
49
APPLICATIONS OF QUALITY RISK ASSESSMENT IN QUALITY BY DESIGN (QbD) DRUG SUBSTANCE PROCESS DEVELOPMENT
Alan Braem
Product Development, Bristol‐Myers Squibb, New Brunswick, NJ, USA
Gillian Turner
Product Development and Supply, GlaxoSmithKline, Stevenage, UK
49.1 WHY RISK ASSESSMENT IS USED IN THE PHARMACEUTICAL INDUSTRY
Pharmaceutical manufacturers and the global regulatory authorities acknowledge that the manufacture of drug substance (the bulk active ingredient) and the drug product (the finished dosage form that the patient uses) both entail some degree of risk to quality. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) establishes regulatory guidance1 for the development and manufacture of pharmaceutical drug substances and drug products that incorporates this understanding of risk. The ICH quality guidelines ICH Q8 [1], Q9 [2], and Q11 [3] describe a quality‐by‐design (QbD) and quality risk management (QRM) approach to pharmaceutical development and manufacturing that has quality risk assessment (QRA) as a core element of the development process. Identifying and managing risks to product quality is crucial to ensuring the safety and efficacy of pharmaceutical products. Chemical engineers often have a key role in conducting or contributing to these QRAs.
QbD is a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and QRM. The goals of QbD typically include:
- The definition of meaningful product quality specifications based on clinical performance.
- Improved process capability and reduction of product variability and defects through enhancing product and process design, understanding, and control.
- Improved development and manufacturing efficiency.
- Enhanced capability for root cause analysis and post‐approval change management.
In particular, QbD is dependent on effective use of risk assessment and management tools (ICH Q9 Quality Risk Management [2]) and needs to be accompanied by good development science (ICH Q8(R2) Pharmaceutical Development [1] and ICH Q11 Development and Manufacture of Drug Substance [3]) and at an appropriate stage integrated into a suitable quality system (ICH Q10 Pharmaceutical Quality System [4]).
Prior to assessing risks to quality, the quality requirements for the drug product and drug substance need to be understood. ICH Q8 [1] indicates that this can be done by “Defining the quality target product profile (QTPP) as it relates to quality, safety and efficacy, considering e.g., the route of administration, dosage form, bioavailability, strength, and stability.” Delineating the QTPP allows definition of potential drug product critical quality attributes (CQAs). Some of the drug product CQAs will be linked only to aspects of the drug product process, but typically several are linked back to drug substance and excipient CQAs, for example, purity and powder properties. Given that the drug substance manufacturing process typically entails a multistep synthetic process, one can in turn identify the quality attributes of synthetic intermediates that have potential to impact the drug substance. Each of these drug product, excipient, drug substance, and intermediate CQAs needs to be controlled within defined limits to ensure product quality.
With these quality targets in hand, a control strategy must be developed to ensure targets are met. This control strategy can consist of:
- Procedural controls – definition of procedural steps and their order.
- Technical requirements – defined limits on facilities and equipment used.
- Parametric controls – limits on process parameters.
- In‐process controls (IPCs) – analysis of process streams against control limits, with an accompanying failure pathway (procedure) that addresses failure modes.
- Analytical test and specifications on both input materials and isolated products.
In order to understand what aspects of the process require control, the process must undergo, as described in ICH Q8 [1], “a systematic evaluation… including
- Identifying (through, e.g., prior knowledge, experimentation, and risk assessment) the material attributes and process parameters that can have an effect on product CQAs
- Determining the functional relationships that link material attributes and process parameters to product CQAs”
Risk assessment is used to distinguish between those process and input material aspects that pose low risk to quality from those that pose moderate or high risk to quality. Thus, risk assessment can be used to focus control strategy development on only those aspects with significant potential to impact quality.
A general framework for QRA is discussed in the ICH Q9 QRM guidance [2]. Q9 defines risk as “the combination of the probability of occurrence of harm and the severity of that harm” and risk assessment as “the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards.”
The fundamental questions that relate to risk assessment therefore include the following:
- What may go wrong?
- What is the likelihood that something will go wrong?
- What are the consequences?
These three questions are addressed by QRA processes through the identification of failure modes that may cause harm, the estimation of probability of failure mode occurrence, and an analysis of the severity of the resulting outcome. The tools used, the level of effort, and the formality and extent of documentation of the process can all vary with the stage of development but should be commensurate with the level of risk. Different companies can use different tools and strategies, but there are many approaches that are commonplace throughout the industry.
In the sections that follow, we will describe common risk assessment approaches and how they are applied at various stages of drug substance development. We will discuss how risk assessments can be used early in the development cycle for risk‐based prioritization of development efforts and identification of potential CQAs and process parameters. For later stages, we will describe how risk assessments enable identification of a robust control strategy and how they are typically used for formal identification of criticality, and we will touch on the associated regulatory implications. We will also cover the assessment of residual risk after control strategies are implemented, risk review, and operational risk assessment. The examples shared are focused on small molecule drug substances rather than biologics that, due to their complexity, have additional challenges to address in the risk management process.
It is important to appreciate that risk assessment is not a single event but a continual process of identification, assessment, mitigation, and reassessment that is driven by increasing knowledge and experience. The effectiveness of the risk management process depends on having the right people contributing sufficient data and knowledge of the process under review and a well‐defined risk assessment process.
49.2 OVERVIEW OF RISK ASSESSMENT PROCESS
The risk assessment process is a structured approach that enables development of a robust control strategy, thereby ensuring manufacturing operations remain in control. It is composed of several steps (see Figure 49.1) including risk identification, risk analysis and evaluation, risk reduction and acceptance, and risk communication and review, and here approaches to each of these steps will be discussed in more detail.

FIGURE 49.1 Risk questions that arise during development and associate risk assessment step.
Risk assessment processes are used throughout the development of the API from early synthetic route and process selection through to definition of a commercial control strategy. Risk assessment can also be used in life‐cycle management of commercial processes to assess changes to the process or manufacturing facility. Before working through the risk process, it is important to define the scope of the activity to ensure that the discussions remain focused and the output is aligned with business needs. The majority of risk assessments focus on the quality of the output API through linkages to the API CQAs, though some will also include elements related to manufacturability too. In particular, risks associated with yield are often included and evaluated though these are not necessarily directly linked to the quality of the API. For more information of the various types of risk assessment, see Section 49.3.
49.2.1 Risk Identification
In early risk assessment, risk identification activities focus on the ability of the potential routes and process to meet both the QTPP and any constraints or opportunities provided by the use of novel and developing technologies. Examples of such technologies are flow and biocatalysis, safety and environmental considerations, and manufacturability aspects including cost, cycle time, and operability. A common approach taken here is structured brainstorms guided by criteria derived from the QTPP in addition to manufacturing considerations.
When route and process have been defined, the later risk identification activities can commence. At this stage, the identification of potential CQAs is usually possible, and this provides one aspect of the scope of the activity. The aim of this step is to identify what could potentially go wrong during the operation of a process and then outline the possible causes and consequences. There are a number of different ways that this can be achieved, and they fall into two main categories, the “top‐down” approach and the “bottom‐up” approach. Ideally each of these will deliver the same end result but the journey will look different. The top‐down approach focuses the initial effort on identification of failure modes that have the potential to impact the quality of the API. This activity can be informed by several different inputs, and those most commonly used include historical data and experience with the specified process, in addition to expert opinions based on information from similar processes and equipment. Once credible failure modes have been identified, the consequences need to be fully explored as the direct impacts can themselves have a secondary impact further through the process. An example of such a situation would be variations in reaction conditions that increase the level of impurity A, and the impurity is not removed in the subsequent downstream operations, resulting in higher levels of the impurity in the next stage of the process. In addition, it may be found that higher levels of impurity A entering the next stage result in a change to the reaction rate or the performance of the crystallization and give rise to elevated levels of a second impurity, impurity B. Increased levels of both impurity A and impurity B are then consequences of the failure mode.
Once a failure mode has been identified, the risk assessment process can then explore the potential causes of the failure. For APIs, these are often variation in parameter settings and levels of impurities, though they can also be due to equipment selection. The detail to which the potential causes are identified can vary depending on the type of risk assessment being carried out and the desired outputs. In some situations, identification of the parameter(s) directly causing the failure modes is sufficient; in other cases, the root cause(s) behind the parameter variation is also recorded. The potential for two or more parameters and attributes to interact and jointly influence a particular failure mode should also be considered. Interactions commonly occur within the same unit operation though they may also include parameters and attributes that are introduced in different parts of the process.
The bottom‐up approach is similar but starts with a list of the parameters and other sources of variation and uses either data or expert opinion to assess what the likely failure modes could be as the parameters vary from high to low values within the range being assessed. In practice, it is often a combination of these two approaches that produces a pragmatic list of the potential risks.
A number of approaches that can be utilized to identify risks and common ones include the following:
- Brainstorming. Working as a group to explore many “what‐if” scenarios before focusing down to the likely/credible/realistic ones.
- Fishbone diagrams, also known as Ishikawa diagrams, provide a structure on which to brainstorm. They ensure that possible causes of a particular failure mode, or variables impacting a CQA, are explored from a variety of perspectives, thus ensuring that the initial list of variables (parameters and attributes) included in the risk assessment is comprehensive. An example of this can be seen in Figure 49.2.
- Input process output (IPO) diagrams. These are visual representations that focus on identifying the inputs and outputs of a process. For a risk assessment, the inputs are typically the starting materials, reagents, solvents, excipients, etc., and the outputs are the CQAs. The parameters, both those that can be controlled and that contribute to the noise, are then added. This is another way to encourage teams to think more broadly than just the process aspects and help to link the causes of failure to the effects. An example of this type of diagram can be seen in Figure 49.3.
- Gemba literally means “the real place” and refers to the place where value is created [5]. The risk identification activity happens as the process operation is observed and provides an excellent way of engaging with the practicalities. It has the advantage of highlighting potential failure modes that may be less obvious from written instructions or equipment diagrams.
- Process mapping describes the individual steps within a process and how they link together and includes information on where substances are added or removed from the process train. Additional information on the equipment and physical state of the process can be added to aid in risk identification during the brainstorm activity. A simple example can be seen in Figure 49.4.
- Mechanism mapping can take a number of formats and uses pictures and diagrams to look in detail at what is happening within the process. It is ideally suited to explaining areas of complexity where a deep understanding of how changes to the material being processed can impact an aspect of the output of the process. In API manufacture, this is typically employed in areas such as particle formation, where understanding the physical mechanisms at play is key to identifying risks. A common application of mechanism mapping for API is in the formation and fate of impurities; visual representations that describe how impurities form and transform, including any mechanistic understanding, and how and where they purge, are used to direct the brainstorming activities.
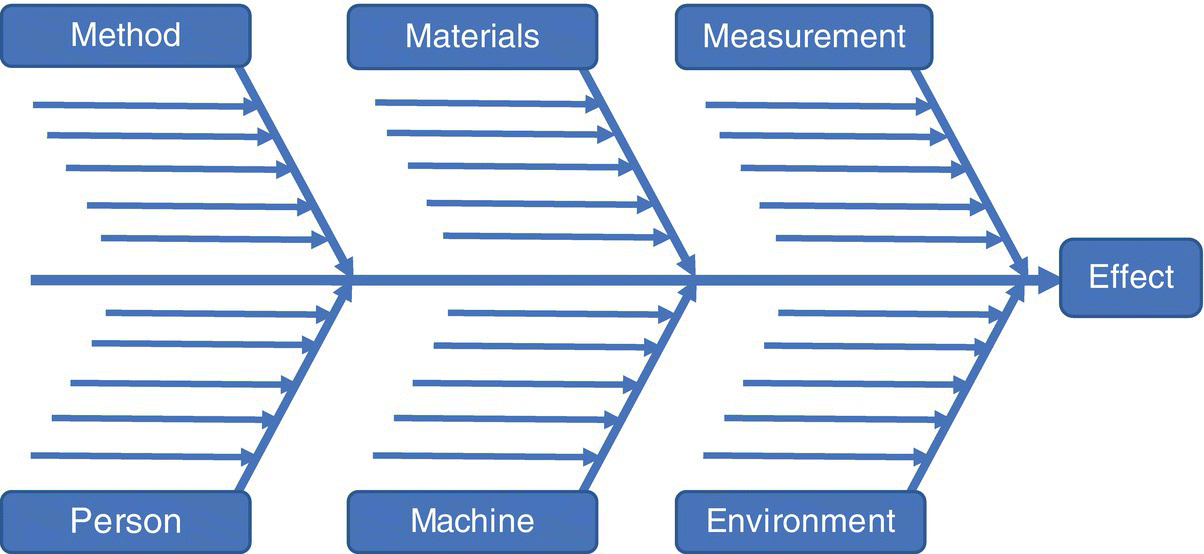
FIGURE 49.2 Fishbone or Ishikawa diagram.
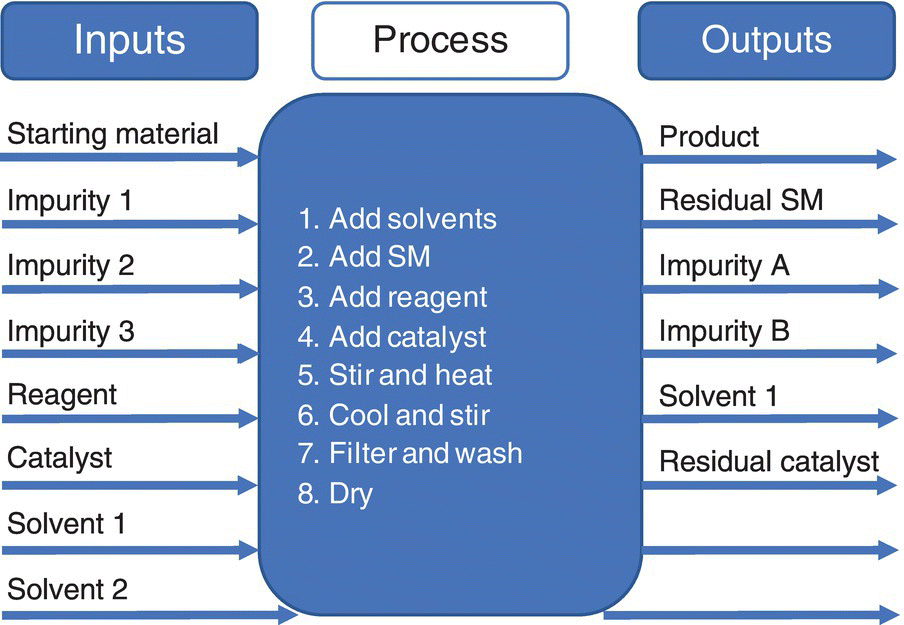
FIGURE 49.3 Input process output diagram.

FIGURE 49.4 Process map.
As with most activities, the quality of the input has a bearing on the quality of the output; the same principles can be applied to the risk assessment process. Having a range of expertise available to work as a team to complete the various stages of the risk assessment will result in a more rigorous assessment and hence ensure that no significant risks are omitted. When assessing an API process, the range of technical experts may typically include representation from engineering, chemistry, analysis, formulation, production, statistics, quality, and regulatory affairs who work together with a facilitator. The value of team members who have practical experience of the process under evaluation cannot be overemphasized; they are in an excellent position to identify any practical constraints that may lead to failure and will have awareness of any previous deviations that have given rise to unanticipated results.
49.2.2 Risk Analysis and Evaluation
Having identified the risks, the next step is to provide an assessment of the adverse impact of the risk and its likelihood of occurrence, in either a quantitative or qualitative manner. In many processes, there is also a consideration of the ability to detect the risk, and in some cases, a knowledge rating may also be included either in the evaluation of the risk or to provide context to the scoring that has been applied. Following these steps, the risks are then ordered, or a particular set of criteria applied, to identify any actions required during risk mitigation.
In the early phases of risk assessment, and in instances where knowledge may be limited, the quantification of risk may not be possible, and in these cases a qualitative system is often used, such as high/low or a traffic light system where red, amber, and green indicate the degree of risk posed by each failure mode. The scoring is often reached through discussion within an expert group, and it is acknowledged that as more information becomes available, the scoring may require reevaluation. It is important to draw the distinction between a lack of knowledge and high risk; not all knowledge gaps result in high risks when data does become available; in fact, it is often the case that those areas where little knowledge exists ultimately prove to be low risk, but this cannot be guaranteed.
As development of the process proceeds and data is generated to support the risk assessment, a more detailed scoring of the risk becomes possible, and at this phase quantitative systems are more prevalent. Typically, tools such as failure mode, effects, and criticality analysis (FMECA) and failure mode effect analysis (FMEA) are used to capture the scoring; see Section 49.4.3 for further information.
Scoring for severity or impact typically considers the link between either the failure mode or the causes of the failure and the ultimate impact that has on the patient. This may be considered as a direct link, by assessment of the impact of the effect of the failure on the patient, for example, or it could be linked through a series of steps as shown in Figure 49.5.
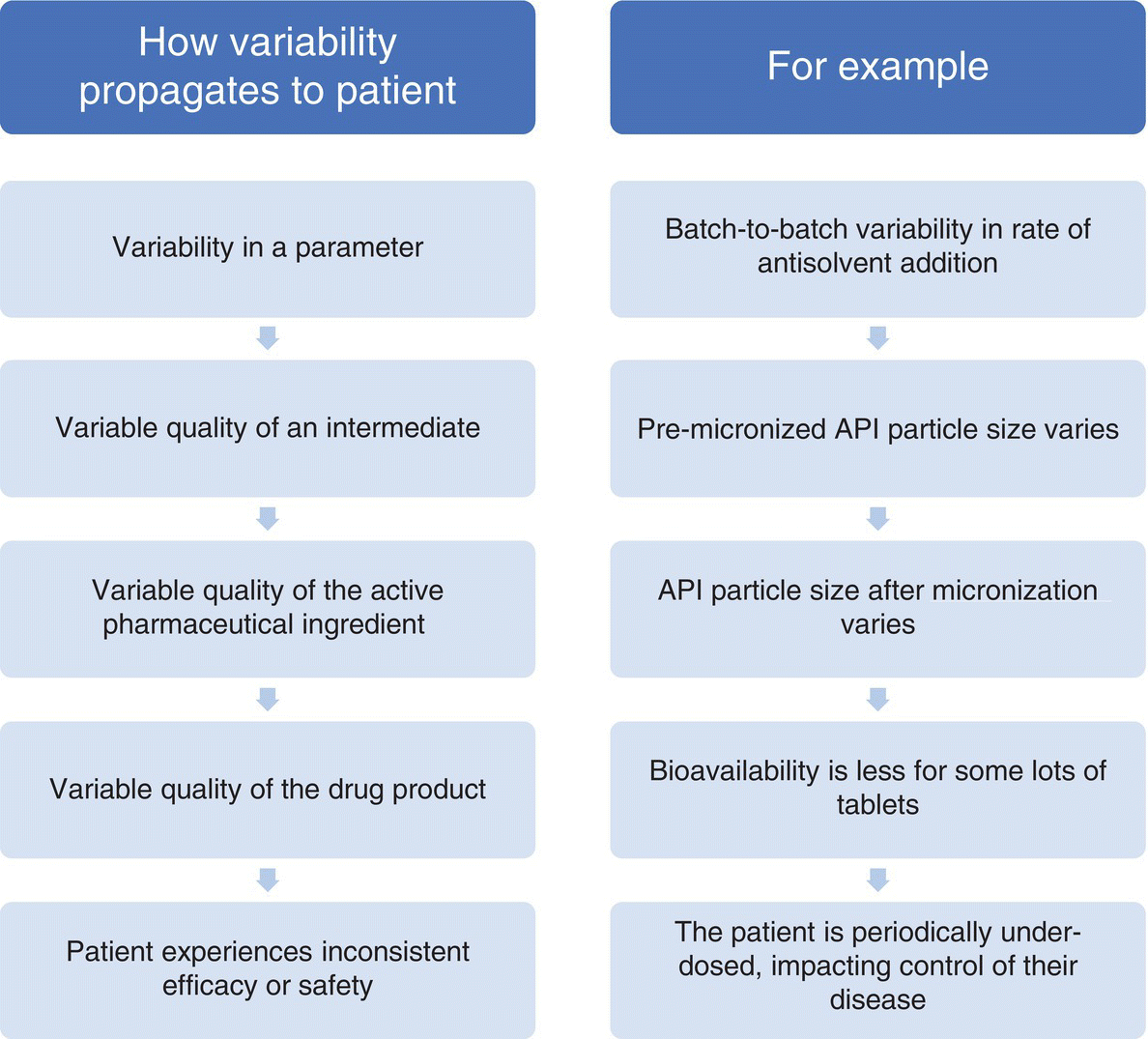
FIGURE 49.5 How upstream variability links to patient impact.
The scoring system may be based on a generic assessment of classes of parameters or attributes or may require an individual assessment of each parameter or material attribute. Inputs to consider when determining the severity score in the latter way include any univariate or multifactorial data and the range of the parameter or material attribute that has been investigated. As is the case with risk identification, risk scoring also requires the right technical input to interpret the data and produce a well‐reasoned score. This is particularly important as once a high level of process understanding has been achieved, the individual severity scores should not be reduced as a means of reducing the risk score.
Scoring the probability or occurrence of the failure mode requires an assessment of how likely it is that the failure mode will occur, either through a direct scoring of the probability of the failure mode occurring or linked to the underlying causes such as parameters varying from their target value or range. This is typically assigned a numerical probability based on the projected frequency of occurrence and may also take into consideration capability of the process or equipment to deliver within a specified range. For example, it could be assumed that the variability in process parameters fits a normal distribution, and plant capability, or normal operating range (NOR), could be defined as one standard deviation; the probability will then be scored directly based on the capability value calculated. More qualitative ways of scoring probability may take account of the type of controls in place and any historical knowledge or data of how these typically perform.
The third element of risk scoring represents detection, and this links directly to the control strategy that is being employed. There are many methods of detection, and common ones can include analysis of starting material, reagents, and solvents to determine if a batch is of the appropriate quality, the use of PAT and in‐line or off‐line analysis during processing to monitor physical and chemical changes, and analysis of the isolated material. This score may reflect either where in a process any detection of the failure mode takes place or how capable the detection method is. Some systems will incorporate both of these elements into the scoring, and emphasis is often placed on whether the detection method can enable an avoiding action to be employed or whether it is just observing, and preventing, onward processing of a batch of insufficient quality. In some cases, where the link between the cause of failure and the failure mode is well established, it may be appropriate to consider detection of the cause in addition to the failure mode as this may offer more opportunity for developing a preventative control strategy.
Following the scoring of the individual components of each risk, the information is combined to provide a risk evaluation. The output of this provides a ranking of the identified risks. The evaluation of the risks often takes one of two common approaches; if quantitative scoring has been applied in the analysis phase, then application of an appropriate formula will result in a numerical scoring of each risk. Where risks have been assessed using qualitative descriptors such as “high,” “medium,” or “low,” an alternative is to make use of a risk matrix. Once the numerical scores have been calculated for each risk, predetermined cutoff values may be applied, or risk ranking tools such as Pareto charts can be used. These enable identification of a specific proportion of the highest scoring risks. The risk matrix approach applies predefined criteria to each combination of scoring for severity, probability, and detection and results in a categorization being applied that feeds into the risk reduction and acceptance phase.
49.2.3 Risk Reduction and Acceptance
Having evaluated the risk, decisions then needs to be made to determine whether the risk will be accepted or whether actions will be required to reduce the risk to an acceptable level. A number of factors may contribute to the decision‐making process including the size of the risk, whether it is practical to implement the changes required to reduce the risk, the cost benefit analysis of making any improvements to the controls, and the potential impact that any risk may have on the patient. For those risks identified as requiring mitigating actions, the choices are the following:
- Reduce the severity of the risk; this can rarely be achieved without introducing a modification to the process as the severity often describes a fundamental relationship.
- Reduce the probability of occurrence of the risk; this is often targeted as a way of reducing risk and relies on improvements either to ways of working or to instrument or equipment capability.
- Improve the detection of the failure mode, typically by the addition of analytical controls or in some cases by moving, or improving, existing controls.
Whenever changes to the control strategy are introduced, it is important to reevaluate the risk assessment to determine if new risks have been created and if the scoring of existing risks is impacted by the change.
Some risks will be of low significance, and the decision may be to accept these risks without making further attempts to reduce them. Elimination of risk entirely is not possible, and there will come a point where it may be impractical to reduce the risk further without committing a disproportionate amount of resource.
49.2.4 Risk Communication and Review
Following a risk assessment exercise, the conclusions concerning the risks and those identified as posing the greatest risk are communicated to the stakeholders. Mitigation plans to reduce the risk and actions to manage any residual risk once the improved controls have been implemented should also be included.
Risk management plays a fundamental part in ensuring the quality of a product or process and needs to be regularly utilized during development to assess any changes and their impact on the control strategy. As the product moves from development into its product life cycle, the risk management process will continue to be used to assess the impact of both planned and unplanned changes.
49.3 RISK ASSESSMENT TYPES
As depicted in Figure 49.6, different risk assessment types and tools are used throughout the development cycle to achieve different purposes. In this section, we will discuss different types of risk assessment, and in Section 49.4 we will discuss risk assessment tools used.
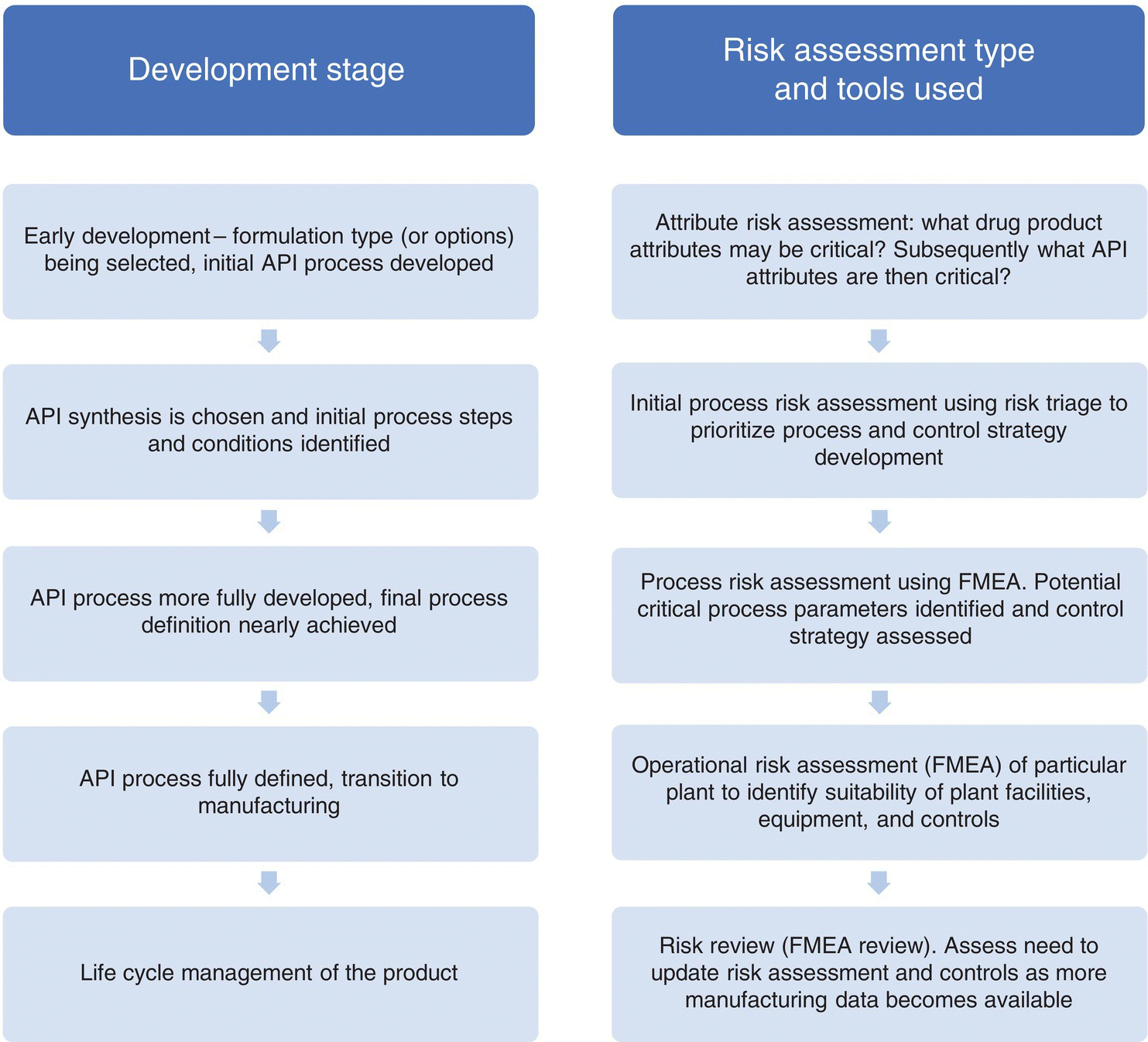
FIGURE 49.6 Risk assessment types and tools used through the development cycle.
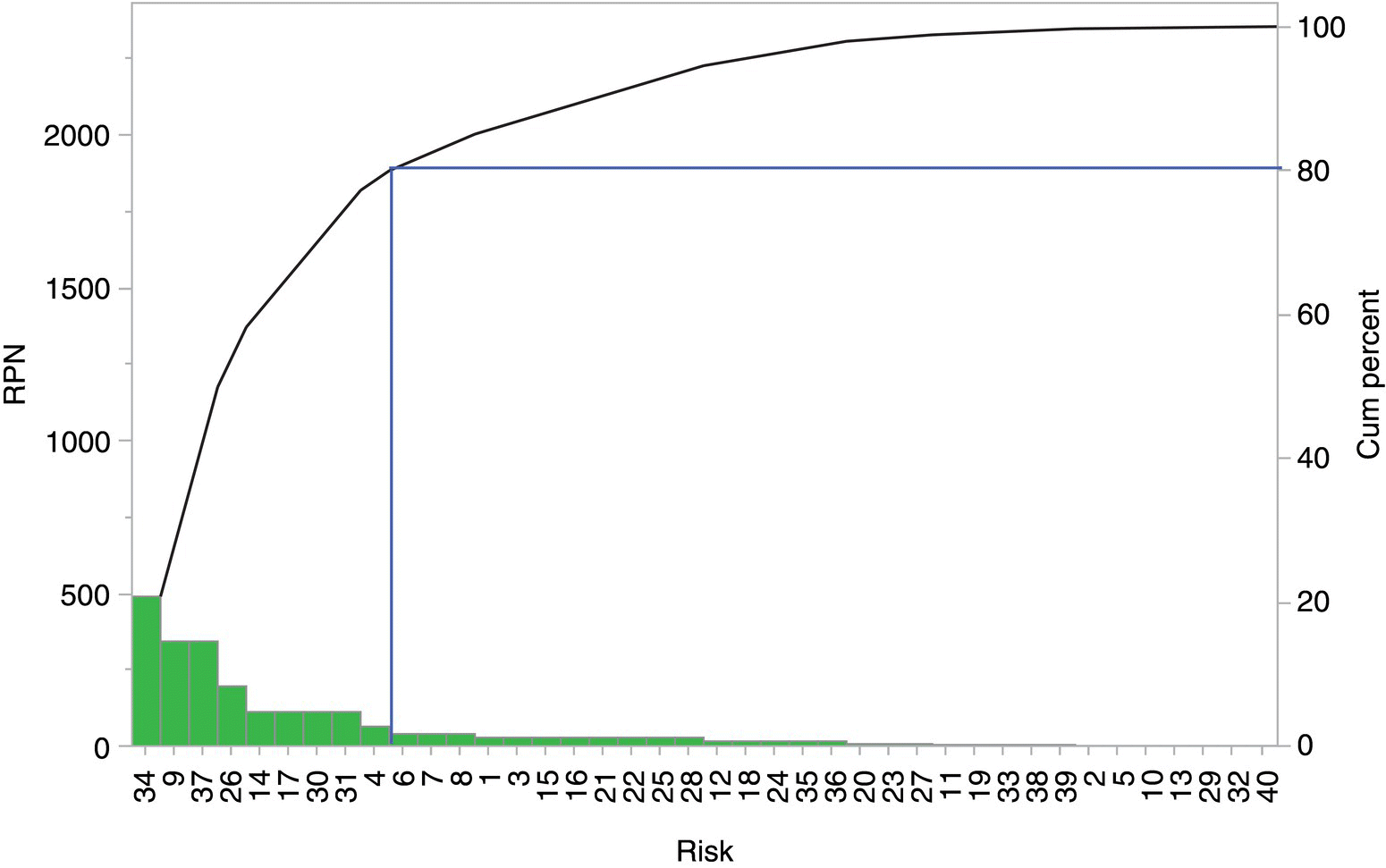
FIGURE 49.7 Example Pareto chart of the relative risk (RPN) and cumulative risk (cum. percent) against risk number ordered by risk size.
49.3.1 Attribute Risk Assessment (Defining CQAs Based on Product/Patient Risk)
As mentioned in the preceding sections, it is necessary to establish which attributes of the API are critical. ICH Q8 [1] defines CQAs as follows: “A CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.” In the case of solid oral dosage forms, CQAs are related to those attributes that impact “product purity, strength, drug release, and stability.” For other dosage forms, additional drug product attributes may be identified, and the starting point for the drug substance risk assessment is to understand the API CQAs that are known to link to these DP CQAs.
Purity is the most straightforward – if impurities are present in the API, they will be present in the drug product and are potentially impactful to the patient. In general, impurities observed in the drug substance are deemed to be CQAs of the API. More specifically, for regions following ICH guidance, non‐mutagenic impurities that are typically found in the API above the ICH Q3A reporting limits [6] are considered to impact the impurity profile of the drug substance and may be deemed as critical impurities. Health authorities require detailed knowledge of impurity formation and purge throughout the synthetic process to make the API. In the case where impurities are related to those formed in synthetic steps prior to the final API step, the precursor impurities may also be designated as critical impurities if they are not completely purged and have the potential to result in impurities in drug substance. Genotoxic impurities and known, or suspected, human carcinogens are special classes of impurities [7] that are typically always deemed critical if they can be present in the API. Typically, a limit may be applied to this, and an example would be that genotoxic impurities potentially present at, or above, 30% of the ICHM7 Threshold of Toxicological Concern are defined as CQAs. Solvents and elemental impurities used in the manufacturing process may also be defined as API CQAs. Assessment of the potential impurities should consider not only the input materials and the process but also the storage of the API. New or raised levels of impurities seen on stability should be included as API CQAs. Water present in API, if shown to increase bioburden or impact the chemical stability, may also be defined as a CQA. The strength or dose of a drug product has two aspects – the average dose and the dose uniformity (also called content uniformity). Each of these is required to be maintained within a range to ensure safe dosing of the patient. The criticality of dose and content uniform may depend on the therapeutic window of the drug (dose range that is effective and safe) and the consequences of chronic or acute under‐ or overdosing. In turn, API properties can impact the effective tablet dose (e.g. API assay) and content uniformity (e.g. particle size distribution). For example, for a low dose drug product, if the particle size distribution includes very large particles, the presence of a few large particles in a tablet can have a large influence on the percent assay of active ingredient in the tablet. In such cases, the particle size distribution can be deemed critical.
Bioavailability of the drug and dissolution performance of the drug product are other typical CQAs of the drug product. API crystal form (which impacts solubility and potentially dissolution) and particle size distribution (which impacts dissolution in most cases) may be designated as critical if they impact bioavailability and dissolution.
An example of how these and a few other aspects are used to generate an attribute risk assessment is given in Table 49.1. In this case, the risk is a simple qualitative risk assessment using expert knowledge of the drug product and drug substance synthesis to arrive at a rationale for criticality or noncriticality of the attributes. In some instances, the link between drug product CQAs and drug substance CQAs needs to be assessed through the generation of data to establish a link; an example of where this is particularly important is in the determination of API physical property CQAs.
TABLE 49.1 Example of an API Attribute Risk Assessment
| Attribute | Critical, Noncritical, or Not Applicable | Rationale |
| Identification | Critical | The patient must receive the correct active ingredient to ensure safety and efficacy |
| Purity/impurity profile | Critical | Impurities impact patient safety |
| Assay | Critical | The drug product has a narrow therapeutic window, and the correct dose is critical |
| Chiral purity | Not applicable | This drug is achiral |
| GTI/potential carcinogen | Critical | There are two Ames‐positive impurities that are potentially present in the API |
| Metals | Critical | Catalytic palladium is used in the final step of the API synthesis |
| Residual solvents | Noncritical | Only less toxic (ICH Class III [8]) solvents are used in the final synthetic steps of the API, and all solvents are reduced to a level where there is no impact to patient safety |
| KF/water content | Noncritical | Water does not impact the form or stability of the API and drug product |
| Form | Critical | Multiple polymorphs of the API have been identified and can potentially be produced in the API process |
| Particle size distribution | Critical | The drug product is very low dose (0.5 mg), and particle size can impact content uniformity |
| Color | Noncritical | The drug product is a film‐coated tablet, and API color does not impact product appearance |
49.3.2 Input Material Quality Risk Assessment
The parameters, the elements of the process that are typically described in the process definition and explored during experimentation, are often the initial focus of the risk assessment in building a comprehensive control strategy. However, consideration of parameters alone will lead to an incomplete control strategy without also considering the quality of the input materials. In an API process the main aspect of input quality comes from the impurities in the starting materials, intermediates, reagents, and solvents. When developing a drug product process or, in some instances, the latter stages of an API process, the impurities and material properties of excipients also need to be included in the risk assessment.
To enable a rigorous assessment of the impurities entering the process, data from multiple representative batches of all inputs should be assembled and then reviewed against any existing specifications. Where possible, knowledge of the synthetic route used to generate the starting materials and more complex intermediates should be obtained for the risk assessment. This data provides the starting point for assessing the probability of encountering these impurities at a level that could impact the quality of the final drug substance. In addition, an understanding of the fate of these impurities in the stage in which they are initially added to the process, and all downstream stages until they are fully purged, is required to determine the severity of impact that they may impose on the process. The ability to detect these impurities is also important to the assessment. If analytical methods are not capable, or are not in use, the risk to the patient may be increased.
With the information outlined above, it is possible to include the quality inputs directly into the same assessment as the parameters. Where there is significant complexity (i.e. numerous potential impurities), it is recommended to complete an initial triage of the impurities using risk‐based tools before combining with the other elements of the process.
Consideration of the input quality should not be limited to impurities alone. Several of the more common material attributes that also require consideration are listed below:
- Physical properties – Variation of physical properties in the inputs can play a significant role in the performance of the process and needs to be carefully considered in the risk assessment. Particle size and the polymorphic form of the input material in particular can impact the rate of dissolution that in turn can affect the rate of the reaction and relative stoichiometry, giving rise to multiple potential failure modes. For heterogeneous reactions, the particle surface area, in addition to PSD, can also impact reaction rate. For catalysts, there is also an influence of surface area and surface activity that can impact both the reaction rate and the reaction mechanism, giving rise to new impurities and varying levels of known impurities. When considering seeded crystallizations, for example, the characteristics of the seed including PSD, form, and even the mode of seed generation should be included in the risk assessment as all have the potential to influence the isolated material.
- Color – A significant number of inputs into reactions either contain colored impurities or are themselves colored when isolated. Depending on the formulation of the drug product selected, there is potential for any color introduced into the process to carry forward with the API into the drug product. In some situations, the color can be controlled through specific impurity control. In other cases, when the link is difficult to establish, the risk assessment will need to draw on data and process experience to assess the risk posed by this input material attribute.
- Bioburden – Bioburden is defined as the combination of microbiological and endotoxin contamination. In processes where bioburden in the drug substance poses a risk to patients, the potential to introduce bioburden in the input material needs to be included in the risk assessment.
49.3.3 Process Risk Assessment/Technical Risk Assessment
Once the CQAs of the drug substance are established via the attribute risk assessment, an assessment of the process or technical risks can occur. In these risk assessments, aspects of the process that may have variability (e.g. process parameters, input material quality) are examined to understand the risk posed by that variability on quality. It is typically taken as a given that the procedural steps that constitute the process will be followed per instructions and that a facility will be used that is suitable for drug substance or intermediate manufacture. The GMP systems that safeguard these aspects can be subject to their own, separate risk assessment and do not need to be included in a risk assessment of a particular process.
In preparation for a risk assessment, the background information is typically compiled in advance of a risk assessment session. This may include the synthetic scheme, a process description, a parameter table and/or Ishikawa diagram, a list of IPCs including procedures to be followed in case of failure to meet control limits, prior performance information, and information on impurities and other quality attributes such as residual solvents, metals, or crystal form. This information is used along with an understanding of the equipment and capabilities of a typical pharmaceutical manufacturing plant to perform the risk assessment. The knowledge of attribute criticality developed in the attribute risk assessment may be used to limit the risk assessment scope to only those items that impact critical attributes.
Either the top‐down or bottom‐up approach (or both), discussed in Section 49.2, may then be used to perform the risk assessment. In the top‐down approach, the individual quality attributes (either all attributes or only the critical ones) are listed, and potential causes are considered. The methods discussed in Section 49.2 (brainstorming, fishbone diagrams, etc.) may be used at this point. An example of top‐down risk identification and enumeration of potential causes is given in Table 49.2. The bottom‐up approach can be particularly effective later in the development cycle when deviations in most or all of the process parameters have been studied, and thus a highly comprehensive examination of failure modes around each is possible. An example of bottom‐up risk assessment is given in Table 49.3.
TABLE 49.2 Identified Risks and Potential Causes Using the Top‐Down Approach
| Failure Mode | Effect | Causes |
| Degradation in the reaction | Impurity A present in the API | High reaction temperature. Long reaction time |
| Dimerization in the reaction | Impurity B present in the API | Too much reagent A charged |
| Insufficient drying | Residual solvent present in the API | Short drying time, poor agitation regimen |
| Seed dissolution during crystallization | Incorrect API crystal form | Amount of seed crystals used in crystallization was too low. Excessive amount of solvent B present |
| Insufficient generation of nucleation sites | Particle size distribution too large | Antisolvent addition rate too slow |
TABLE 49.3 Identified Failures and Associated Risks of a Reaction Step Using the Bottom‐Up Approach
| Potential Failure | Risk |
| Too little solvent used | Reduced stability of the reaction stream resulting in formation of impurity A |
| Too much solvent used | No impact |
| Too little reagent A used | Competitive side reaction occurs resulting in formation of impurity B |
| Too much reagent A used | No impact |
| Low reaction temperature | Incomplete reaction resulting in residual starting material present in the downstream steps |
| High reaction temperature | Decomposition of product to impurity A |
| Reaction time too short | Incomplete reaction resulting in residual starting material present in the downstream steps |
| Reaction time too long | None, no additional side reactions anticipated |
Once the failure types and potential causes are identified, the risk associated with each can be assessed. The manner of assessment depends on the risk assessment tool being used. These tools and examples using them will be provided in Section 49.4.
A final step in the risk assessment is to describe any risk mitigations that have been, or will be, put into place to mitigate the risk and to evaluate their effectiveness in risk reduction. These controls, which can be of the various types discussed in Section 49.1, collectively can be referred to as the control strategy. The level of risk that exists after controls are put in place is known as the residual risk. In general, mitigations are put into place until the residual risk falls either into the “low risk” category or a “moderate risk, but as low as reasonably practicable (ALARP).” The ALARP concept acknowledges that with infinite resources, a risk could be driven very low; however it is not appropriate to spend excessive resources to affect a minor reduction in risk. In instances where the residual risk after controls are put in place is high, many companies will deem the risk as unacceptable and look to avoid the risk altogether, for example, by redesign of the process or use of different process chemistry or technology.
Effective application of risk management requires that the risk assessment process is regularly updated throughout development. As a greater understanding of the process is formed, and knowledge gaps are filled, the assessment of severity can be refined. Increasing experience in operation of the process leads to a better estimate of the probability of a failure mode occurring, and with evolving analytical controls, and enhanced monitoring of the process, changes to the detection scoring may also be required.
49.3.4 Manufacturing/Operational Risk Assessment
Manufacturing or operational risk assessments differ from process risk assessments in that they are generally focused on a specific plant or equipment train. The risk assessment may still be a step‐by‐step examination of the process; however the probability of failure, failure detection systems, and controls will be specific to the facility. As examples, the precision of the weigh scales used, accuracy of temperature probes and control capability of the heat transfer system, and specific alarms and interlocks could all influence the probability of failure and ability to detect and control the risk.
Often, the general process or technical risk assessment will be taken as the input to the operational risk assessment. The risk rating (if applicable, the probability and detectability ratings) can be determined for the particular plant implementation. A brief example of operational risk assessment is provided in Table 49.4.
TABLE 49.4 Example of an Operational Risk Assessment
| Failure Mode (From Process Risk Assessment) | Risk (From Process Risk Assessment) | Probability (From Process Risk Assessment) | Operational Controls | Operational Risk Assessment of Probability | Operational Risk Assessment of Detectability | Residual Risk |
| Temperature too high | Decomposition of product to form impurity A | High |
|
Medium | High detectability | Medium (ALARP) |
| Amount of reagent A too low | Side reaction occurs to form impurity B | Medium |
|
Low | Moderate detectability | Low |
49.4 RISK ASSESSMENT TOOLS
Different tools can be applied at the various stages of risk assessment, depending on the objective, the amount of information available prior to risk assessment, and the stage of development. Two relatively simple qualitative tools are risk triage and risk relationship matrices. In later‐stage development, when more development information is available, failure mode and effects analysis (FMEA) is very commonly used for thoroughly assessing process risk and determining critical process parameters (CPPs) and control strategy. Each of these tools is discussed in the following section.
49.4.1 Risk Triage/Three‐Level Qualitative Risk Assessment
The simplest method of risk assessment may be the risk triage. This qualitative risk assessment approach has the advantage of being expedient, and it results in concise, easily digestible output. The main disadvantage is that it is less thorough and nuanced than tools such as FMEA.
A typical risk triage can consider the impact or severity of the risk and the likelihood or probability. These together can be used to define an overall risk level. A scoring guide is typically used to ensure consistent risk rating between assessors. An example scoring system is given in Table 49.5.
TABLE 49.5 Example of a Qualitative Risk Assessment (Risk Triage) Scoring Guide
| Risk Rating | Impact Guide | Probability Guide |
| Low | Non‐detectable impact or detectable but not significant impact to CQAs | Not likely (occurrence rate <1 in 100 batches) |
| Medium | Significant impact to CQAs – potential to impact patient safety | Moderately likely (occurs in 1 in 10 batches to 1 in 100 batches) |
| High | Impact to CQAs with potential for severe harm to patient | Likely (more frequent than 1 in 10 batches) |
Using this guide and the example failure information from Table 49.3, Table 49.6 is an example qualitative risk assessment. In this example, four failures with moderate or high risk were identified, which necessitate the implementation of controls.
TABLE 49.6 Example Qualitative Risk Assessment
| Potential Failure | Risk | Impact Rating | Probability Rating | Overall Risk Rating |
| Too little solvent used | Reduced stability of the reaction stream resulting in formation of impurity A | High | Low | Moderate |
| Too much solvent used | No impact | Low | Low | Low |
| Too little reagent A used | Competitive side reaction occurs resulting in formation of impurity B | Moderate | Moderate | Moderate |
| Too much reagent A used | No impact | Low | Moderate | Low |
| Low reaction temperature | Incomplete reaction resulting in residual starting material present in the downstream steps | Moderate | Moderate | Moderate |
| High reaction temperature | Decomposition of product to impurity A | High | High | High |
| Reaction time too short | Incomplete reaction resulting in residual starting material present in the downstream steps | Moderate | Low | Low |
| Reaction time too long | None, no additional side reactions anticipated | Low | Low | Low |
49.4.2 Risk Relationship Matrices
Relationship matrices can be used throughout the risk management process and provide a visual way to convey the areas of potential risk that need to be considered. They are structured to enable an assessment of risk, or a known relationship, to be recorded and typically have a list of CQAs on one axis. The other axis can be populated at various levels depending on the information available and may include, for example, each stage of a process, or each unit operation, or each parameter as shown in the example in Table 49.7. The strength of relationship, or size of the risk, can be represented by a categorical or numeric input. These can be particularly useful as a quick way to brainstorm where there may be risks that need further analysis and which parts of a process present lower risk.
TABLE 49.7 Example of a Risk Relationship Matrix
| Impurity A | Genotoxic Impurity B | Form | Particle Size Distribution | |
| Reaction temperature | Strong relationship | Moderate relationship | No known relationship | No known relationship |
| Amount of reagent | No known relationship | Strong relationship | No known relationship | No known relationship |
| Crystallization temperature | Moderate relationship | No known relationship | No known relationship | Moderate relationship |
| Crystallization seed amount | No known relationship | No known relationship | Moderate relationship | Moderate relationship |
Similar matrices can also be used to represent the relationships between API CQAs and drug product CQAs.
49.4.3 Failure Mode and Effects Analysis (FMEA)
Failure mode and effects analysis is a tool that was developed in the late 1950s to study potential malfunctions of military systems. It is a highly structured and systematic method of failure analysis that is well suited to detailed assessment of pharmaceutical (and other industries') manufacturing processes. There are several types of FMEA analysis that are used in different contexts, but in this chapter, we will focus on process FMEAs. Process FMEAs are ubiquitous in pharmaceutical process development and manufacturing.
In later‐stage development, failure mode and effect analysis (FMEA) is recommended as a risk management tool given its thorough and detailed approach and broad acceptance by global health authorities. FMEA provides for a numerical evaluation of various potential failure modes and their likely effect on product performance and safety. Failure modes are identified and scored according to the mode severity, probability of occurrence, and detectability. Risk is then calculated as a product (Risk Priority Number or RPN) by
(Detectability may not always be employed as a separate step; see Section 49.4.3.3.)
49.4.3.1 Severity
Severity (S) is defined as the measure of the possible consequence of the hazard. Two different example systems for assessing severity are provided in Tables 49.8 and 49.9; one links to patient impact, and the other reflects the relationship (or lack thereof) and sensitivity to variability between the cause and the resultant effect on an API CQA. An example of this would be to determine the strength of the relationship between a process parameter, such as temperature, and the formation of an impurity. In addition, the part played by a parameter in an interaction should also be considered in the assessment of severity. Some parameters can show little or no impact when evaluated alone but have a more significant impact as part of an interaction with other parameters or attributes.
TABLE 49.8 Example Severity Levels and Terminology Based on Patient Safety Impact
| Severity Level (Score) | Potential Impact to Patient Safety |
| Disastrous (5) | Will cause permanent impairment or damage of a body structure or function. Could lead to patient death |
| Critical (4) | Could cause permanent impairment or damage to a body structure or function, but is not fatal or life threatening. Likely to impact product efficacy |
| Moderate (3) | May cause significant temporary unintended impairment of a body function. May impact product efficacy |
| Minor (2) | May cause transient, self‐limiting, unintended impact to a body function. May cause dissatisfaction to the patient and customer complaint |
| Irrelevant (1) | No performance impact to patient. May have cosmetic defect that is unlikely to cause dissatisfaction to the patient |
TABLE 49.9 Example of a Different Severity Scoring System Based on Sensitivity of CQAs to the Failure Mode
| Score | Description | Criteria |
| 1 | Not severe | The attribute or process parameter has no impact on CQAs |
| 4 | Slightly severe | Large change of this attribute or process parameter in combination with other factors has a significant impact on a CQA |
| 7 | Moderately severe | Large change of this attribute or process parameter or a small change in this attribute or parameter in combination with other factors has a significant impact on a CQA |
| 10 | Extremely severe | Small to moderate change of this attribute or process parameter has a significant impact on a CQA |
49.4.3.2 Probability
Probability (P) is defined as the likelihood of the occurrence of the failure mode. In some instances, the occurrence of the harm is distinct from the occurrence of the failure mode and is also taken into account.
Different risk assessments need to account for different magnitudes of failure frequency: drug substance and some drug product risk assessments consider failure modes that would cause an entire batch to fail, whereas device risk assessments and some other drug product risk assessments consider the failure rate of individual units in a production run. The former case has a context where only 5–20 batches may be run per year, whereas in the latter case hundreds of thousands or millions of units may be manufactured per batch. In light of these different contexts, it is possible that multiple probability scales can be defined, e.g. one for occurrence rates impacting the batch and one for rates impacting individual unit occurrence. An example of such scoring is given in Table 49.10.
TABLE 49.10 Example Probability Scoring
| Probability Term | Score | Probability (For Batch Occurrence) | Probability (For Unit Occurrence) |
| Improbable | 1 | ≤1 in 1000 | ≤1 in 100 000 |
| Remote | 2 | >1 in 1000 | >1 in 100 000 |
| Occasional | 3 | >1 in 100 | >1 in 10 000 |
| Probable | 4 | >1 in 10 | >1 in 1 000 |
| Frequent | 5 | >1 in 5 | >1 in 100 |
Where insufficient data exists to enable any numerical estimate of probability, it may be more pragmatic to consider other approaches such as comparison of NORs with proven acceptable ranges (PARs) or design space ranges or a generic assessment of individual operations to determine their inherent variability based on, for example, the level of automated versus manual operations. In these cases, the probability of occurrence may not score the same for each end of a range, and an understanding of the cause and mechanism of the failure being assessed is key to defining an appropriate probability score.
49.4.3.3 Detectability
Detectability (D) is the ability to discover or determine the existence, presence, or fact of a failure mode and provide a control action on the failure mode and/or effect. An example detectable scoring system is given in Table 49.11.
TABLE 49.11 Example Detectability Scoring System
| Detectability (D) | Ranking Score | Explanation |
| High degree of detectability | 1 | Controls will almost certainly detect potential failure modes to enable a control action, e.g. validated automated detection system that is a direct measure of failure. Two or more manual operated validated detection systems, direct or indirect (e.g. monitored control range and IPC) |
| Good detectability | 2 | Controls will have a good chance of detecting potential failure modes to enable a control action, e.g. single non‐automated, validated detection system that is a direct measure of failure (e.g. IPC of failure, validated PAT) |
| Likely to detect | 3 | Controls may detect the existence of failure to enable a control action, e.g. single manually operated validated detection system that is not a direct measure of failure (e.g. PAT measurements or IPCs not directly linked to failure) |
| Fair detectability | 4 | Controls may not detect failure modes, e.g. non‐validated (manual or automated) detection such as a visual level check, visual inspection) |
| Low or no detectability | 5 | Controls will very likely not detect the existence of potential failure mode, e.g. no detection system of any kind in use |
TABLE 49.12 RPN Matrix Example
| RPN Scores | |||||
| Severity (Severity of Failure Increases from Left to Right) | |||||
| Probability | Irrelevant (1) | Minor (2) | Moderate (3) | Critical (4) | Disastrous (5) |
| Frequent (5) | 25 | 50 | 75 | 100 | 125 |
| Probable (4) | 20 | 40 | 60 | 80 | 100 |
| Occasional (3) | 15 | 30 | 45 | 60 | 75 |
| Remote (2) | 10 | 20 | 30 | 40 | 50 |
| Improbable (1) | 5 | 10 | 15 | 20 | 25 |
A case where detectability = 5.
The use of a separate detectability element is not always mandated. For example, detectability may not be employed in early‐stage (design) assessments where detection systems have not been developed or where inherent risk is required to be assessed before corrective measures are put in place.
Where a Risk Prioritization Number (RPN) is to be calculated and detectability is not utilized, a score of 5 should be assumed for D, i.e. to reflect no separate detection measures.
49.4.3.4 RPN
The composite risk score for each risk assessed is called a Risk Priority Number (RPN) that is the product of its three individual component ratings: severity, probability, and detectability. This composite risk score can be reduced in two ways, mitigation (see Section 49.4.3) or detection.
Table 49.12 shows an example risk matrix with acceptability criteria. A “not‐acceptable” (dark shaded) composite score indicates that the anticipated failure mode is likely to produce an unacceptably severe effect on the patient or the manufacturing site. Further development to reduce the risk by redesign, mitigation, or detection is required. An “ALARP” (lightly shaded) composite score indicates that all reasonable efforts should be made to reduce the risk by mitigation (reduction of probability or severity) or detection. An “acceptable” (unshaded) composite score indicates that the risk has been reduced to acceptable levels and no further development actions are needed.
TABLE 49.13 Example FMEA Risk Assessment
| Failure Mode | Effect | Detection/Mitigation Available | Severity | Probability | Detectability | RPN | Action Plan |
| Too little solvent used | Reduced stability of the reaction stream resulting in formation of impurity A | None | 4 | 1 | 5 | 20 | Determine if additional controls can be developed |
| Too much solvent used | No impact | None | 1 | 1 | 5 | 5 | |
| Too little reagent A used | Competitive side reaction occurs resulting in formation of impurity B | None | 3 | 3 | 5 | 45 | Additional in‐process control to be developed |
| Too much reagent A used | No impact | None | 1 | 3 | 5 | 15 | |
| Low reaction temperature | Incomplete reaction resulting in residual starting material present in the downstream steps | In‐process control point (analytical test and control actions) | 3 | 3 | 1 | 9 | |
| High reaction temperature | Decomposition of product to impurity A | Temperature alarm with control action | 4 | 4 | 3 | 48 | Additional process development needed |
| Reaction time too short | Incomplete reaction resulting in residual starting material present in the downstream steps | In‐process control point (analytical test and control actions) | 3 | 2 | 1 | 6 | |
| Reaction time too long | None, no additional side reactions anticipated | None | 1 | 1 | 5 | 5 |
An alternative way to identify the areas of greatest risk is through the use of a Pareto chart. This displays the risks in size order, and then a suitable RPN cutoff can be defined that accounts for a proportion of the total risk. For example, a cutoff figure of 80% is shown in Figure 49.7. Risks that are above the cutoff are further examined to determine any additional controls that can be reasonably employed to reduce the risk. Treating risk in this way ensures that 80% of the cumulative risk is being evaluated for further action. Typical examples of mitigating actions for the detection score include additional analysis of materials prior to use, additional parametric monitoring, and the introduction of improved analytical methods. Opportunities to reduce the probability score typically come from modification in the way the process is executed, introduction of equipment with enhanced control, and modification of the parameter settings.
One additional consideration in risk assessment scoring is that at earlier stages of development, knowledge of the failure modes may be incomplete. This can be handled either by incorporating an explicit knowledge score or by defaulting the severity and/or the probability to a high level until further knowledge is obtained. The risk assessment can be repeated and finalized later in the development cycle when better information is available to assess the true risk.
49.4.3.5 FMEA Assessment Tool
With such scoring system in place, the FMEA exercise is undertaken by considering each failure mode, the effect of the failure mode, the associated severity, probability, and detectability, and the RPN number. Often an additional action plan is documented along with the risk assessment. An example FMEA risk assessment, following the risks previously given in Table 49.3, is presented in Table 49.13.
In this example, it becomes clear where additional development efforts are required to address the unacceptable or ALARP items. Another outcome of the risk assessment can be the designation of CPPs. In the above example, reagent A quantity and reaction temperature could be designated as CPPs. When taking an enhanced QbD approach to product development, identification of any CPPs in the process is required by the health authorities. For all batches manufactured, the manufacturer must track whether the CPPs have been maintained with a range that was proven to result in an acceptable outcome (PAR). Deviation of a CPP from a PAR in a given batch triggers a deviation investigation by the quality unit, and in some instances a notification to some health authorities must be provided.
49.5 RISK ASSESSMENT BEST PRACTICES
There are several common best practices for risk assessment programs that help ensure risk assessments will effectively meet the business needs of pharmaceutical development and pass regulatory scrutiny. Among these, the use of expert facilitation may be the most important. Given that risk assessment methodologies necessarily have some room for differing interpretations and flexibility in scoring, it is common for technical teams to tend toward excessive discussion of how to assess each individual item. Expert facilitators can help to move the discussion forward, can ensure a risk rating is well founded without being overanalyzed, and can also ensure consistent application of the system.
A second aspect is the composition of the team involved in the risk assessment. By bringing together members of different functional areas with different risk perspectives, the team can ensure a complete risk picture is reflected in the assessment. For example, chemistry, engineering, analytical, and manufacturing plant experts can all provide a valued perspective.
Another important aspect is to have a predetermined, clear, and aligned scoring system in place in advance of the risk assessment. Having the scoring system distributed to participants in advance, with clear guidance and examples of how severity, probability, and detectability (in the case of FMEA) are scored, minimizes unconstructive debate on the rating of the risk.
A final element is that it is very useful to have data and findings from the commercial manufacture fed back to the risk assessment teams. This allows the teams to add missed findings to the risk assessment documentation. Further, it allows those that manage the risk assessment process and facilitate the risk assessments to continuously improve the risk assessment system to ensure it accurately captures the true risk profile found in routine manufacturing.
49.6 RISK ASSESSMENTS THROUGH THE PRODUCT LIFE CYCLE
Producing a comprehensive risk assessment that supports the transfer of the process from R&D to manufacturing, in addition to the regulatory submissions, is not the end of the process. During the life cycle of the product, a regular assessment of risk and the performance of the control strategy are highly recommended. This activity should consider a review of recent batch performance, any additional laboratory data that has been generated, equipment changes, and experience of additional batches of starting material, reagent, and solvent supplies. A more comprehensive reassessment is required if post‐approval changes to the process are planned such as changing the site of manufacture or the introduction of a new processing step or different technology. If any of these changes result in the identification of a new CQA, then the whole process must be reassessed. It is also possible that additional experience gained in commercial manufacturing results in the definition of new CPPs when a rigorous risk management process is applied.
49.7 CONCLUDING REMARKS
In this chapter, we have outlined the role of QRA in QbD development, various risk assessment approaches and tools, and examples of how these tools may be applied during the development cycle. In our exemplification, we have focused on technical risks to quality, leading to a robust control strategy based on QbD principles. Many pharmaceutical companies use these or similar approaches in a structured or formal way throughout product development, and they can provide an excellent framework for in‐depth scientific discussion in addition to managing risk. A final aspect to acknowledge is that as well as such codified work practices, less formal risk assessment also has application as part of sound process development. Individual scientists, or teams, can make use of informal risk assessment to prioritize their efforts and convey risk to their management as a justification of resource spend or development timelines. Chemical engineers are often called upon to lead or perform these risk assessments, particularly in later stages of development. Thus, an understanding of risk assessment principles, terminology, and practices is fundamental to any process development engineer in the pharmaceutical industry.
REFERENCES
- 1. ICH (2009). Q8(R2): Pharmaceutical development. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf.
- 2. ICH (2005). Q9: Quality risk management. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
- 3. ICH (2012). Q11: Development and manufacture of drug substances. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf.
- 4. ICH (2008). Q10: Pharmaceutical quality system. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf.
- 5. Womack, J. (2011). Gemba Walks, 348. Lean Enterprise Institute, Inc.
- 6. ICH (2006). Q3A(R2): Impurities in New Drug Substances. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3A_R2/Step4/Q3A_R2__Guideline.pdf.
- 7. ICH (2017). M7: Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M7/M7_R1_Addendum_Step_4_2017_0331.pdf.
- 8. ICH (2018). Q3C(R7): Impurities: guideline for residual solvents. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3C/Q3C‐R7_Document_Guideline_2018_1015.pdf.