‐win solution in oral delivery of lipophilic drugs: supersaturation via amorphous solid dispersions increases apparent solubility without sacrifice of intestinal membrane permeability. Molecular Pharmaceutics 9 (7): 2009–2016.
- 17. Tao, J., Sun, Y., Zhang, G.G.Z., and Yu, L. (2009). Solubility of small‐molecule crystals in polymers: D‐mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharmaceutical Research 26 (4): 855–864.
- 18. Sun, Y., Tao, J., Zhang, G.G.Z., and Yu, L. (2010). Solubilities of crystalline drugs in polymers: an improved analytical method and comparison of solubilities of indomethacin and nifedipine in PVP, PVP/VA, and PVAc. Journal of Pharmaceutical Sciences 99 (9): 4023–4031.
- 19. Kyeremateng, S.O., Pudlas, M., and Woehrle, G.H. (2014). A fast and reliable empirical approach for estimating solubility of crystalline drugs in polymers for hot melt extrusion formulations. Journal of Pharmaceutical Sciences 103 (9): 2847–2858.
- 20. Lehmkemper, K., Kyeremateng, S.O., Heinzerling, O. et al. (2017). Long‐term physical stability of PVP‐ and PVPVA‐amorphous solid dispersions. Molecular Pharmaceutics 14 (1): 157–171.
- 21. Lehmkemper, K., Kyeremateng, S.O., Bartels, M. et al. (2018). Physical stability of API/polymer‐blend amorphous solid dispersions. European Journal of Pharmaceutics and Biopharmaceutics 124: 147–157.
- 22. Raina, S.A., Zhang, G.G.Z., Alonzo, D.E. et al. (2014). Enhancements and limits in drug membrane transport using supersaturated solutions of poorly water soluble drugs. Journal of Pharmaceutical Sciences 103 (9): 2736–2748.
- 23. Taylor, L.S. and Zhang, G.G.Z. (2016). Physical chemistry of supersaturated solutions and implications for oral absorption. Advanced Drug Delivery Reviews 101: 122–142.
- 24. Ilevbare, G.A. and Taylor, L.S. (2013). Liquid‐liquid phase separation in highly supersaturated aqueous solutions of poorly water‐soluble drugs: implications for solubility enhancing formulations. Crystal Growth & Design 13 (4): 1497–1509.
- 25. Jackson, M.J., Kestur, U.S., Hussain, M.A., and Taylor, L.S. (2016). Dissolution of danazol amorphous solid dispersions: supersaturation and phase behavior as a function of drug loading and polymer type. Molecular Pharmaceutics 13 (1): 223–231.
- 26. Abramov, Y.A., Loschen, C., and Klamt, A. (2012). Rational coformer or solvent selection for pharmaceutical cocrystallization or desolvation. Journal of Pharmaceutical Sciences 101 (10): 3687–3697.
- 27. Cruz‐Cabeza, A.J., Reutzel‐Edens, S.M., and Bernstein, J. (2015). Facts and fictions about polymorphism. Chemical Society Reviews 44 (23): 8619–8635.
- 28. Caira, M.R. (1998). Crystalline polymorphism of organic compounds. In: Design of Organic Solids (ed. E. Weber), 163–208. Berlin: Springer.
- 29. Liu, J. and Rasmuson, A.C. (2013). Influence of agitation and fluid shear on primary nucleation in solution. Crystal Growth & Design 13 (10): 4385–4394.
- 30. Abramov, Y.A. (2016). Computational Pharmaceutical Solid State Chemistry. Hoboken, NJ: Wiley.
- 31. Etter, M.C. (1991). Hydrogen bonds as design elements in organic chemistry. The Journal of Physical Chemistry 95 (12): 4601–4610.
- 32. Etter, M.C. and Reutzel‐Edens, S.M. (1991). Hydrogen bond directed cocrystallization and molecular recognition properties of acyclic imides. Journal of the American Chemical Society 113 (7): 2586–2598.
- 33. Reutzel‐Edens, S.M. and Etter, M.C. (1992). Evaluation of the conformational, hydrogen bonding and crystal packing preferences of acyclic imides. Journal of Physical Organic Chemistry 5 (1): 44–54.
- 34. Desiraju, G.R. (1991). The C–H···O hydrogen bond in crystals. What is it? Accounts of Chemical Research 24 (10): 290–296.
- 35. Aakeröy, C.B. and Seddon, K.R. (1993). The hydrogen bond and crystal engineering. Chemical Society Reviews 22 (6): 397–407.
- 36. Dunitz, J.D. and Gavezzotti, A. (2005). Molecular recognition in organic crystals: directed intermolecular bonds or nonlocalized bonding? Angewandte Chemie International Edition 44 (12): 1766–1787.
- 37. Desiraju, G.R., Vittal, J.J., and Ramanan, A. (2011). Crystal Engineering: A Textbook. Singapore: World Scientific Publishing Co. Pte. Ltd.
- 38. Allen, F.H. (2002). The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallographica Section B 58 (3): 380–388.
- 39. Groom, C.R. and Allen, F.H. (2014). The Cambridge Structural Database in retrospect and prospect. Angewandte Chemie International Edition 53 (3): 662–671.
- 40. Galek, P.T.A., Fábián, L., Motherwell, W.D.S. et al. (2007). Knowledge‐based model hydrogen‐bonding propensity in organic crystals. Acta Crystallographica Section B 63 (5): 768–782.
- 41. Galek, P.T.A., Fábián, L., and Allen, F.H. (2010). Truly prospective prediction: inter‐ and intramolecular hydrogen bonding. CrystEngComm 12 (7): 2091–2099.
- 42. Galek, P.T.A., Allen, F.H., Fábián, L., and Feeder, N. (2009). Knowledge‐based H‐bond prediction to aid experimental polymorph screening. CrystEngComm 11 (12): 2634–2639.
- 43. Abramov, Y.A. (2013). Current computational approaches to support pharmaceutical solid form selection. Organic Process Research & Development 17 (3): 472–485.
- 44. Wood, P.A., Olsson, T.S.G., Cole, J.C. et al. (2013). Evaluation of molecular crystal structures using Full Interaction Maps. CrystEngComm 15 (1): 65–72.
- 45. Bruno, I.J., Cole, J.C., Rowland, R.S. et al. (1997). Isostar: a library of information about nonbonded interactions. Journal of Computer‐Aided Molecular Design 11 (6): 525–537.
- 46. McKinnon, J.J., Spackman, M.A., and Mitchell, A.S. (2004). Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallographica Section B 60 (6): 627–668.
- 47. Spackman, M.A. and Jayatilaka, D. (2009). Hirshfeld surface analysis. CrystEngComm 11 (1): 19–32.
- 48. Dunitz, J.D. (2003). Are crystal structures predictable? Chemical Communications 0 (5): 545–548.
- 49. Day, G.M. (2011). Current approaches to predicting molecular organic crystal structures. Crystallography Reviews 17 (1): 3–52.
- 50. Lommerse, J.P.M., Motherwell, W.D.S., Ammon, H.L. et al. (2000). A test of crystal structure prediction of small organic molecules. Acta Crystallographica Section B 56 (4): 697–714.
- 51. Gavezzotti, A. and Filippini, G. (1995). Polymorphic forms of organic crystals at room conditions: thermodynamic and structural implications. Journal of the American Chemical Society 117 (49): 12299–12305.
- 52. Motherwell, W.D.S., Ammon, H.L., Dunitz, J.D. et al. (2002). Crystal structure prediction of small organic molecules: a second blind test. Acta Crystallographica Section B 58 (4): 647–661.
- 53. Day, G.M., Motherwell, W.D.S., Ammon, H.L. et al. (2005). A third blind test of crystal structure prediction. Acta Crystallographica Section B 61 (5): 511–527.
- 54. Day, G.M., Cooper, T.G., Cruz‐Cabeza, A.J. et al. (2009). Significant progress in predicting the crystal structures of small organic molecules – a report on the fourth blind test. Acta Crystallographica Section B 65 (2): 107–125.
- 55. Bardwell, D.A., Adjiman, C.S., Arnautova, Y.A. et al. (2011). Towards crystal structure prediction of complex organic compounds –– a report on the fifth blind test. Acta Crystallographica Section B 67 (6): 535–551.
- 56. Reilly, A.M., Cooper, R.I., Adjiman, C.S. et al. (2016). Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallographica Section B 72 (4): 439–459.
- 57. Price, S.L. (2009). Computed crystal energy landscapes for understanding and predicting organic crystal structures and polymorphism. Accounts of Chemical Research 42 (1): 117–126.
- 58. Neumann, M.A. (2008). Tailor‐made force fields for crystal‐structure prediction. The Journal of Physical Chemistry B 112 (32): 9810–9829.
- 59. Neumann, M.A., Leusen, F.J.J., and Kendrick, J. (2008). A major advance in crystal structure prediction. Angewandte Chemie International Edition 47 (13): 2427–2430.
- 60. Rosso, L., Mináry, P., Zhu, Z., and Tuckerman, M.E. (2002). On the use of the adiabatic molecular dynamics technique in the calculation of free energy profiles. The Journal of Chemical Physics 116 (11): 4389–4402.
- 61. Chen, M., Yu, T.Q., and Tuckerman, M.E. (2015). Locating landmarks on high‐dimensional free energy surfaces. PNAS 112 (11): 3235–3240.
- 62. Yu, T.Q. and Tuckerman, M.E. (2011). Temperature‐accelerated method for exploring polymorphism in molecular crystals based on free energy. Physical Review Letters 107 (1): 015701.
- 63. van Eijck, B.P. and Kroon, J. (2000). Structure predictions allowing more than one molecule in the asymmetric unit. Acta Crystallographica Section B 56 (3): 535–542.
- 64. Linifanib (2010). Drugs R&D 10 (2): 111–122.
- 65. Desiraju, G.R. (1995). Supramolecular synthons in crystal engineering – a new organic synthesis. Angewandte Chemie International Edition 34 (21): 2311–2327.
- 66. Reddy, L.S., Chandran, S.K., George, S. et al. (2007). Crystal structures of N‐aryl‐N′‐4‐nitrophenyl ureas: molecular conformation and weak interactions direct the strong hydrogen bond synthon. Crystal Growth & Design 7 (12): 2675–2690.
- 67. Bruno, I.J., Cole, J.C., Edgington, P.R. et al. (2002). New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallographica Section B 58 (3): 389–397.
- 68. Hulme, A.T. and Price, L.S. (2007). Toward the prediction of organic hydrate crystal structures. Journal of Chemical Theory and Computation 3 (4): 1597–1608.
- 69. Takieddin, K., Khimyak, Y.Z., and Fábián, L. (2016). Prediction of hydrate and solvate formation using statistical models. Crystal Growth & Design 16 (1): 70–81.
- 70. Bajpai, A., Scott, H.S., Pham, T. et al. (2016). Towards an understanding of the propensity for crystalline hydrate formation by molecular compounds. International Union of Crystallography 3 (6): 430–439.
- 71. Abramov, Y.A. (2015). Virtual hydrate screening and coformer selection for improved relative humidity stability. CrystEngComm 17 (28): 5216–5224.
- 72. Tilbury, C.J., Chen, J., Mattei, A. et al. (2018). Combining theoretical and data‐driven approaches to predict drug substance hydrate formation. Crystal Growth & Design 18 (1): 57–67.
- 73. Klamt, A. (2011). The COSMO and COSMO‐RS solvation models. WIREs: Computational Molecular Science 1 (5): 699–709.
- 74. Klamt, A. (1995). Conductor‐like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. The Journal of Physical Chemistry 99 (7): 2224–2235.
- 75. Pearson, R.G. (2005). Chemical hardness and density functional theory. Journal of Chemical Sciences 117 (5): 369–377.
- 76. Nangia, A. (2010). Hydrogen bonding and molecular packing in multi‐functional crystal structures. In: Organic Crystal Engineering Frontiers in Crystal Engineering (ed. E.R.T. Tiekink, J. Vittal and M. Zaworotko), 151–189. Chichester, UK: Wiley.
- 77. Noyes, A. and Whitney, W. (1897). The rate of solution of solid substances in their own solutions. Journal of the American Chemical Society 19 (12): 930–934.