
7
QUANTITATIVE APPLICATIONS OF NMR SPECTROSCOPY
Brian L. Marquez*
Pfizer, Inc., Groton, CT, USA
R. Thomas Williamson**
Roche Carolina, Inc., Florence, SC, USA
7.1 INTRODUCTION
7.1.1 General Principles of NMR
Nuclear magnetic resonance (NMR) is an analytical method that takes advantage of the magnetic properties of certain atomic nuclei. This approach is similar to other types of spectroscopy in that the absorption or emission of electromagnetic energy at characteristic frequencies provides analytical information. However, NMR differs from other types of spectroscopy in that the discrete energy levels and the transitions between them are created by placing the samples in a strong magnetic field (B0).
When an atom is placed in a magnetic field, its electrons circulate about the direction of the applied magnetic field. The circulation of these nuclei generates a very small magnetic field, which is generally on the order of 1–20 ppm of the total applied magnetic field for 1H and 1–200 ppm for 13C. This field opposes the applied magnetic field and can be detected through the same Rf coil that is used to excite the nuclei of interest. When nuclei spin about the axis of this externally applied magnetic field, they possess an angular momentum. This angular momentum can be expressed as a function of a proportionality constant, I, which can be either an integer or half‐integer. I is referred to as the spin quantum number or more simply as the nuclear spin. It is possible for some isotopes to have a spin quantum number I = 0. These nuclei are not considered magnetic and cannot be detected by NMR. In order for a nuclei to have a spin quantum I = 0, it must have an even atomic number and even mass. Commonly occurring non‐NMR active nuclei include 12C, 16O, and 32S. The group of nuclei most commonly observed by NMR methods is nuclei with a spin of ½. These include 1H, 13C, 19F, 31P, and 15N. Other spins with I = 1 or I > 1 nuclei can be observed with slightly more difficulty. These nuclei include 2H and 14N (I = 1) and 10B, 11B, 17O, and 23Na (I > 1) (Figure 7.1).

FIGURE 7.1 A spinning magnetic nuclei in an externally applied magnetic field with its axis precessing around the direction of the applied field (B0) analogous to a gyroscope.
The rate at which a particular nuclei spins in a particular magnetic field is known as its precession frequency. This frequency is both a function of the externally applied field, the nucleus of interest, and the environment in which it resides. For a proton (1H), in an applied 2.35 T magnetic field, the reference precession frequency is approximately 100 MHz. In the same externally applied field, other nuclei have different gyromagnetic ratios. For example, 13C with a gyromagnetic ratio of 4 will precess at 25 MHz in the same magnetic field. This characteristic precession frequency is known as the Larmor frequency of the nucleus. An NMR sample may contain many different magnetization components, each with its own Larmor frequency. Therefore, an NMR spectrum may be made up of many different frequency lines.
Nuclei aligned with the axis of the externally applied magnetic field will be in the lowest possible energy state. Thermal processes oppose this tendency, such that there are two populations of nuclei in an externally applied magnetic field. One is aligned with the axis of the field, and another, which is only slightly smaller, is aligned opposite to the direction of the applied field. The distribution of spins between these two energy levels is referred to as the Boltzmann distribution, and it is this population difference between the two levels that provides the observable collection of spins in an NMR experiment. This difference is very small compared to the total number of spins present, which leads to the comparative inherent insensitivity of NMR.
In the modern NMR experiment, pulsed radio frequency (Rf) energy is used to excite all frequencies at once. In a simplistic sense, a certain amount of energy from an Rf pulse is absorbed by each nuclei. As these nuclei relax back from the excited state to the ground state, a corresponding amount of Rf energy is emitted. The frequency and the amplitude of this emitted energy contain important information about the nucleus from where it originated. In other words, the application of a radio frequency (Rf) pulse of energy orthogonal to the axis of the applied magnetic field perturbs the Boltzmann distribution, thereby producing an observable event that is governed by the Bloch equations. When the application of that pulse is completed, the vector that has been rotated into the x,y plane will continue to precess about the axis of the externally applied field (z‐axis), generating an oscillating signal in a receiver coil as the vector rotates about the z‐axis at its characteristic Larmor frequency. The signal from the magnetization vector will decay back to an equilibrium condition along the B0 axis as a function of two time constants, the spin–spin or transverse relaxation time, T2, and the spin–lattice relaxation time, T1. Once equilibrium is reestablished after a short delay, the process can be repeated, and multiple acquisitions can be added to increase S/N. Immediately after the original Rf pulse, a receiver is turned on, and a signal known as a time‐domain interferogram is acquired via an Rf receiver. With the help of an analogue to digital convertor, this data is saved to a computer. This so‐called interferogram contains information on all signals emitted by the sample at various NMR frequencies. This information is present as a sum of all damped sinusoid signals emitted by the sample at various nuclear resonance frequencies. The specific resonance frequencies of these signals vary by the strength of the corresponding magnetic field. For instance, at 11.7 Tesla (T) the 1H resonance frequency is approximately 500 MHz, and at 18.8 T it is 800 MHz. The collected interferogram is called a free induction decay (FID). An example of FID can be found in Figure 7.2.
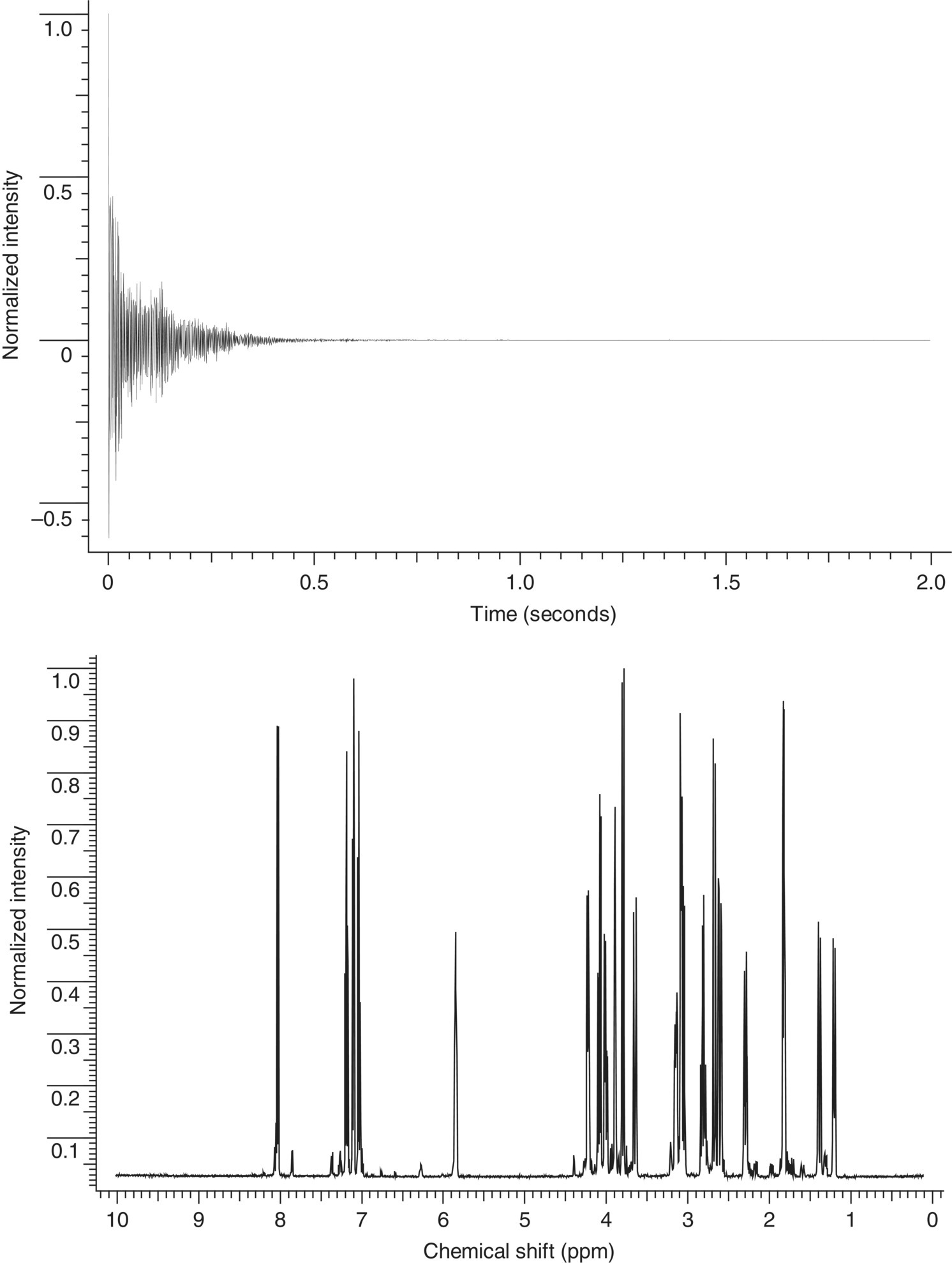
FIGURE 7.2 The top panel shows the free induction decay (FID) acquired for a sample of strychnine [1] at an observation frequency of 500 MHz. The spectrum was digitized with 16 K points and an acquisition time of approximately 2 seconds. Fourier transforming the data from the time domain to the frequency domain yields the spectrum of strychnine presented as intensity vs. frequency shown in the bottom panel.
Once acquired and saved, these data are converted from the time domain to frequency data to make them more meaningful and easier to interpret for the end user. This conversion is typically done through a mathematical manipulation known as the Fourier transform named for the mathematician Jean‐Baptiste Joseph Fourier. This mathematical transformation also provides signal intensity information that is a key to the usefulness of NMR data. Typically, in common practice, a derivation of the Fourier transform is done by computer using the Cooley–Tukey fast Fourier algorithm otherwise known as the fast Fourier transform (FFT).
The frequency of the Rf energy absorbed by a particular nucleus is strongly affected by its chemical environment. These variables in the chemical environment can lead to changes in its so‐called chemical shift. Three basic components of an NMR spectrum reveal its extreme usefulness. These include (i) the chemical shift, (ii) the amplitude of the signal at that chemical shift, and (iii) the splitting of the signal in response to its interaction with neighboring nuclei.
The location of an NMR signal in a spectrum is known as the signal's chemical shift. The location of this chemical shift is a function of the chemical environment of the sampled nuclei and is designated with a scale value referenced to an internal standard. For solution‐state 1H and 13C NMR experiments, the reference standard is tetramethylsilane, or TMS, which has an accepted chemical shift of 0.00 ppm Most proton signals appear to the left or “downfield” of TMS. Aliphatic hydrocarbon signals will generally be grouped nearer the position of TMS and are said to be “shielded” relative to vinyl or aromatic signals that are as a group referred to as “deshielded” (Figure 7.3). Chemical moieties involving heteroatoms, e.g. –OCH3, –NCH2–, typically will be located in a region of the NMR spectrum between the aliphatic and vinyl/aromatic signals.

FIGURE 7.3 Image showing the proton chemical shift range depending on chemical environment.
In addition to chemical shift information, an NMR spectrum may also contain scalar coupling information. For NMR of liquids, scalar (J) couplings in proton spectra provide information about the local chemical environment of a given proton resonance. Proton resonances are split into multiplets related to the number of neighboring protons. For example, an ethyl fragment will be represented by a triplet with relative peak intensities of 1 : 2 : 1 for the methyl group, the splitting due to the two neighboring methylene protons, and a 1 : 3 : 3 : 1 quartet for the methylene group, with the splitting due to the 3 equiv. methyl protons. This concept is easily illustrated with the geometrical arrangement of binomial coefficients known as Pascal's triangle shown in Figure 7.4. More complex molecules, of course, lead to considerably more complicated spin‐coupling patterns as shown in Figure 7.5.
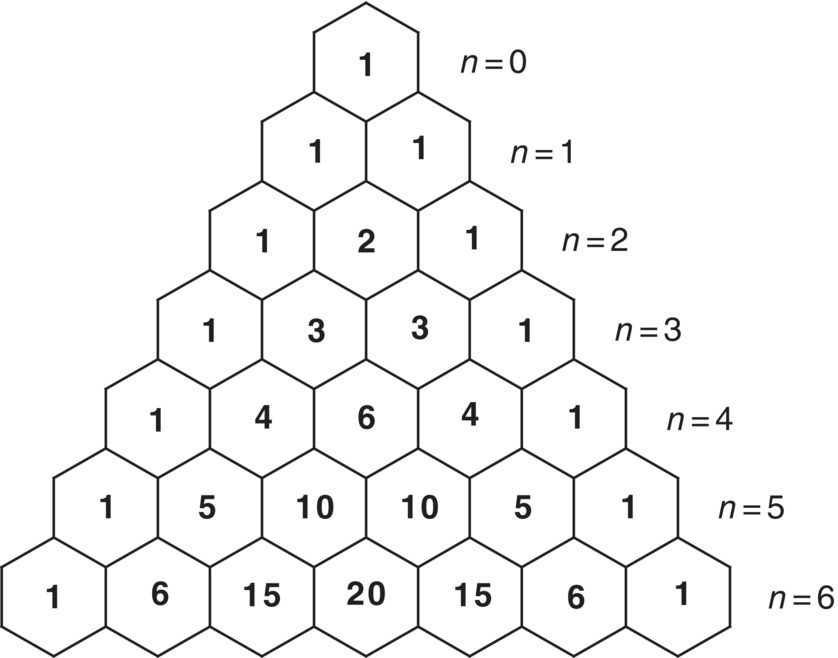
FIGURE 7.4 Illustration of Pascal's triangle only showing ratios to n = 6 according to M = (n + 1) where M is the multiplicity and n is the number of scalar coupled nuclei. For example, a proton adjacent to three protons (n = 3) would appear as a quartet (M = 4) with relative peak intensities of 1 : 3 : 3 : 1.
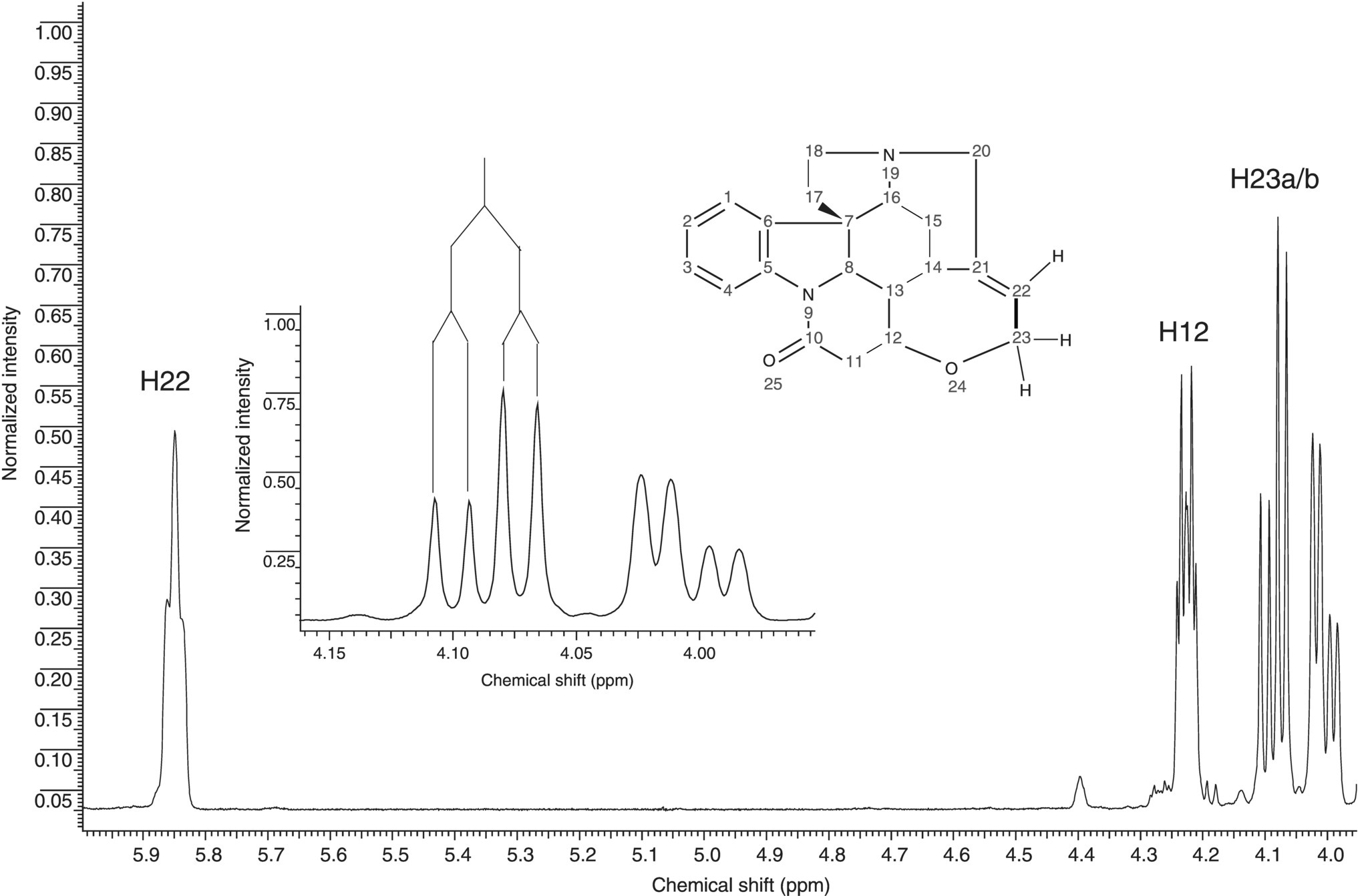
FIGURE 7.5 Expansion of a portion of the proton NMR spectrum of strychnine (inset structure). The full proton spectrum is shown in Figure 7.1. The resonances for the H22 vinyl proton and the H12 and H23 oxygen‐bearing methine and methylene resonances, respectively, are shown. The inset expansion of the H23 methylene protons shows a splitting diagram for this resonance. The larger of the two couplings is the geminal coupling to the other H23 resonance and the smaller coupling is the vicinal coupling to the H22 vinyl proton.
Prior to the advent of homonuclear two‐dimensional (2D) NMR experiments, it was necessary to rigorously interpret a proton NMR experiment and identify all of the homonuclear couplings to assemble the structure. Alternatively, there are multidimensional NMR experiments that provide similar information in a more readily interpretable way. These techniques will be discussed in more detail later.
Beyond the qualitative molecular information afforded by NMR, one can also obtain quantitative information. Depending on the sample, NMR can measure relative quantities of components in a mixture as low as 0.1–1% in the solid state. NMR limits of detection are much lower in the liquid state, often as low as 1 000 : 1 down to 10 000 : 1. Internal standards can be used to translate these values into absolute quantities. Of course, the limit of quantization is not only dependent on the type of sample but also on the amount of sample. While not as mass sensitive as other analytical techniques, NMR has dramatically improved in sensitivity in recent years.
Although sample volumes and tube sizes can vary greatly for specialized applications, the most often analyzed NMR sample format contains approximately 500–1000 μl of solvent in a 5 mm diameter glass tube. Typical amounts of sample for this configuration range from 1 to 20 mg depending on the amount and solubility of the sample available. NMR hardware accommodating smaller diameter sample tubes enable the detection of much smaller samples. Common commercially available liquid NMR tubes range from one to 11 mm in diameter. Amounts of detectable sample for these configurations can be as low as hundreds of nanograms for 1H NMR detection. Carbon sensitivity is on the order of 100 times worse, so detection limits are usually limited to tens of micrograms.
Essentially all magnetic nuclei can be observed by NMR, but the sensitivity of each is determined by the relative sensitivity and the natural abundance of the nuclei. For example, not only is 1H (with a spin of ½) one of the most sensitive nuclei, but it also has a natural abundance of 99.985%. Alternatively, 13C is fairly sensitive but only has a natural abundance of 1.108%. Other nuclei like 15N (spin I = −1/2) have both low natural abundance (0.37%) and low sensitivity, making them doubly difficult to detect. These types of nuclei are very difficult to observe directly and are most commonly detected using what is known as “inverse detection” through a more sensitive nucleus. This technique can dramatically increase sensitivity for insensitive nuclei and can make available chemical shift and coupling information available that otherwise would have never been possible with existing technologies.
For the NMR analysis of solid samples, the amount of sample required is much greater in part because the apparent signal‐to‐noise ratio is significantly reduced. This is due to much broader line shapes, easily an order of magnitude wider than those observed in equivalent spectra of liquids. A standard solid NMR sample is a powder packed tightly into a small zirconia rotor and sealed with end caps. As for liquids, solid sample configurations are described in terms of their diameter, in this case the diameter of the sample rotor. Common commercially available rotors range from 2.5 to 11 mm in diameter. Amounts of sample for these configurations depend on the sample and its density and typically range from 30 to 500 mg.
In common practice, most 1H NMR spectra are recorded in solutions prepared with deuterated solvents. The advantages of this customary practice of sample preparation are twofold. Firstly, the lack of protons in the solvent enables observation of the protons of interest without interference from solute protons. Secondly, the presence of deuterium in the sample allows the NMR instrument to “lock” onto a reference frequency outside of those frequencies being observed. The ability to lock on a signal as the magnetic field drifts slightly over time can substantially improve the quality of spectra in experiments with long acquisitions. This can be very important when observing small amounts of sample or when the highest quality spectra with narrow lines are required. References [1–9] are listed as suggested reading for a deeper understanding of the theory, application, and experimental setup of many commonplace experiments [1–9].
7.2 ONE‐DIMENSIONAL NMR METHODS
7.2.1 1D Proton NMR Methods
7.2.1.1 Magnetically Equivalent Nuclei
A critical element to one of the fundamental phenomenon that is used for structure characterization is that of J‐coupling (spin–spin coupling, scalar coupling, or often referred to as simply “coupling”). Of particular importance is the understanding of magnetic equivalency and this coupling interaction. Magnetically equivalent nuclei are defined as nuclei having both the same resonance frequency and spin–spin interaction with neighboring atoms. The spin–spin interaction does not appear in the resonance signal observed in the spectrum. It should be noted that magnetically equivalent nuclei are inherently chemically equivalent, but the reverse is not necessarily true. An example of magnetic equivalence is the set of three protons within a methyl group. All three protons attached to the carbon of a methyl group have the same resonance frequency and encounter the same spin–spin interaction with its vicinal neighbors. As a result, the resonance signal will integrate for three protons split into the appropriate coupling pattern (see next section) to adjacent nuclei.
7.2.1.2 NOE Experiments
The acronym NOE is derived from the nuclear Overhauser effect. This phenomenon was first predicted by Overhauser in 1953 [10]. It was later experimentally observed by Solomon in 1955 [11]. The importance of the NOE in the world of small molecule structure elucidation cannot be overstated. As opposed to scalar coupling described above, NOE allows the analysis of dipolar coupling. Dipolar coupling is often referred to as through‐space coupling and is most often used to explore the spatial relationship between two atoms experiencing zero scalar coupling. The spatial relationship of atoms within a molecule can provide an immense amount of information about a molecule, ranging from the regiochemistry of an olefin to the three‐dimensional solution structure.
A simplified explanation of the NOE is the magnetic perturbations induced on a neighboring atom, resulting in a change in intensity. This is usually an increase in intensity but may alternatively be zero or even negative. One can envision saturating a particular resonance of interest for a time t. During the saturation process a population transfer occurs to all spins that feel an induced magnetization.
There are two types of NOE experiments that can be performed. These are referred to as the steady‐state NOE and the transient NOE. The steady‐state NOE experiment is exemplified by the classic NOE difference experiment [12]. Steady‐state NOE experiments allow one to quantitate relative atomic distances. However, there are many issues that can complicate their measurement, and a qualitative interpretation is more reliable [13]. Spectral artifacts can be observed from imperfect subtraction of spectra. In addition, this experiment is extremely susceptible to inhomogeneity issues and temperature fluctuations.
One‐dimensional (1D) transient NOE experiments employing gradient selection are more robust and therefore are more reliable for measuring dipolar coupling interactions [14]. Shaka et al. published one such 1D transient NOE experiment that has, in most cases, replaced the traditional NOE difference experiment [15]. The sequence dubbed the double pulsed field gradient spin echo (DPFGSE) NOE employs selective excitation through the DPFGSE portion of the sequence [16]. Magnetization is initially created with a 90° 1H pulse. Following this pulse are two gradient echoes employing selective 180° pulses. The flanking gradient pulses are used to dephase and recover the desired magnetization as described above. This selection mechanism provides very efficient selection of the resonance of interest prior to the mixing time where dipolar coupling is allowed to build up.
7.2.1.3 Relaxation Measurements
Relaxation is an inherent property of all nuclear spins. There are two predominant types of relaxation processes in NMR of liquids. These relaxation processes are denoted by the longitudinal (T1) and transverse (T2) relaxation time constants. When a sample is excited from its thermal equilibrium with an RF pulse, its tendency is to relax back to its Boltzmann distribution. The amount of time to re‐equilibrate is typically on the order of seconds to minutes. T1 and T2 relaxation processes operate simultaneously. The recovery of magnetization to the equilibrium state along the z‐axis is longitudinal or the T1 relaxation time. The loss of coherence of the ensemble of excited spins (uniform distribution) in the x,y plane following the completion of a pulse is transverse or T2 relaxation. The duration of the T1 relaxation time is a very important feature as it allows us to manipulate spins through a series of RF pulses and delays. Transverse relaxation is governed by the loss of phase coherence of the precessing spins when removed from thermal equilibrium (e.g. an RF pulse). The transverse or T2 relaxation time is visibly manifested in an NMR spectrum in the line width of resonances; the line width at half height is the reciprocal of the T2 relaxation time. In liquids NMR many of the same physical mechanisms dictate these two rate constants and in most cases are equal. These two relaxation mechanisms can provide very important information concerning the physical properties of the molecule under study, tumbling in solution, binding or interaction with other molecules, etc.
T1 relaxation measurements provide information concerning the time constant for the return of excited spins to thermal equilibrium. For spins to fully relax, it is necessary to wait a period of five times T1. To accelerate data collection, in most cases one can perform smaller flip angles than 90° and wait a shorter time before repeating the pulse sequence. Knowing the value of T1 proves to be very useful in some instances, and it is quite simple to measure. The pulse sequence used to perform this measurement is an inversion recovery sequence [17]. The basic linear sequence of RF pulses (an NMR pulse sequence) consists of a 180‐τ‐90‐acquire. Knowledge of the T1 is of paramount importance when looking to increase the accuracy of quantitative NMR (qNMR) experiments. This will be described in greater detail below in the section on qNMR.
7.2.2 1D Carbon NMR Methods
7.2.2.1 Proton Decoupling
Carbon‐13, or 13C, is a rare isotope of carbon with a natural abundance of 1.13% and a gyromagnetic ratio, γC, that is approximately one quarter that of 1H. Early efforts to observe 13C NMR signals were hampered by several factors. First, the 100% abundance of 1H and the heteronuclear spin coupling, nJCH where n = 1–4, split the 13C signals into multiplets, thereby making them more difficult to observe. The original efforts to observe 13C spectra were further hampered by attempts to record them in the swept mode, necessitating long acquisition times and computer averaging of scans. These limitations were circumvented, however, with the advent of pulsed Fourier transform NMR spectrometers with broadband proton decoupling capabilities [18].
Broadband 1H decoupling, in which the entire proton spectral window is irradiated, collapses all of the 13C multiplets to singlets, vastly simplifying the 13C spectrum. An added benefit of broadband proton decoupling is NOE enhancement of protonated 13C signals by as much as a factor of three.
Early broadband proton decoupling was accomplished by noise modulation that required considerable power, typically 10 W or more, and thus caused significant sample heating. Over the years since the advent of broadband proton decoupling methods, more efficient decoupling methods have been developed including GARP, WURST, and others [19]. The net result is that 13C spectra can now be acquired when needed with low power pulsed decoupling methods and almost no sample heating.
7.2.2.2 Standard 1D Experiments
Of the multitude of 1D 13C NMR experiments that can be performed, the two most common experiments are a simple broadband proton‐decoupled 13C reference spectrum, and a DEPT sequence of experiments [20]. The latter, through addition and subtraction of data subsets, allows the presentation of the data as a series of “edited” experiments containing only methine, methylene, and methyl resonances as separate subspectra. Quaternary carbons are excluded in the DEPT experiment and can only be observed in the 13C reference spectrum or by using another editing sequence such as APT [21]. The individual DEPT subspectra for CH, CH2, and CH3 resonances of santonin (1) are presented in Figure 7.6.

FIGURE 7.6 Multiplicity edited DEPT traces for the methine, methylene and methyl resonances of santonin [1]. Quaternary carbons are excluded in the DEPT experiment and must be observed in the 13C reference spectrum or through the use of another multiplicity editing experiment such as APT.
7.3 TWO‐DIMENSIONAL NMR METHODS
7.3.1 Basic Principles of 2D
2D NMR methods are highly useful for structure elucidation. Jeener described the first 2D NMR experiment in 1971 [22]. In standard NMR nomenclature, a data set is referred to by one less than the total number of actual dimensions, since the intensity dimension is implied. The 2D data matrix therefore can be described as a plot containing two frequency dimensions. The inherent third dimension is the intensity of the correlations within the data matrix. This is the case in “1D” NMR data as well. The implied second dimension actually reflects the intensity of the peaks of a certain resonance frequency (which is the first dimension). The basic 2D experiment consists of a series of 1D spectra. These data are run in sequence and have a variable delay built into the pulse program that supplies the means for the second dimension. The variable delay period is referred to as t1.
All 2D pulse sequences consist of four basic building blocks: preparation, evolution (t1), mixing, and acquisition (t2). The preparation and mixing times are periods typically used to manipulate the magnetization (aka “coherence pathways”) through the use of RF pulses. The evolution period is a variable time component of the pulse sequence. Successive incrementing of the evolution time introduces a new time domain. This time increment is typically referred to as a t1 increment and is used to create the second dimension. The acquisition period is commonly referred to as t2. The first dimension, generally referred to as F2, is the result of Fourier transformation of t2 relative to each t1 increment. This creates a series of interferograms with one axis being F2 and the other the modulation in t1. The second dimension, termed F1, is then transformed with respect to the t1 modulation. The resultant is the two frequency dimensions correlating the desired magnetization interaction, most typically scalar or dipolar coupling. Also keep in mind that there is the “third dimension” that shows the intensity of the correlations.
When performing 2D NMR experiments, one must keep in mind that the second frequency dimension (F1) is digitized by the number of t1 increments. Therefore, it is important to consider the amount of spectral resolution that is needed to resolve the correlations of interest. In the first dimension (F2), the resolution is independent of time relative to F1. The only requirement for F2 is that the necessary number of scans is obtained to allow appropriate signal averaging to obtain the desired S/N. These two parameters, the number of scans acquired per t1 increment and the total number of t1 increments, are what dictate the amount of time required to acquire the full 2D data matrix. 2D homonuclear spectroscopy can be summarized by three different interactions, namely, scalar coupling, dipolar coupling, and exchange processes.
7.3.2 Homonuclear 2D Methods
7.3.2.1 Scalar‐Coupled Experiments: COSY and TOCSY
The correlated spectroscopy (COSY) experiment is one of the simplest 2D NMR pulse sequences in terms of the number of RF pulses it requires [23]. Once the time‐domain data are collected and Fourier transformed, the data appear as a diagonal in the spectrum that consists of the 1H chemical shift centered at each proton's resonance frequency. The off‐diagonal peaks are a result of scalar coupling evolution during t1 between neighboring protons. The data allow one to visualize contiguous spin systems within the molecule under study.
In addition to the basic COSY experiment, there are phase‐sensitive variants that allow one to discriminate the active from the passive couplings, allowing clearer measurement of the former. Active couplings give rise to the off‐diagonal cross‐peak. However, the multiplicity of the correlation has couplings inherent to additional coupled spins. These additional couplings are referred to as passive couplings. One such experiment is the double quantum‐filtered (DQF) COSY experiment [24]. Homonuclear couplings can be measured in this experiment between two protons isolated in a single spin system. Additional experiments have been developed that allow the measurement of more complicated spin systems involving multiple protons in the same spin system [25]. The 2D representation of the scalar coupled experiment is useful when identifying coupled spins that are overlapped or are in a crowded region of the spectrum. An example of a DQF COSY spectrum is shown in Figure 7.7. This data set was collected on astemizole (2).
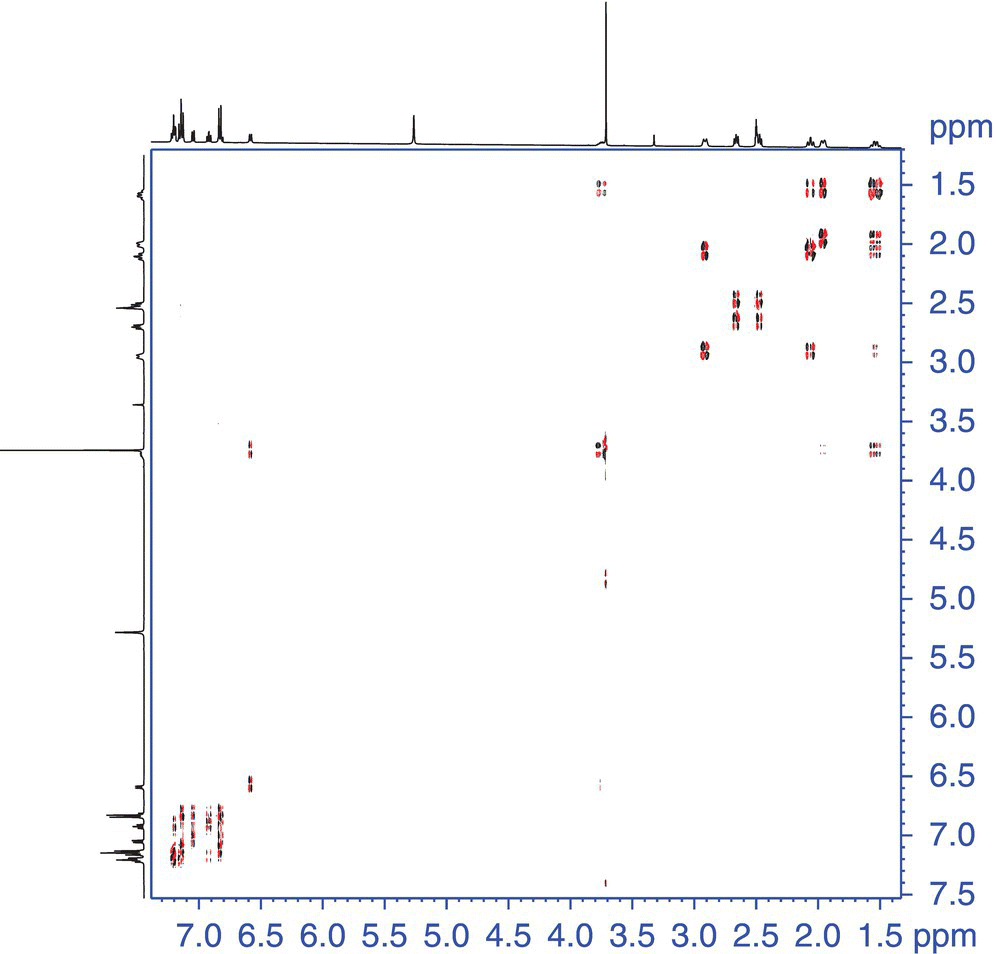
FIGURE 7.7 DQF COSY data of astemizole [2]. The black bars indicate the contiguous spin‐system drawn with arrows on the relevant portion of astemizole.
Total correlation spectroscopy (TOCSY) is similar to the COSY sequence in that it allows observation of contiguous spin systems [26]. However, the TOCSY experiment additionally will allow observation of many coupled spins simultaneously (contiguous spin system). The basic sequence is similar to the COSY sequence with the exception of the last pulse, which is a “spin‐lock” pulse train.
7.3.2.2 Scalar Coupled Experiments: INADEQUATE
2D homonuclear correlation experiments are typically run using 1H as the nucleus in both dimensions. This is advantageous, as the sensitivity for proton is quite high. However, there are 2D homonuclear techniques that detect other nuclei. One such experiment is the INADEQUATE experiment [27]. Its insensitive nature arises from the low natural abundance of 13C and the fact that one is trying to detect an interaction between two adjacent 13C atoms. The chance of observing this correlation is 1 in every 10 000 molecules. Given sufficient time or isotope‐enriched molecules, the INADEQUATE provides highly valuable information. The data generated from this experiment allow one to map out the entire carbon skeleton of the molecular structure. The only missing structural features occur where there are intervening heteroatoms (e.g. O, N).
7.3.2.3 Dipolar Coupled Experiments: NOESY
The 2D experiments described thus far rely solely on the presence of scalar coupling. There are other sequences that allow one to capitalize on the chemical shift dispersion gained with the second dimension for dipolar coupling experiments as well. One example in this category is the 2D NOESY pulse sequence.1 The pulse sequence for the 2D NOESY experiment is essentially identical to that of the DQF COSY experiment with the exception of an element that allows the buildup of dipolar couplings. The processed data is reminiscent of a COSY spectrum in that there is a diagonal represented by the 1D 1H spectrum in both frequency dimensions. In sharp contrast to the scalar coupled cross‐peaks in the COSY experiment, NOESY provides off‐diagonal responses that correlate spins through space. This sequence is used extensively in the structure characterization of small molecules for the same reason as its 1D counterparts. The spatial relationship of 1H atoms is an invaluable tool.
The similarity of the NOESY to the COSY also causes some artifacts to arise in the 2D data matrix of a NOESY spectrum. The artifacts arise from residual scalar coupling contributions that survive throughout the NOESY pulse sequence. These artifacts are usually quite straightforward to identify, as they have a similar antiphase behavior as can be seen for the DQF COSY data, Figure 7.8.
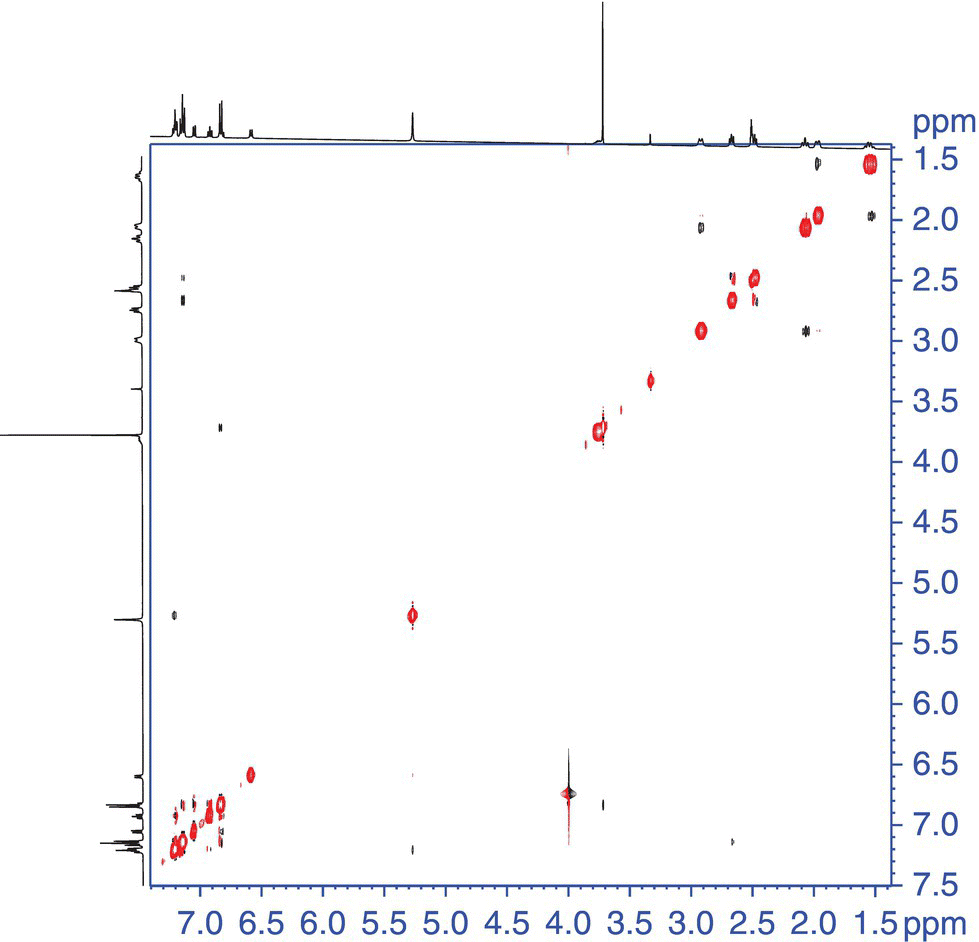
FIGURE 7.8 2D NOESY data of astemizole [2]. The mixed phase correlations in the aromatic region (boxed) are examples of the artifacts described in the text.
7.3.3 Heteronuclear 2D Methods
7.3.3.1 Direct Heteronuclear Chemical Shift Correlation
There are many numbers of heteronuclear correlation experiments that reach into the spin systems of many different chemical environments. This chapter will focus on the two major types of experiments that are used for structure elucidation. These are the 1JCH scalar coupled experiments and the long‐range (nJCH, where n is >1) scalar coupled experiments. The two predominant experiments for 1JCH are the heteronuclear multiple quantum coherence (HMQC) and the heteronuclear single quantum coherence (HSQC) methods. Both of these methods rely on the sensitive and time‐efficient proton, or so‐called “inverse”‐detected heteronuclear chemical shift correlation experiments are preferable [29]. For molecules with highly congested 13C spectra, 13C rather than 1H detection is desirable due to high resolution in the F2 dimension [30].
Using strychnine (Structure 7.3) as a model compound, a pair of HSQC spectra is shown in Figure 7.9. The top panel shows the HSQC spectrum of strychnine without multiplicity editing. All resonances have positive phase. In contrast, in the opinion of the authors, the much more useful multiplicity‐edited variant of the experiment is shown in the bottom panel. The multiplicity‐editing feature allows one to phase the data so that the correlations representing methyl and methine groups are in the same direction and methylenes are the opposite phase. Other less common direct heteronuclear shift correlation experiments have been described in the literature [31].
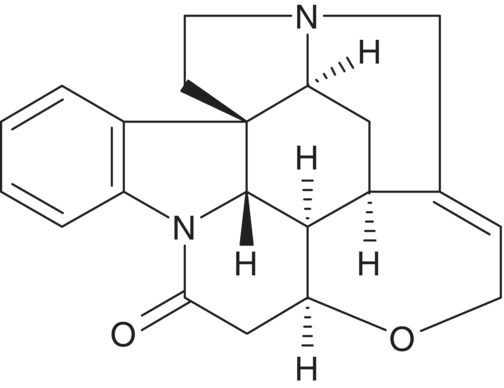
STRUCTURE 7.3 Strychnine.

FIGURE 7.9 (a) GHSQC spectrum of strychnine [3] using the pulse sequence shown in Figure 7.15 without multiplicity editing. (b) Multiplicity‐edited GHSQC spectrum of strychinine showing methylene resonances (gray contours) inverted with methine resonances (black contours) with positive phase. (Strychnine has no methyl resonances.)
7.3.3.2 Long‐Range Heteronuclear Shift Correlation Methods
There are numerous 13C‐detected long‐range heteronuclear shift correlation methods developed [32]. The primary reason that these methods have largely fallen into disuse is because of the heteronuclear multiple bond correlation (HMBC) experiment [33]. The proton‐detected HMBC experiment and its gradient‐enhanced variant offer considerably greater sensitivity than the original heteronucleus‐detected methods.
7.4 QUANTITATIVE NMR SPECTROSCOPY (qNMR)
7.4.1 The Basics
One of the tremendous qualities of NMR spectroscopy is its intrinsic quantitative attributes. A few recent publications have done an excellent job of detailing the basic principles and examples of the use of qNMR as well as pertinent parameters and reference standards [34–36]. The ability to quantitatively determine relative concentrations of multiple components in a mixture, determine potency, and product recovery, to name a few, has led this technique to gain tremendous popularity in the last 15 years. As with all things, popularity can lend itself to usage of the technique without carefully considering the physical phenomenon that allow these properties to be measured. This chapter will deal with the typical uses of qNMR, including the key parameters to consider and examples of applications.
With respect to qNMR the most relevant relationship is that between the integrated signals to the number of nuclei responsible for the resonance:
where
- I = the integrated resonance that is directly correlated to the number of nuclei responsible for the integrated resonance
- K = a spectrometer constant.
The factor K cancels as the substances are undergoing the same experimental conditions, assuming proper care is taken to ensure appropriate parameters are in place [34]. The utilization of NMR for quantitative explanations is also given in the USP‐NF 28, Chapter 761.2 There are two primary ways in which qNMR is used. These are a relative approach and an absolute method.
7.4.2 Relative and Absolute Methods
The most frequently used method, often in a pseudo‐qualitative mode (not optimal acquisition parameters), is the relative approach. For a quick analysis of two components of similar chemical properties (e.g. similar molecular weight and atomic composition), one can select two resonances of similar hybridization and integrate and get a rough estimate of molar ratios ![]() using Eq. (7.2). One can clearly solve this equation to provide the mole fraction of either of the two components:
using Eq. (7.2). One can clearly solve this equation to provide the mole fraction of either of the two components:
If the chemical compositions of both components are known, then the weight percent is inherent. However, one must be careful when using this “crude” method with components of an unknown chemical composition as there are many factors that can lead to ever increasingly erroneous results. One of the most important of these is the intrinsic relaxation rates of the compounds under study. If little is known of the compound being measured, then large differences in this chemical property can lead to a poor result. To make a rudimentary comparison, it would be equivalent to measuring the relative response of two compounds by UV at a single wavelength with no knowledge of the extinction coefficient/relative response factors.
The above description provides a very fast approach, yet it is not a rigorous quantitative method. To make this a rigorous relative quantitative method, one needs to measure a few critical NMR parameters that are intrinsic to the compounds in the mixture. These parameters, especially relaxation times, are well defined in the suggested reading list. In addition, there have been a few papers that have been published that detail general sets of parameters to be used to achieve a particular level of confidence in the measurement [35, 36]. Publications by Malz et al. go into a very good level of detail concerning the validation of quantitative methods [34, 35]. Utilizing careful experiment parameterization, one can get approximately 0.5–2% accuracies in their quantitative measurements. A “validated” parameter setup is shown below. Table 7.1 has been taken directly from the paper published by Malz et al. [35]. The reference by Malz et al. also deals a great deal with attributes related to specificity and selectivity as well as accuracy, precision, measurement uncertainty, and sensitivity. It is the authors opinion that this reference should serve as the primary reference to begin ones exploration into the use of NMR as a quantitative tool.
TABLE 7.1 Summary of the Universal Spectrometer Parameters for qNMR
Source: Taken from Ref. [35].
| Parameter | Bruker | JEOL | Varian | Value |
| 90° pulse strength | Pl1 | x_atn | tpwr | Instrument specific |
| 90° pulse length | P1 | X_90_width | pw90 | Instrument specific |
| Spin rotation | Spin_set | spin | Optional | |
| Measurement temperature | TE | Temp_get | temp | 300 K |
| Frequency of excitation | o1 | Irr_freq | tof | Middle of spectrum |
| Pulse angle | X_angle | pw | 30° | |
| Preacquisition delay | DE | Initial_wait | alfa | 5 μs |
| Acquisition time | AQ | X_acq_time | at | 3.41 s |
| Relaxation delay | D1 | Relaxation_delay | D1 | ≥(7/3)x longest T1 |
| Sweep width | SW | X_sweep | sw | 16 ppm |
| Filter width | FW | Filter_width | fb | ≥20 ppm |
| Number of FID points | TD | X_points | np | 32 k |
| Number of scans | ns | Scans | nt | Declined of reached S/N |
| Signal‐to‐noise ratio | S/N | Sn_ratio | dsn | ≥150 |
| Line broadening (em) | lb | Width | lb | 0.3 Hz |
| Number of frequency points | SI | X_points | fn | 64 k |
TABLE 7.2 Data Showing the Variation of Potency Determination with Variable Wavelength and NMR
| Method | % BHT | % API |
| HPLC‐UV Area% (200 nm) | 16.8 | 83.2 |
| HPLC‐UV Area% (214 nm) | 11.2 | 88.8 |
| HPLC‐UV Area% (224 nm) | 2.7 | 97.3 |
| HPLC‐UV Area% (282 nm) | 0.98 | 99.0 |
| HPLC‐UV RF wt/wt % | 2.5 | |
| qNMR wt/wt % | 1.94 | 94.2 |
The use of the absolute method traditionally involves the use of internal standards that are added into the NMR sample and used as an internal reference to use the relative method described above. The amount of care one takes in both sample preparation as well as experiment setup will dictate the level of accuracies that are obtainable. The primary literature also provides several good reviews that include a vast number of potential internal standards to utilize. Standards should be selected for the sample at hand based on several factors including chemical shift, similar molecular weight, relaxation rates, etc. One should also consider the use of solvents that allow for reasonable manipulation without evaporation as this can also lead to inaccurate measurements (Figure 7.10).
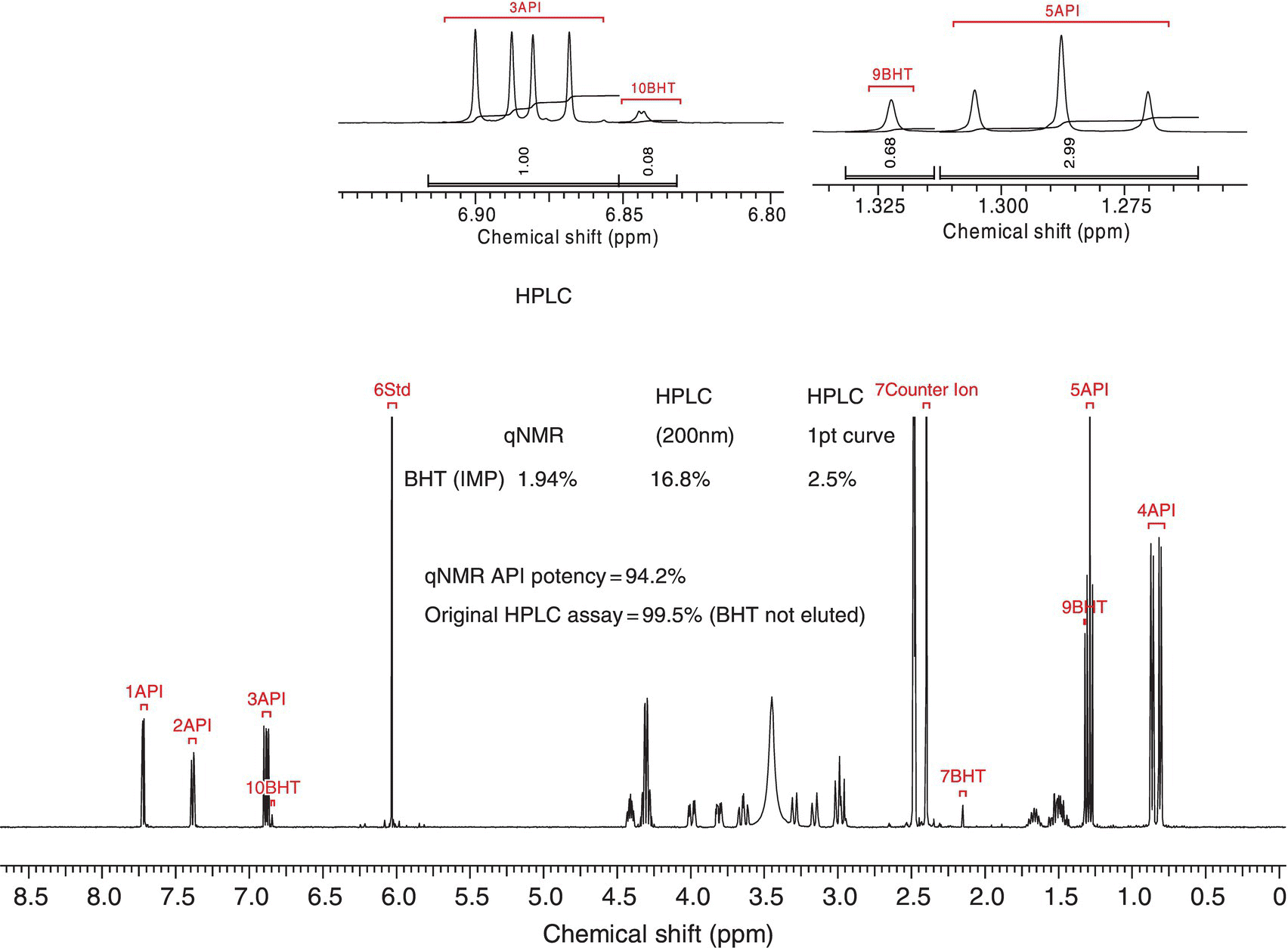
FIGURE 7.10 Example of the “absolute” method for qNMR experiments. In this case BHT was used as an internal reference with a know active pharmaceutical (API). See Table 7.2 to observe the impact of using NMR as opposed to UV for quantitation.
One thing that has not been discussed is the criticality of processing of the data once it is required. Several steps should be taken to ensure proper data processing. Three of the most important features involve the use of baseline correction, proper phasing of the resultant Fourier transformed spectrum, and choosing appropriate integration ranges to ensure the majority of the signal is encompassed in the integration [34–36].
7.4.3 Electronic Referencing in qNMR
The use of internal reference materials has been the method of choice for many years to do quantitative measurements. In the pharmaceutical industry this is particularly true as it relates to potency measurements, mass balance, etc. An alternative approach has been gaining traction for those that work in this area. This approach utilizes an electronic signal that is generated artificially and is calibrated to a particular value and subsequently inserted into the collected NMR spectrum and used as a “standard” for comparison. The method was published under the acronym of ERETIC [37]. The original application was developed for imaging. It was later elaborated for use in high resolution NMR [38]. In effect, an electronic signal is routed through a “spare” coil in the probe. The amplitude of the signal is set by the operator, and this signal can be inserted at a user‐defined resonance frequency so as to not interfere with any of the signals related to the compound under study. This signal can then be used with a reference standard independently and then related back to the sample of interest using the same amplitude.
Subsequent to the ERETIC method, there have been alternate approaches developed. One such method uses software to simulate a signal that is incorporated into a reference spectrum [39]. The simulated signal is then integrated relative to the reference. As with the ERETIC the signal is then placed in the spectrum containing the compound/chemistry under investigation as a reference. The simulated signal can be placed anywhere in the spectrum and can be crafted to have similar lineshape and intensity to the resonance/species of interest.
7.4.4 Quantitation in Flow NMR
The use of quantitation under flow NMR conditions requires a bit more consideration than for a static NMR tube arrangement. The theory and practicality of flow NMR has been described thoroughly in the literature, including several comprehensive reviews.3,4,5,6 Unlike static tube‐based experiments where all spins in the sample are uniformly experiencing B0 and are therefore at Boltzmann distribution prior to excitation with a radio frequency pulse, flow experiments require the scientist to adjust the flow rate in such a manner to ensure that the sample flowing through has enough residence time within the B0 field that they reach Boltzmann distribution prior to the active region of the flow cell for excitation. To ensure quantitation this condition must be met (Figure 7.11).

FIGURE 7.11 Diagram showing the key components within the flow path of the flow cell located in the NMR probe. This diagram is showing the key elements along the flow path that must be considered when designing flow‐NMR experiments under quantitative condition.
Equation (7.3) shows the interdependency of the flow of the system to the intrinsic relaxation processes as well as the geometry of the probe ensuring quantitative conditions, where Ti represents either T1 or ![]() and τ is flow dependent [34]. Ensuring the sample is at or near Boltzmann's conditions prior to excitation is critical. The typical amount of time that the sample must reside in the magnetic field prior to irradiation is about five times T1. It should be noted that this time should be calculated based on the entity with the slowest T1. The experimental parameters employed for the measurement of T1 values are described in detail elsewhere [9]. Calculation of the maximum flow rate while accomplishing optimal premagnetization of the spins within the sample can be done through Eq. (7.4):
and τ is flow dependent [34]. Ensuring the sample is at or near Boltzmann's conditions prior to excitation is critical. The typical amount of time that the sample must reside in the magnetic field prior to irradiation is about five times T1. It should be noted that this time should be calculated based on the entity with the slowest T1. The experimental parameters employed for the measurement of T1 values are described in detail elsewhere [9]. Calculation of the maximum flow rate while accomplishing optimal premagnetization of the spins within the sample can be done through Eq. (7.4):
Within a standard 1D proton NMR experiment, there are several factors that must be taken into account. Two of these are the acquisition time (to ensure proper digitization), taq, and the relaxation time to ensure the vast majority of spins “re‐achieve” Boltzmann distribution, td. Both of these parameters (see Table 7.1) together are known as the pulse repetition time tp. One can envision the interdependency of the flow and the repetition time in Eq. (7.5):
An example would be a compound with a T1 of 5 seconds, and a 100 μl premagnetization volume would have a maximum flow rate (Vflow,maximum) of approximately 0.24 ml/min.
7.4.5 Reaction Kinetics
One important and sometimes overlooked application of qNMR is the ability to monitor the progress and kinetics of a chemical reaction directly by NMR. A recent publication does a very good job of reviewing this topic and provides many references to the primary literature. This section will give highlights of the methodology and a few examples [40]. A simplistic example of the use of NMR to monitor reactions is shown for the hydrolysis of acetic anhydride to acetic acid in the presence of D2O. Figure 7.12 shows the reaction progression under two different experimental conditions. One is static in an NMR tube, and the other is measured in real‐time through the application of flow NMR. Both of these methods give good accuracy in the measurement of this first‐order rate constant.

FIGURE 7.12 (a) NMR Tube Kinetics the reagent is injected, NMR tube shaken and placed in the magnet and spectra are recorded. (b) Flow NMR Kinetics, the reactor is integrated to the NMR and run in SemiBatch mode. The conditions of the reaction were 20 ml acetic anyhydride (AcOAc) dosed over 20 minutes into 200 ml of D2O. The flow rate through the probe was 3 ml/min.
Source: am Ende, et. al. AIChE 2010 [41].
The practice of monitoring reactions in NMR tubes has been used since the inception of NMR and involves preparing reactions on a small scale and initiating them in an NMR tube with a suitable deuterated solvent. Although there are certain and obvious advantages to following this practice, the use of solvents lacking protons (CDCl3, D2O, CCl4, etc.) is by no means a necessity. Recently, with the advent of more stable magnets and improved instrument electronics, it has become routine practice to monitor reactions by collecting NMR spectra in non‐deuterated solvents [42]. The applications of the technique are wide ranging and can be used to directly monitor or assay most any reaction mixture or reagent solution. As mentioned previously, no deuterium solvents are used so the spectra are recorded in an “unlocked mode.” One factor that facilitates the acquisition of NMR data in this way is that the concentration of most commonly used neat organic solvents is usually somewhere around 10 M. The concentration of the reactants in most reaction solutions is in the range of 0.1–1 M (and of solutions of most commercial reagents ca. 0.5–2.5 M). Thus, the ratio of solvent to solute/analyte molecules is usually between 100 : 1 and 10 : 1 in most solutions of interest. It is a straightforward matter for most NMR spectrometer hardware to handle dynamic ranges of proton intensities of greater than 4 orders of magnitude. If needed, techniques can be applied that use special “pulse sequences” to suppress unwanted signals from the solvent [34]. By doing so, the dynamic range and spectral quality of the reactions species can be improved (Figure 7.13).
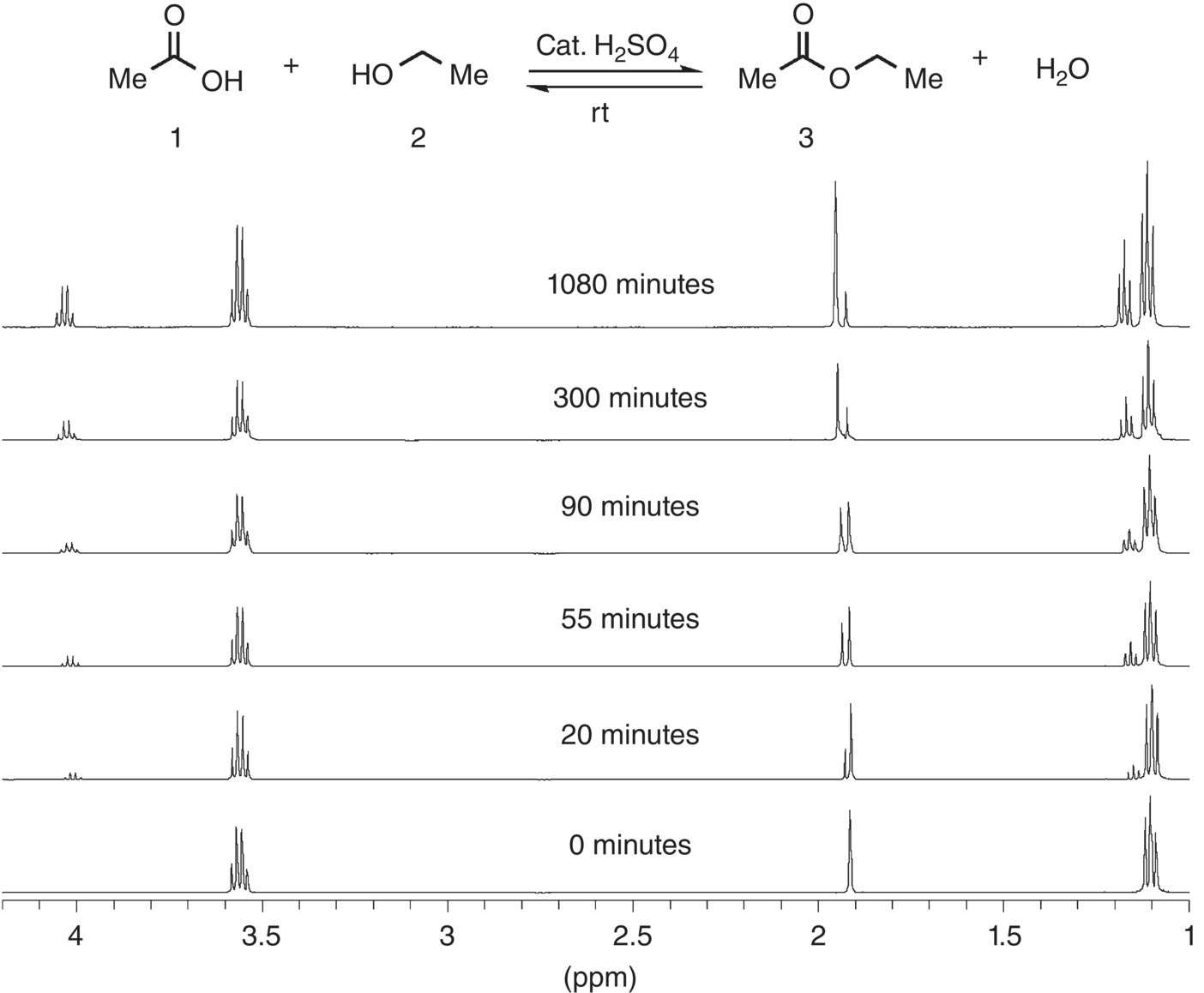
FIGURE 7.13 Example of the No‐D NMR method Fischer esterification of acetic acid in ethanol (1 : 4 M ratio).
Source: Reprinted with permission from Hoye et al. [42]. Copyright (2004) American Chemical Society.
Other more advanced techniques such as flow NMR can be utilized to monitor larger‐scale reactions in real time. The use of flow NMR has been around for many years and has been used to monitor everything from reaction completion, kinetics, combinatorial chemistry, etc. [5]. Of late there has been a great deal of activity in the area of monitoring process chemistry using flow NMR [4042–45].7,8
There have been many different designs (instrument setup) to accomplish real‐time reaction monitoring. However, one that works very well is that published by Maiwald et al. and is used by many others in the field [40]. The basic premise is to have a fast loop that carries the reaction mixture from a reactor to ensure that the loop is just a “real‐time” extension of the reactor. From this loop there is a split (many ways to accomplish this) that allows a slow loop to flow into the NMR flow cell. One must keep in mind that the criteria must be met as described in Section 7.4.5 to be under quantitative conditions if quantitation is desired. Obviously if one is interested in strictly observing the reaction for gross features (e.g. reaction completion and/or reaction optimization), the quantitative conditions are not needed, and a less rigorous approach can be taken. A general schematic is described in Ref. [40} (Figure 7.14).
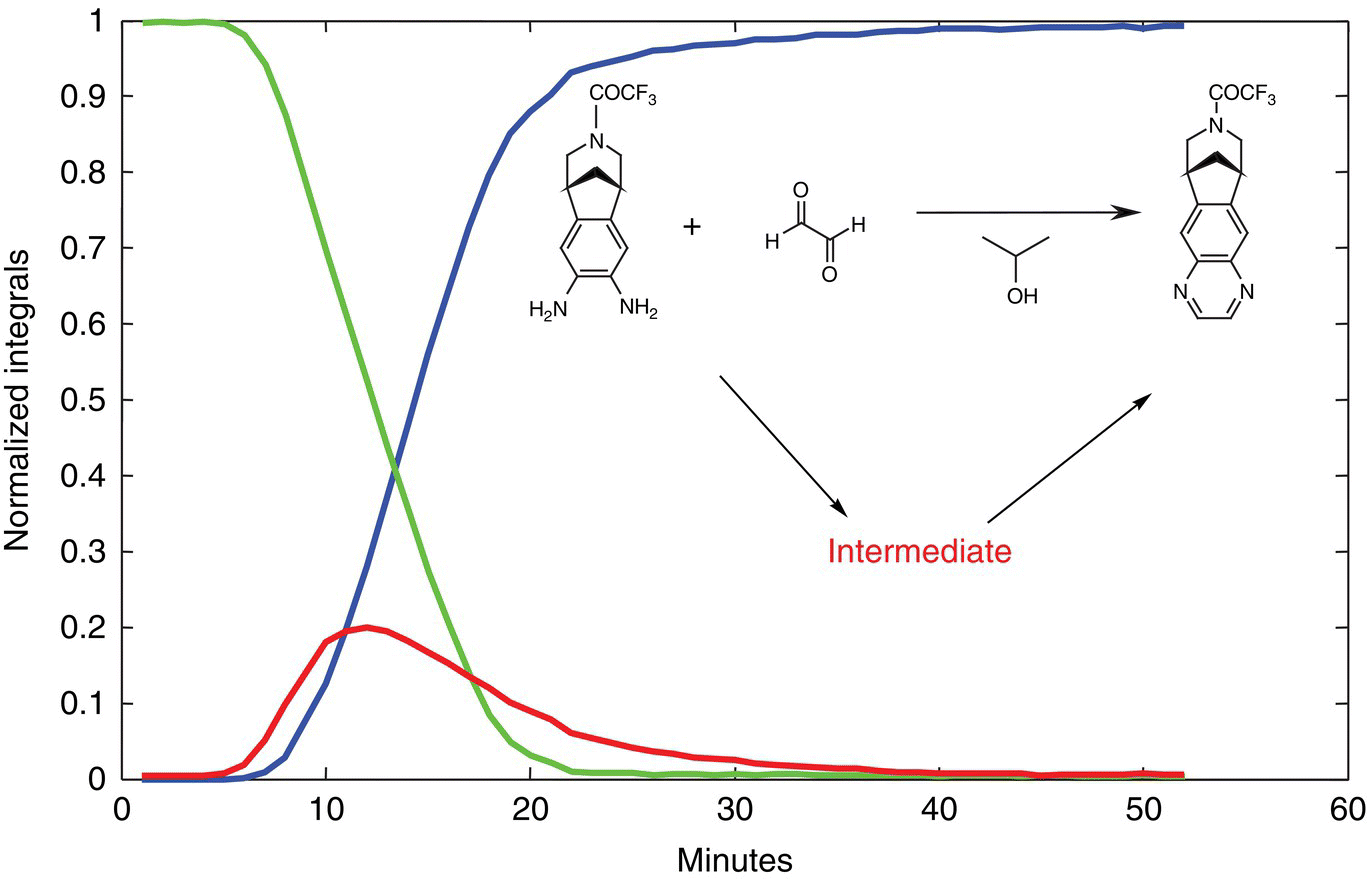
FIGURE 7.14 A graph showing the presence of an intermediate in the formation of veranicline. This intermediate was readily observed by NMR.
REFERENCES
- 1. Friebolin, H. (1993). Basic One‐ and Two‐Dimensional NMR Spectroscopy. New York: VCH Publishers.
- 2. Gunther, H. (1995). NMR Spectroscopy, 2e. New York: Wiley.
- 3. Bloch, F., Hensen, W.W., and Packard, M. (1946). Phys. Rev. 69: 127.
- 4. Derome, A.E. (1987). Modern NMR Techniques for Chemistry Research. New York: Pergamon Press.
- 5. Claridge, T.D.W. (1999). High Resolution NMR Techniques in Organic Chemistry. New York: Pergamon Press.
- 6. Keeler, J. (2006). Understanding NMR Spectroscopy. New York: Wiley.
- 7. Levitt, M.H. (2002). Spin Dynamics: Basics of Magnetic Resonance. New York: Wiley.
- 8. Croasmun, W.R. and Carlson, R.M.K. (1994). Two‐Dimensional NMR Spectroscopy: Applications for Chemists and Biochemists, 2e. New York: VCH Publishers.
- 9. Berger, S. and Braun, S. (2004). 200 and More NMR Experiments: A Practical Course. Weinheim: Wiley‐VCH Verlag GmbH and Co. KGaA.
- 10. (a) Overhauser, A.W. (1953). Phys. Rev. 89: 689. (b) Overhauser, A.W. (1953). Phys. Rev. 92: 411.
- 11. Solomon, I. (1955). Phys. Rev. 99: 559.
- 12. Richarz, R. and Wuthrich, K. (1978). J. Magn. Reson. 30: 147.
- 13. Neuhaus, D. and Williamson, M. (2000). The Nuclear Overhauser Effect in Structural and Conformational Analysis, P., 2e. New York: Wiley.
- 14. Claridge, T.D.W. (1999). High‐Resolution NMR Techniques in Organic Chemistry, Tetrahedron Organic Chemistry Series, vol. 19. Oxford: Elsevier Science.
- 15. Stott, K., Keeler, J., Van, Q.N., and Shaka, A.J. (1997). J. Magn. Reson. 1125: 302.
- 16. Stott, K., Stonehouse, J., Keeler, J. et al. (1995). J. Am. Chem. Soc. 117: 4199.
- 17. Hahn, E.L. (1949). Phys. Rev. 76: 145.
- 18. (a) Levy, G.C. and Nelson, G.L. (1972). Carbon‐13 Nuclear Magnetic Resonance for Organic Chemists. New York: Wiley‐Interscience. (b) Stothers, J.B. (1972). Carbon‐13 NMR Spectroscopy. New York: Academic Press.
- 19. Shaka, A.J. and Keeler, J. (1987). Prog. Nucl. Magn. Reson. Spectrosc. 19: 47–129.
- 20. (a) Doddrell, D.M., Pegg, D.T., and Bendall, M.R. (1982). J. Magn. Reson. 48: 323–327. (b) Doddrell, D.M., Pegg, D.T., and Bendall, M.R. (1982). J. Chem. Phys. 77: 2745–2752.
- 21. Patt, S.L. and Shoolery, J.N. (1982). J. Magn. Reson. 46: 535–539.
- 22. Jeener, J. (1971). Ampere International Summer School, Basko Polje, (proposal).
- 23. (a) Aue, W.P., Bartholdi, E., and Ernst, R.R. (1976). J. Chem. Phys. 64: 2229–2246. (b) A. Bax, R. Freeman, and G. A. Morris, J. Magn. Reson., 42, 164–168 (1981).
- 24. Piantini, U., Sorensen, O.W., and Ernst, R.R. (1982). J. Am. Chem. Soc. 104: 6800.
- 25. Griesinger, C., Sorensen, O.W., and Ernst, R.R. (1985). J. Am. Chem. Soc. 107: 6394.
- 26. Braunschweiler, L. and Ernst, R.R. (1983). J. Magn. Reson. 53: 521.
- 27. Bax, A., Freeman, R., and Frenkiel, T.A. (2102). J. Am. Chem. Soc. 1981: 103.
- 28. Jeener, J., Meier, B.H., Bachmann, P., and Ernst, R.R. (1979). J. Chem. Phys. 71: 4546.
- 29. (a) Müller, L. (1979). J. Am. Chem. Soc. 101: 4481. (b) Bodenhausen, G. and Ruben, D.J. (1980). Chem. Phys. Lett. 69: 185–189.
- 30. Reynolds, W.F., MacLean, S., Jacobs, H., and Harding, W.W. (1999). Can. J. Chem. 77: 1922–1930.
- 31. Martin, G.E. (2002). Qualitative and quantitative exploitation of heteronuclear coupling constants. In: Annual Report NMR Spectros, vol. 46 (ed. G.A. Webb), 37–100. New York: Academic Press.
- 32. Martin, G.E. and Zektzer, A.S. (1988). Magn. Reson. Chem. 26: 631.
- 33. Bax, A. and Summers, M.F. (1986). J. Am. Chem. Soc. 108: 2093–2094.
- 34. Malz, F. (2008). Quantitative NMR in the solution state. In: NMR Spectroscopy in Pharmaceutical Analysis, 1e (ed. U. Holzgrabe, I. Wawer and B. Diehl), 43–60. Amsterdam, The Netherlands: Elsevier.
- 35. Malz, F. and Jancke, H. (2005). J. Pharm. Biomed. Anal. 38: 813.
- 36. Pauli, G.F., Jaki, B.U., and Lankin, D.C. (2005). J. Nat. Prod. 68: 133.
- 37. Barantin, L., Akoka, S., and LePape, A. (1995). French Patent CNRS no.95 07651, 26 June 1995.
- 38. Akoka, S., Barantin, L., and Trierweiler, M. (1999). Anal. Chem. 71: 2554–2557.
- 39. Wider, G. and Drefer, L. (2006). J. Am. Chem. Soc. 128 (8): 2571–2576.
- 40. Maiwald, M., Steinhof, O., Sleigh, C. et al. (2008). Quantitative NMR in the solution state. In: NMR Spectroscopy in Pharmaceutical Analysis, 1e (ed. U. Holzgrabe, I. Wawer and B. Diehl), 471–491. Amsterdam, The Netherlands: Elsevier.
- 41. am Ende, D., Dube, P., Gorman, E., Marquez, B., and Mark Zell and R. Krull, D. Piroli, M. Fey and K. Colson, Reaction NMR: A Quantitative Kinetic Analysis “Probe” for Process Development, presented at AIChE National Meeting, Salt-Lake City November 6-10, 2010; The reported kinetic isotope ratio is from Batts, B.D. and Gold, V. (1969). J. Chem. Soc. A (6): 984.
- 42. Hoye, T.R., Eklov, B.M., Ryba, T.D. et al. (2004). Org. Lett. 6: 953–956.
- 43. Maiwald, M., Fischer, H.H., Kim, Y.K. et al. (2004). J. Mag. Reson. 166: 135–146.
- 44. Albert, K., Hasse, H. et al. (2005). Chem. Eng. Process. 44: 653–660.
- 45. Horvath, I.T. and Millar, J.M. (1991). Chem. Rev. 91: 1339–1351.