
30
MULTISCALE ASSESSMENT OF API PHYSICAL PROPERTIES IN THE CONTEXT OF MATERIALS SCIENCE TETRAHEDRON CONCEPT
Raimundo Ho, Yujin Shin, Yinshan Chen, Laura Poloni*, Shuang Chen, and Ahmad Y. Sheikh
Solid State Chemistry, AbbVie Inc., North Chicago, IL, USA
30.1 INTRODUCTION
Materials science and engineering is an interdisciplinary field focusing on the relationships between material's structure, properties, processing, and performance. The interplay between these elements of materials science is best represented as the materials science tetrahedron (MST) (Figure 30.1) [1, 2]. The ultimate goal of materials science and engineering is to enable the design and manufacture of product for a targeted performance criterion via application of engineering principles to control material properties from fundamental understanding of the structure–property relationships. Characterization, the core of MST, provides the bridge between the four interdependent elements of the MST that form the fundamental basis for engineering materials to meet target performance needs. A thorough understanding of material properties, process, and performance is an essential part of the pharmaceutical development process. Such insights are crucial to understand (i) material structure–property relationships, (ii) sensitivity of processing on material attributes, and (iii) the influence of material attributes on product performance such as safety, bioavailability, efficacy, manufacturability, stability, etc.

FIGURE 30.1 Materials science tetrahedron. The materials science paradigm depicts correlation of the structure of materials, their properties and processing, and subsequently performance of applications.
In the context of small molecule active pharmaceutical ingredient (API), the properties of materials at the bulk scale are intrinsically related to the molecular structure and its packing across multiple length scales (Figure 30.2). This multiscale structure–property relationship originates from the molecular flexibility of chemical structure that gives rise to polymorphism of the solid crystalline phase. The molecular arrangement and conformation of molecules in the crystal lattice are dictated by thermodynamic and kinetic factors, which determine the crystal properties at the single particle level. The API properties measured at the bulk level and, therefore, its bulk powder behavior are a consequence of the distribution of single‐crystal properties from each individual particulate that make up the bulk material as well as the interactions between single particles. Needless to say, API physical/chemical properties across scales can significantly influence formulation choice/composition, manufacturing route, and performance of the pharmaceutical product.

FIGURE 30.2 Multiscale relationships of API physical properties.
It remains an aspiration in the field of pharmaceutical materials science to predict the API bulk powder behavior from the fundamental structural properties of the crystalline species. The challenge lies in the complexity to link and model multilevel structure–property relationships. Although molecular modeling has been used to predict crystal properties from the crystal structure, correlating single‐crystal properties to the bulk scale is not straightforward due to crystal anisotropy and potential interparticle interactions. Despite the challenge, characterization tools are becoming increasingly refined to study material properties at different scales and can be used to develop practical structure–property–performance relationships. These advanced characterization tools provide deep understanding of structural, surface, bulk, and mechanical properties of materials and enable our understanding of the correlations between structural parameters and physical/chemical properties of APIs at different length scales, and ultimately facilitate the design of drug substance and drug product (DP) processes to achieve targeted performance criterion.
In this chapter, a broad range of characterization techniques that can be used to elucidate the structure–property–performance relationships of APIs are summarized. Additionally, two case studies are presented to demonstrate the application of the MST concept in process development and optimization of APIs to meet required performance targets. The case studies are related to (i) modeling of API powder flowability from fundamental API physical properties and (ii) impact of API intrinsic mechanical properties on process‐induced disorder.
30.2 STRUCTURAL, SURFACE, BULK, AND MECHANICAL CHARACTERIZATION TOOLS
API physical and chemical properties can be broadly classified into four major categories – structural, surface, bulk, and mechanical properties. These properties and the associated characterization techniques contribute to a comprehensive and in‐depth understanding of the APIs for process optimization and formulation development. Table 30.1 provides an overview of characterization tools for API properties and will be further discussed in this section.
TABLE 30.1 API Multiscale Characterization Tools for Systematic Assessment of API Attributes
| Solid‐State Properties | API Physical Properties | Characterization Tools |
| Structure |
|
|
| Surface/interface |
|
|
| Bulk |
|
|
| Mechanical properties |
|
|
30.2.1 Understanding Crystal Structure and Molecular Interactions
API molecule can crystallize in a number of crystal forms, including hydrates, solvates, salts, and co‐crystals (Figure 30.3). The crystal form and polymorph that is observed is dependent on thermodynamic and kinetic factors, including solvent composition, concentration of counterions or coformers, and temperature/supersaturation [3]. Many of the emergent properties of the powder, including surface, mechanical, and bulk properties, have their origins in the packing of molecules within the crystalline lattice. A thorough understanding of structural features such as hydrogen bonding network, van der Waals interactions, planar chemistry, and slip planes can therefore help understand and rationalize behavior and performance of drug substance.

FIGURE 30.3 An API molecule can have many different crystal forms and polymorphs of these forms, including (a) anhydrous forms, (b) hydrates (water molecules shown as circles), (c) solvates (solvent molecules shown as ellipsoids), (d) salt forms (counterion shown as crosses), (e) cocrystals (conformer shown as thin arrows), and (f) hydrates and solvates of salts and co‐crystals.
The packing of molecules in the solid state is a result of intermolecular interactions between constituents comprising a crystal, also known as synthonic interactions or intrinsic synthons, and includes ionic interactions, π–π stacking, hydrogen bonding, and van der Waals interactions. The strength and three‐dimensional arrangement of these interactions determine the bulk properties of the crystalline material. Intermolecular interactions in molecular crystals are typically anisotropic, leading to anisotropic properties of single crystals, e.g. direction‐dependent mechanical properties resulting from preferred slip planes for deformation and propensity for cleavage and fracture [4–6]. In a review by Reddy et al. on the structural–mechanical property correlations in molecular crystals, it was noted that polymorph with a layered structure is subjected to slipping and shearing, commonly due to the weakness of the interlayer interaction, which leads to bending and twining of the crystals [7]. Molecular crystals that show bending phenomenon occur when the packing is anisotropic in a way that strong and weak interaction patterns are in nearly perpendicular directions, and they tend to be highly plastic and ductile. In contrast, hard and brittle crystals that fracture and break down into pieces on application of stress are generally due to isotropic interactions in all three dimensions of the structure, where the interactions can be strong (H‐bonding) or weak (van der Waals).
As a single crystal grows and surfaces are formed, intrinsic synthons are terminated and exposed. The molecules associated with these “broken” intermolecular interactions, also known as extrinsic synthons, are partially unsaturated and govern many of the interfacial physical and chemical properties of a crystalline particle. The strength and arrangement of extrinsic synthons at a given crystal surface (i.e. the surface chemistry) governs the crystal growth processes. The anisotropic nature of these interactions leads to face‐specific growth rates, and the relative growth rates of different crystal faces in turn determine the crystal morphology/shape. Crystal shape can significantly affect the surface and mechanical properties of single crystals and thereby their bulk powder properties [8, 9]. For example, crystals with strong hydrogen bonding primarily along one crystallographic direction will often form a needlelike morphology with a low aspect ratio (AR). A bulk powder composed of needlelike particles will likely have poor flowability [10].
Extrinsic synthons will also govern the interactions between a crystal and its environment. Unsatisfied intermolecular interactions on a surface can be reactive in certain environments. For example, a surface with unsatisfied hydrogen bonds might preferentially adsorb moisture when exposed to air, which could be detrimental to the stability and performance of an API powder. Extrinsic synthons can affect crystal–crystal interfacial interactions in an API powder by mediating the processes that can lead to cohesion and agglomeration of particles. In a study on the sticking propensity of ibuprofen, unsatisfied hydrogen bonds due to exposed surface carboxylic acid group were found to dominate the specific surface energy and stickiness of the ibuprofen [11]. It should also be noted that the extrinsic synthons play a role in the dissolution of a given crystal surface, akin to their role in growth phenomena at the same surface [12]. Depending on the formulation of the final DP, API wettability and dissolution plays an important role in the design of the DP for drug delivery or may impact the bioavailability of the API in vivo. Extrinsic synthons significantly influence the wettability of API crystal [13]. As the hydrophobicity of extrinsic synthons increases the contact angle, it may compromise the dissolution performance of the API [9].
Overall, by understanding packing of molecules within the crystalline lattice and the molecular interactions at the surface, it can lead to rational design and production of crystalline particles with desirable shape and size distributions – properties that stem from the crystal structure of the API – for consistent physical properties of the API powder and subsequent streamlined downstream processing to manufacture the DP.
30.2.2 Surface/Interfacial Interactions and Characterization
The surface properties are known to profoundly impact interfacial behavior of materials including dissolution [14], cohesion, and adhesion [15] and a variety of processing behavior including crystallization [16], wet granulation [17], milling [18], drug‐excipient compatibility and mixing [19], coating, tableting [20] and aerolization performance of dry powder inhaler (DPI) formulations [21]. Due to anisotropy of crystalline solids, their surface properties also display direction‐dependent characteristics relative to the orientation of the crystal unit structure. Some of the commonly studied surface attributes include specific surface area, surface chemistry, surface morphology/topography, and surface energy of particles. Common methods of characterizing the surface properties of particulate pharmaceuticals rely on indirect approaches involving the use of characterized or known vapors, liquids, or solids as probes, such as inverse gas chromatography (IGC), wettability methods (such as sessile drop contact angle, Wilhelmy plate method, etc.), and atomic force microscopy (AFM), respectively.
Vapor probe techniques rely on two principal modes of vapor sorption to characterize surfaces, namely, physisorption and chemisorption, which are distinct by the nature of the intermolecular attractions. The type of adsorption on a material is strongly dependent on the interfacial intermolecular interactions, vapor pressure, and temperature. Surface adsorption models, such as Langmuir, Brunauer–Emmett–Teller (BET), and Guggenheim–Anderson–de Boer (GAB), were developed for monolayer and multilayer gas adsorption from which enthalpy of adsorption and monolayer capacity (and therefore specific surface area) can be determined. Other methods such as Kelvin's equation, Barrett–Joyner–Halenda (BJH), and Dollimore–Heal (DH) are used to model capillary condensation, where micro/meso porosity and pore size distribution can be determined.
IGC, which is a vapor probe technique, is an evolving technology in the field of pharmaceutical R&D, and its utility spans a variety of applications including batch‐to‐batch variability, solid–solid transitions, physical stability, interfacial behavior in powder processing, and more. Figure 30.4a shows the schematic setup of IGC. In IGC, probe molecules in the vapor phase (adsorbate with known properties) are passed over a sample solid (stationary phase adsorbent of interest), which is packed in a column of inert surface, via a carrier gas (mobile phase). The partition coefficient, KR, of the adsorbate between the mobile and stationary phase at a particular temperature and pressure is directly related to the net retention volume, VN, which is the volume of carrier gas required to elute the injected adsorbate through the column. Both KR and VN are indications of the interaction strength between the adsorbate and the solid sample of interest. Together, they form the fundamental bases for the derivations of surface‐specific thermodynamic parameters such as surface energies, surface glass transition temperature, free energy of adsorption, enthalpy of adsorption, electron‐donating or electron‐accepting properties, etc. When the concentration of adsorbate is increased to a level such that adsorption goes beyond Henry's law region, IGC can be used to characterize surface energy/chemistry heterogeneity, specific surface area, and bulk properties such as solubility parameters (e.g. Hansen) and interaction parameters (e.g. Flory–Huggins). An extensive review of IGC and its utility in pharmaceutical development can be found elsewhere [22].
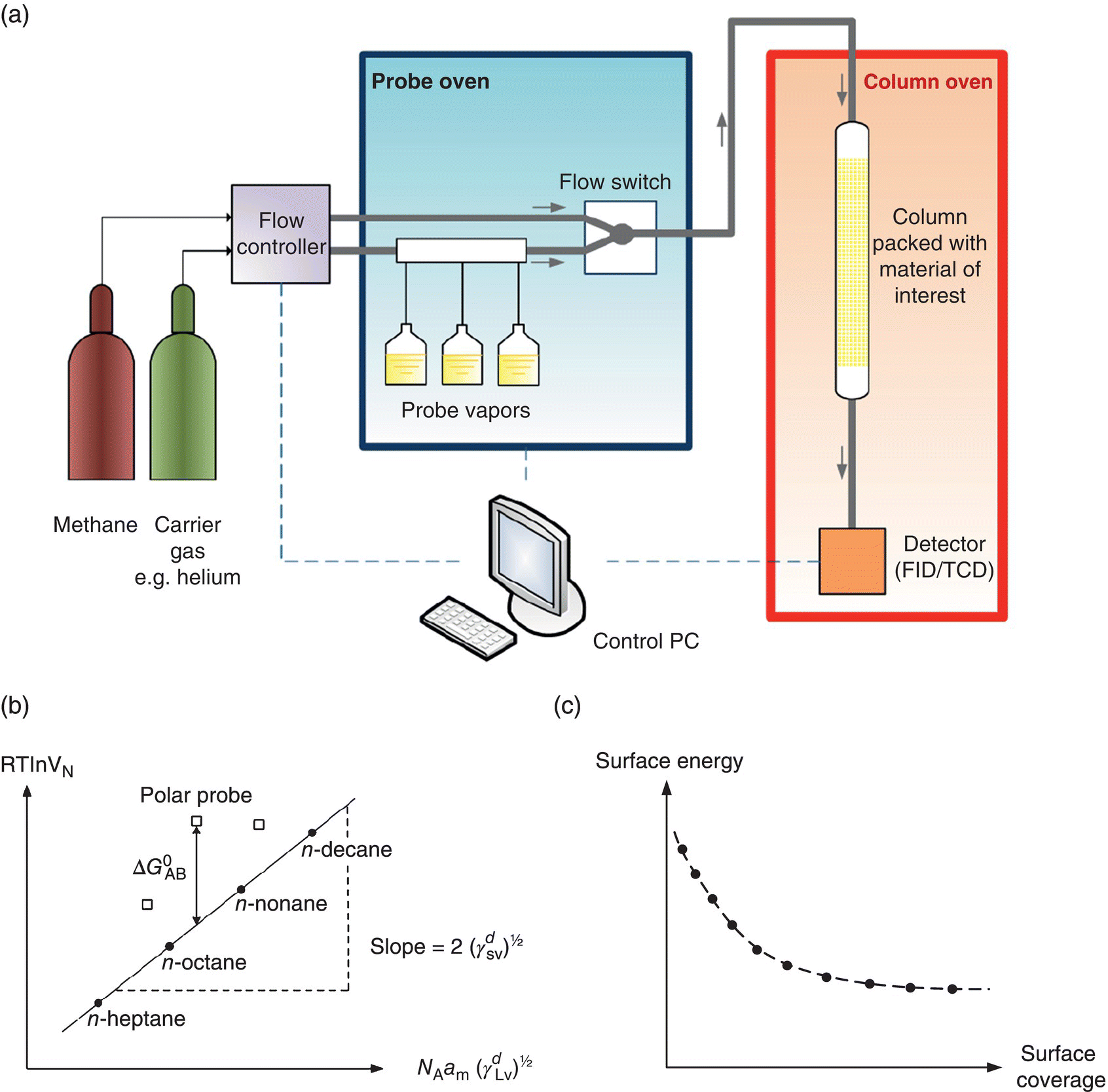
FIGURE 30.4 (a) Schematic of inverse gas chromatography. (b) Schultz approach for determination of the dispersive surface energy, γd, and specific free energy of the material (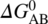 ). (c) Surface energy heterogeneity profile of the material as a function of probe surface coverage.
). (c) Surface energy heterogeneity profile of the material as a function of probe surface coverage.
For surface energetics, a common methodology employs the Schultz approach by calculating the dispersive and specific components using a series of n‐alkane and polar probes, respectively (Figure 30.4b). For n‐alkane probes, the dispersive component of the surface energy is derived from the slope of the linear regression (i.e. alkane line) of the retention volumes as a function of the dispersive liquid surface tension of the probes. The vertical distance between the polar probe points and the alkane straight line represents specific free energy (![]() , which can be used to further derive the specific or acid–base surface energy component and surface chemistry [22, 23]. In addition to surface energy determination, IGC at finite concentration can also provide heterogeneity of the surface energy (Figure 30.4c) [24, 25].
, which can be used to further derive the specific or acid–base surface energy component and surface chemistry [22, 23]. In addition to surface energy determination, IGC at finite concentration can also provide heterogeneity of the surface energy (Figure 30.4c) [24, 25].
Dynamic vapor sorption (DVS) is another vapor probe technique that provides rapid measurements of gravimetric moisture and organic vapor uptake and loss in a sample. The gravimetric solvent sorption/desorption profile of the sample can be determined as a function of time at fixed relative humidities or partial pressures. Aside from hydration and solvation studies of solid materials including hydration, solvation, and drying kinetics [26], DVS can be also used to study solid‐state reactions and phase transitions such as surface adsorption, bulk sorption, deliquescence, sorption/desorption hysteresis, crystallization of amorphous solids, glass transition temperatures, and moisture‐induced phase transformations.
Microscopy is an important tool in visualizing particle size, morphology, and surface topography of pharmaceutical materials. Their vast utility and applications are outside the scope of this chapter, but common types of microscopy in pharmaceutical R&D include optical [27], electron [28, 29], and scanning probe microscopes [30, 31]. Polarized light microscopy (PLM) is often used to verify API crystallinity as well as crystal morphology. Anisotropy in molecular crystals results in orientation‐dependent differences in the refractive index of the crystallites. When polarized light enters a molecular crystal (not incident along the optical axis of the crystal), the two components of light that are mutually perpendicular travel at different velocities, resulting in double refraction or birefringence. PLM is a simple technique to differentiate between amorphous and crystalline API and, for crystalline API, can be used to observe crystal size and morphology. Advances in confocal microscope combined with laser light sources and high‐sensitivity photomultiplier also offer new ways to study pharmaceutical systems, including phase‐separated systems, colloidal systems, tablets, and film coatings [32].
Electron microscopy is a powerful technique to study organic crystalline materials [33]. Among the many types of electron microscopy, scanning electron microscopy (SEM) is the most widely used in the pharmaceutical industry. SEM uses a focused electron beam that is raster‐scanned across the specimen, and high‐energy backscattered electrons are measured using a secondary electron detector to produce images. SEM is commonly used to observe crystal and particle morphology and size and can be combined with elemental analysis. Conventional SEM is performed under high vacuum and with high voltage, and so molecular crystalline samples typically need to be sputter coated with a conductive coating to prevent charging of the sample. Metal coatings are typically used because they are good secondary electron emitters.
AFM is a versatile technique that can be used to explore many properties of pharmaceutical materials. AFM is a type of scanning probe microscopy (SPM) and measures the interaction forces between the surface of interest (i.e. an API crystal surface) and a sharp tip mounted on a cantilever. The sharp tip (radius of curvature at the apex ~10 nm) is raster‐scanned over the sample surface, and the deflection of the cantilever is monitored using a laser that is reflected from the upper side of the cantilever onto a photodiode detector. The most common imaging modes are contact mode, in which a feedback system maintains a constant tip deflection and vertical movement of the tip enables measurement of topographical information, and tapping mode, in which the cantilever oscillates near the substrate while a feedback system maintains a constant amplitude of oscillation to report topographical information.
AFM can be performed in a sealed fluid cell, which enables the observation of API crystal growth kinetics in situ by introducing supersaturated solutions of the crystallizing solute into the fluid cell [34]. Mature faceted crystals are affixed to a substrate, and growth on crystal surfaces can be observed following introduction of the supersaturated solution. Either contact mode or tapping mode can be used to observe crystal growth in situ. This technique can be used to assess the effects of various process parameters, including solvent composition, concentration of crystallizing compound, and effect of impurities or additives [35], on crystal growth modes (i.e. spiral growth, rough growth) and relative crystal growth rates along different crystallographic directions [36].
AFM can also be used to measure mechanical properties of surfaces using force spectroscopy, in which force–distance curves are measured by monitoring the deflection of the cantilever as a function of probe–sample separation distance (Figure 30.5a and b). The appearance of the force–distance curve provides insights into the nature of the tip–sample interaction and enables the measurement of mechanical properties including adhesion, dissipation, deformation, and Young's modulus (Figure 30.5c). In particular, Young's modulus can be obtained by fitting the retraction curve (Figure 30.5c) with Derjaguin–Muller–Toporov (DMT) model by [37]

where
- Ftip is the force on the cantilever.
- Fadh is the adhesion force between the AFM tip and sample.
- R is the AFM tip radius.
- d is the deformation of the sample.
- Er is the reduced Young's modulus.
Young's modulus of the sample can then be further determined from the reduced modulus. The recent introduction of the PeakForce imaging mode has increased the ease with which force curves can be obtained and analyzed, enabling real‐time simultaneous mapping of several mechanical properties with high spatial resolution.
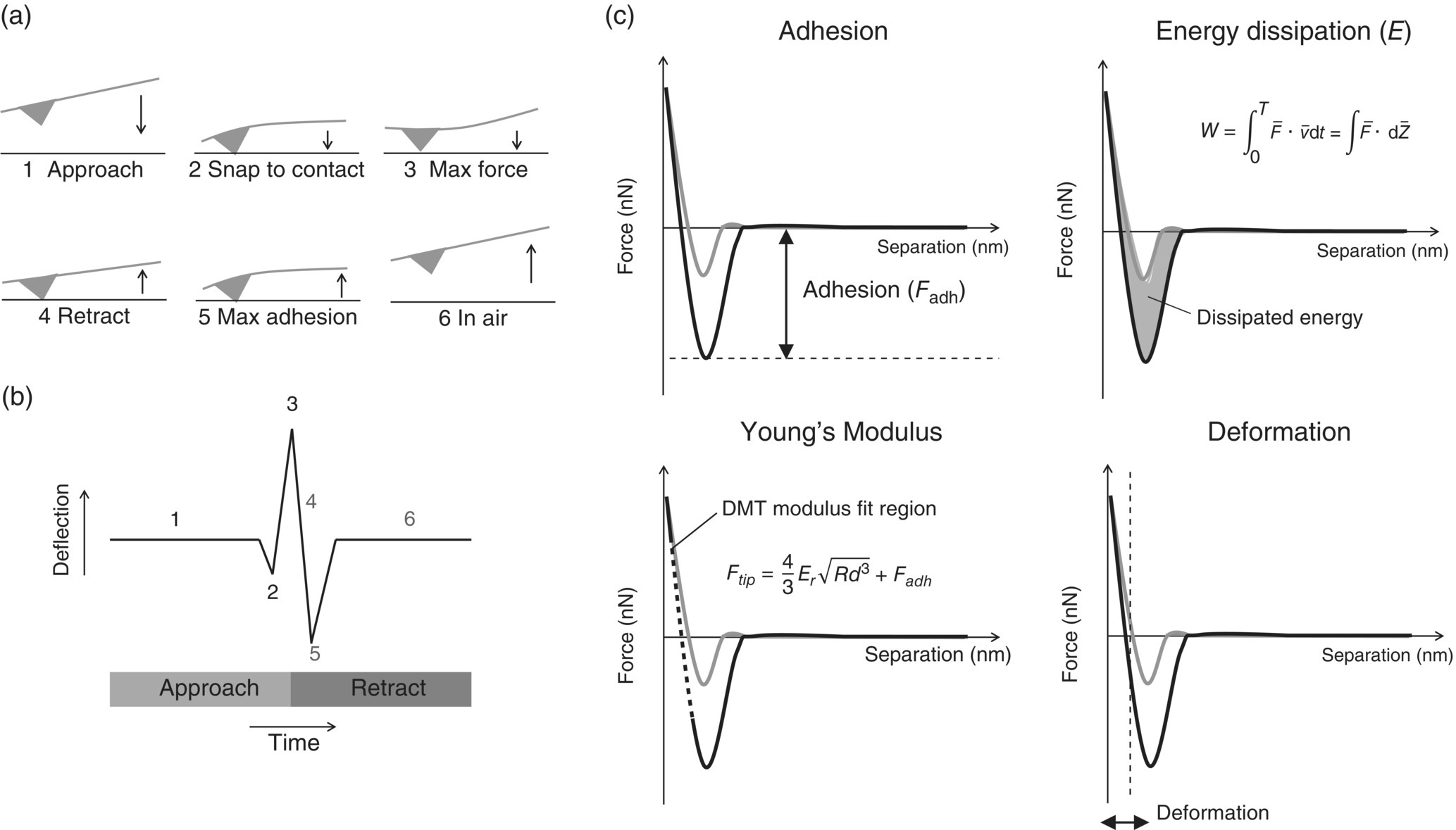
FIGURE 30.5 Force vs. distance curves obtained upon approach (gray line) and retraction (black line) of a cantilever from a surface. (a) Movement of the cantilever and (b) corresponding measured deflection, which is converted into a force vs. separation curve (c) from which mechanical properties can be determined.
Chemical force microscopy (CFM) is an application of force spectroscopy in which gold‐coated AFM tips are functionalized with alkanethiols to investigate interactions between specific functional groups and crystal surfaces [38]. For example, AFM tips can be coated in alkanethiols with terminal COO−, NH3+, OH, and CH3 groups, and tip–crystal adhesion forces can be measured on crystallographically unique crystal surfaces. These interactions can provide insights to crystal–crystal aggregation and the effects of crystal morphology on crystal agglomeration.
30.2.3 Bulk Properties and Characterization
Crystalline API particles undergo various formulation processes including but not limited to mixing, granulation, drying, blending, tableting, and coating. In many of these unit operations, API bulk physical properties are key determinants of the process performance. Therefore, API process optimization is traditionally focused on API bulk property improvements. The bulk physical properties are also key elements of the API physical property control strategy. As a consequence both from quality and regulatory perspectives, it is important to understand and monitor consistency of API bulk physical properties to ensure robust DP manufacturing based on alignment of drug substance and DP control strategies.
Among the bulk properties, particle size distributions (PSD) are considered most pertinent to ensure the final DP quality. The ICH guidelines explicitly state that particle size is one of the physical properties that influences DP processability and performance. Further regulatory guidance on setting particle size acceptance criterion is described in ICH Q6A. Laser diffraction and sieve analysis are widely used in PSD analysis. Other techniques including image analysis, sedimentation, and electrozone sensing are also used, depending on material particle size range and scope of measurement needs for the particular formulation. Since there is no single “right” way to determine PSD, considerations should be paid to (i) selection of fundamental methodology that is most suited for the targeted formulation; (ii) intent of measurements such as primary particles versus associated particles; (iii) sampling procedure, method development, and validation challenges; (iv) ease of implementation in a quality control (QC) setting; and (v) regulatory requirements. A detailed review of particle size analysis in pharmaceutical development is provided by Shekunov et al. [39].
In laser diffraction, the angular intensity of the scattered light from a particle stream is often dispersed by means of dry air or a liquid medium. The accumulated light diffraction data is then analyzed to calculate the size of the particles using an optical model, such as Mie theory, which generally assumes particles are spherical in shape, are nonporous, and have consistent true density. In recent years, particle shape distribution analysis has also become a routine characterization during DS development [40, 41]. Image analysis can provide rapid and accurate particle size and shape distributions for dry powders and wet dispersions. Image analysis can be categorized into static and dynamic image analysis (DIA). Static techniques generally utilized microscopy equipped with motorized stage that moves stepwise to capture images of dispersed particles. DIA uses a pulse light source to illuminate dry or wet dispersions that are streamed continuously through the measuring zone and captured by high‐speed camera. Based on the two‐dimensional (2D) images of particles, a number of particle size and shape factors including circular equivalent diameter, Feret diameters, circularity, solidarity, area, aspect ratio, etc. can be quantified for nonspherical‐shaped particles most commonly encountered for the APIs. In the static techniques, the preferred orientation (e.g. the longest dimensions) can be easily captured because particles are typically deposited on the microscope stage, whereas dynamic techniques allow more random particle orientations and a statistically larger number of particles can be analyzed quickly. In many cases, particle size and shape distributions can also indirectly capture bulk powder attributes, such as flowability. This topic is the focus of the first case study.
Other bulk properties that are routinely characterized for APIs are true/apparent density, bulk density, and tapped density. True density is the density of solid material excluding any open and closed pores and can be calculated from the lattice parameters for a given crystal form. It can be measured to high accuracy by gas pycnometry. When the materials contain inaccessible pores, the measured density can be referred as apparent density. The bulk density, ρbulk, is the ratio of the untapped powder sample and its volume including contribution of interparticulate void volume. The tapped density, ρtapped, presents the increased bulk density attained after the sample bed has reached a consolidated state by mechanical tapping. The compressibility index, (ρtapped − ρbulk)/ρtapped, and Hausner ratio, ρtapped/ρbulk, are also frequently used to characterize powder compressibility.
30.2.4 Mechanical Properties and Characterization
During the manufacturing of drug substance and DP, crystalline API particles undergo various formulation processes including milling, blending, tableting, etc., which can subject the API to mechanical stresses of varying magnitudes. The mechanical response of API particle under such external stress can originate from the intrinsic mechanical properties of the isolated primary crystalline particles and their associated structures. Fundamental understanding of the intrinsic mechanical properties facilitates the design, optimization, and control of relevant unit operations.
Under external mechanical stress, crystalline organic solid undergoes elastic deformation and then plastic deformation, although some excipients, such as starch and microcrystalline cellulose and amorphous solids, may also exhibit viscoelastic properties. During elastic deformation, the particle returns to its original shape once the applied force is removed. Young's modulus (E) is a measure of the resistance to elastic deformation. During plastic deformation, the particle experiences a permanent deformation by gliding, twining, and kinking of molecular layers. Hardness (H) is the measure of resistance to plastic deformation. When the strain is high enough, fracture intervenes, and a material experiences a failure into pieces. Fracture toughness (KIC) is a parameter to measure the resistance to propagation of a fracture. This parameter is related to the brittleness of a material. Brittleness index (BI), which is defined as H/KIC, is used to describe the overall brittleness of a material. In the full strain–stress curve, tensile strength is typically defined as the stress at which a material begins to deform plastically or develops macroscopic damage. In addition to nano/microscale elastic and plastic deformation, irreversible defects/imperfections at a molecular level can also occur under mechanical stress. Total diffraction analysis, where both the Bragg and diffuse scattering elements of the data are thoroughly assessed, provides an avenue to assess these molecular level imperfections in bulk solids at angstrom‐level resolution. The latter can be further improved by using diffraction data from low wavelength synchrotron‐based radiation sources to better understand the impact of processing on local‐, short‐ and long‐range order in pharmaceutical solids. The influence of structural and mechanical properties of APIs in process‐induced defects is the subject of the second case study.
30.2.4.1 Micro/Macroscale Mechanical Assessment
Micro‐indentation and macro‐indentation are used to characterize the E, H, KIC, and brittleness of macroscopic particles, such as agglomerates and granules. Such characterization is important in understanding the processing and the influence on consolidation, extrusion, and attrition of particles [42].
The strength of a granule can be interpreted as its resistance to permanent deformation and fracture during a stressing event, and it is normal to consider granule strength as the maximum stress value of the granule before fracture occurs [43]. Granules are not continuum solid particles; instead they are clusters of small particles held together by interparticle bonds. Stress applied at a local point may cause the granule to shear apart at the point of application before the load is transmitted to the rest of the solid volume. The strength of a granule is, therefore, determined by the nature and concentration of its internal bonds and granule microstructure. Apart from the tensile strength, mechanical properties of granular materials can also be described by Young's modulus, Poisson's ratio, tensile yield stress, shear strength, fracture toughness, and hardness [43, 44], which can be measured by various techniques such as confined compression tests, indentation, shearing tests, three‐point bending test, single particle compression, and wall impact tests depending on the choice of the mechanical parameter one wish to obtain.
Texture analyzer is an emerging technique in studying mechanical properties at macroscale that are suitable for both crystals and granular materials. It achieves multifunctional applications by taking advantages of the versatility of the indenters. In a typical measurement, as the indenter approaches and withdraws from the specimen, the force is monitored against travel distance in either compression or tensile testings. Similar to three‐point bending method, when a tablet is compressed, the breaking force measured can be used to characterize the tensile strength of APIs from the compactibility plot (i.e. tensile strength at zero porosity) [45]. Under confined uniaxial compression, mechanical strength of agglomerates can be characterized. When agglomerated powder is compressed in the uniaxial bed, the force–displacement data are measured. According to Kawakita's model, agglomerate strength can be extrapolated from the fitting of volume–pressure relationship [46]. If the single agglomerate strength needs to be assessed, Adam's model could be applied to extrapolate the agglomerate strength based on the fitting of pressure–strain relationship [47]. Measured strength reflects the initial cracking of agglomerates. Together with the macroscopic mechanical properties mentioned above, agglomerate strength can be used to predict the tableting behavior and impact of agglomerated particles on downstream processes and product performance [42].
Additional dynamic testing of API can be done using hydraulic pressing, single station tablet press, compaction simulator, etc. From the compression profile of solid fraction‐compaction pressure, yield strength of material under the dynamic condition can be determined as a measure of deformability [48]. Other measured information like compatibility, tabletability, and compressibility are significantly applied in understanding and process design of API and DP.
30.2.4.2 Nanoscale Mechanical Assessment
Quasi‐static nano‐indentation has emerged as an effective material sparing technique to characterize the mechanical properties of molecular crystals. In the measurement, the applied load and the corresponding depth of the penetration of the indenter are continuously monitored. When the indenter is withdrawn, the unloading displacement is also monitored until the zero load is reached and a final penetration depth is measured. From the loading–unloading curve, E and H can be easily calculated. Nano‐indentation has also been applied to measure KIC of molecular crystals. The method is based on the radial cracking when brittle material is indented with a sharp indenter. KIC can be calculated from a semiempirical relation of peak load, E, H, and the length of the radial cracks. These intrinsic properties measured by nano‐indentation have been widely applied in the study of milling, and various practical performance‐indicating models have been developed. In Vogel and Peukert's model of breakage probability, the rate of particle breakage during milling is described as a function of E, crack propagation velocity, fracture energy (KIC), elastic energy wave propagation, particle size, and flaw size [49–51]. A similar model derived by de Vegt et al. shows that the breakage rate is a function of E, H, and KIC [52]. These models have been applied and validated by the quantitative nano‐indentation measurement of E, H, KIC, and H/KIC in Meier et al.'s experimental study [53]. In Taylor et al.'s study, an elegant empirical correlation has been derived between milling effectiveness (% size reduction ratio) and BI (H/KIC) measured with nano‐indentation by studying five model drug systems [54]. Nano‐indentation is also used to study the mechanical anisotropic properties and long‐range molecular/layer migration (e.g. slip plane) of organic crystals [55, 56]. By measuring the slip plane, the fracture behavior and plasticity of the crystal can be predicted. The information is used to investigate the tableting performance of the crystal particles [57]. Finally, nano‐indentation equipped with a lateral force transducer can also be extended to characterize friction and wear. Besides, cutting‐edge techniques, such as the AFM, can also greatly broaden the scope in the assessment of material intrinsic mechanical properties. For example, AFM can achieve mapping of mechanical properties across the surface in nanoscale. It is possible to study the heterogeneity of mechanical properties, such as stiffness and elastic modulus, across a surface at previously unattainable resolutions. When choosing between nano‐indentation equipment for intrinsic mechanical property characterization, factors such as the nature of material (i.e. hardness and its response to force), dynamic force range, load and displacement sensitivity, and the need for high resolution imaging should be considered for the desired application. For example, specialized nano‐indentation device with a maximum force load in the mN range may be more suitable for pharmaceutical organic crystals, whereas the AFM nano‐indentation, due to its tip size (usually <10 nm) and force range, may be more applicable to mechanical characterization of surface layers and for conducting high resolution imaging.
30.2.4.3 Molecular‐Level Assessment of Process‐Induced Defects
From a biopharmaceutical perspective, smaller particle size is desirable, because it leads to an increase in specific surface area and surface to volume ratio, thereby improving dissolution rate. Improvements in dissolution rates can improve bioavailability when the overall rates are not permeability limited. Particle size reduction processes are therefore often employed during API production, such as wet milling, screen milling, and impact milling. The milling processes provide small PSD but can also generate surface‐ or structural‐level defects in the particles. These process‐induced imperfections can manifest as increased chemical reactivity, loss of crystallinity/physical stability, increase in hygroscopicity, loss of mechanical integrity, and increased propensity for aggregation and agglomeration. Many of these consequences negatively impact downstream processing performance. The defects generated by milling processes can be localized and are not easy to detect with conventional characterization techniques.
A high‐energy powder X‐ray diffraction (PXRD) method known as total scattering (TS) analysis is a powerful tool that utilizes Bragg and diffuse scattering content of diffraction data to establish a relationship between the real‐space arrangement of atoms and X‐ray diffraction intensity through the atomic pair distribution function (PDF). When the data is collected at low wavelength over wide diffraction angles, atomic distance resolutions of less than 0.3 Å can be achieved. Such resolution is ideally suited to study existence and emergence of local, short, and long order/disorder in pharmaceutical solids. This structural information is obtained by mathematically treating the TS data to obtain PDF. The pair distribution function, G(r), provides information on local molecular packing arrangement by giving the number of atoms in a spherical shell of unit thickness at a distance r from any reference atom. An advantage of PDF G(r) analysis is that the amplitude of the oscillations in PDF plot gives a direct measure of the structural coherence of the sample. In crystalline samples with some degree of disorder, the amplitude of G(r) falls off faster than dictated by the resolution of scattered wave factor Q (Q = 4π sin(θ)/λ). As the positions of the PDF peaks give the separations of pairs of atoms in the structure directly and the width and shape of the PDF peaks contain information about the real atomic probability distribution, the combined assessment of amplitude and peak position/shape therefore provides a complete understanding of local, short‐ and long‐range order/disorder in materials.
30.3 MODELING POWDER FLOWABILITY FROM FUNDAMENTAL PHYSICAL PROPERTIES
30.3.1 Introduction
The powder flow properties of API are very important for high drug loading formulations because they affect downstream processing, such as mixing/blending, capsule and die filling, hopper transfer, fluidization, and coating [58]. Optimization of bulk powder flowability therefore becomes a fairly routine objective during the development of API crystallization and isolation processes. In the practical industrial application, powder flowability is determined by the driving force relative to the flow constraints [59]. The driving force is provided by the unit operation arising from agitation, pressure, shear, gravitational forces, etc. The flow constraints include both extrinsic constraints, which are defined by physical volumes and stresses (e.g. hopper dimension, consolidation stress due to weight of materials, etc.), and intrinsic constraints, which are related to the fundamental material characteristics and properties. Processing challenges related to powder flow can often be overcome by increasing the driving force or by equipment design and selection to relieve extrinsic constraints. However, by understanding the intrinsic material properties that affect powder flowability, crystal engineering solutions can be chosen and tailored to maximize target flow property performance criterion that is dominated by intrinsic constraints of powder flow.
Many examples of modeling powder flowability from particle size and shape distributions have been reported [60–63]. In our case study, we present a rigorous modeling approach based on partial least squares regression (PLS) to predict powder flowability from comprehensive particle physical properties covering both bulk powder properties and surface properties. The latter addresses very important aspects related to interparticle cohesion. The surface properties are important contributors to powder flowability in the context of nongranular materials such as API, as they often exhibit higher surface area to volume ratios due to their relatively smaller particle size compared to commonly used pharmaceutical excipients. The main purpose of this study is to establish and gain fundamental insights into the key material characteristics that would give rise to better powder flowability for more efficient API process development.
30.3.2 Physical Properties that Impact Powder Flowability
Eight APIs and seven excipients with various particle size and shape distributions were used in this study. The flow behavior of each individual material was quantified using the flow function coefficient ffc (preconsolidated at 1 kPa) obtained from shear cell tests (ring shear tester, Dietmar Schulze, Germany). The ffc, which is the ratio of consolidation stress to unconfined yield strength, is a commonly used flowability performance indicator in drug process development [58]. Materials with better flow performance are associated with higher ffc. The ffc of the model materials were modeled by PLS regression with their bulk and surface properties as tabulated in Table 30.2. The particle size and shape distributions were obtained using DIA (QICPIC, Sympatec, Germany) operated with a wet dispersion system. In DIA, the particle size is characterized by the circular equivalent diameter (CED or D), which is calculated by taking the 2D projected area of the particle image and then calculating the diameter of a circle with the same area. The aspect ratio (AR) is calculated by dividing the minimum Feret's diameter by the maximum Feret's diameter taken perpendicular to the minimum. The circularity (C) is calculated from the ratio of the CED perimeter to the perimeter of the projected area. The solidarity (S), which also describes the macroscale roughness of the particle, is calculated by dividing the projected area by the area of the particle created by smoothing edges and filling in voids. The volume‐based (v) size and shape factors were used in this case study. The surface properties of the model compounds were characterized by their dispersive (van der Waals) surface energies, γd, and acid–base surface energies, γab, using IGC (Surface Energy Analyzer, Surface Measurement Systems, United Kingdom). The total surface energy, γTOTAL, is the summation of both dispersive and acid–base components. The dispersive surface energies were measured following the Schultz method using decane, nonane, octane, and heptane as probes, and the acid–base surface energies were calculated using monopolar probes, ethyl acetate and dichloromethane [22]. The surface energies were measured at a probe surface coverage of 5%, a background RH of 32%, and a temperature of 30 °C. All samples were stored at approximately 32% RH in a controlled humidity chamber for at least seven days before characterization.
TABLE 30.2 Physical Properties That Impact Powder Flowability
| Material Physical Properties | |||
| Parameter | Definition | Equation | Illustration |
| CED (Dv) | Diameter of a circle with equivalent projected area |  |
|
| Aspect ratio (ARv) | Feret min divided by Feret max |  |
|
| Circularity (Cv) | CED perimeter divided by real perimeter |  |
|
| Solidarity (macro‐roughness) (Sv) | The object area divided by the area enclosed by the convex hull |  |
|
| Surface energies | Surface van der Waals and acid–base interactions | γTOTAL = γd + γab | |
| Flowability Performance Indicator | |||
| Parameter | Definition | Equation | |
| Flow function coefficient (ffc) | Ratio of major principal stress and unconfined yield strength/stress at a defined normal load | ||
30.3.3 Relationships Between Powder Flowability and Physical Properties
The model APIs and excipients exhibited a broad range of particle size and shape distributions as shown in Figure 30.6, with Dv50 (median volume‐based CED) in the range of 21–301 μm. The morphologies of the model materials include acicular, columnar, blade, plate, irregular, and granular morphologies with ARv50 (median volume‐based aspect ratio) in the range of 0.21–0.75. The total surface energies varied from 42.5 mJ/m2 (low‐energy materials) to 77.6 mJ/m2 (representing highly energetic and cohesive organic solids), with γd ranging between 33.1 and 61.9 mJ/m2 and γab between 3.8 and 15.7 mJ/m2. The range of ffc varied between 1.4 (representing very cohesive, poor flowing material) and 12 (representing free flowing material).

FIGURE 30.6 SEM images of model APIs and excipients.
The relationships between particle size, shape, surface energies, and ffc of the compounds are shown in Figure 30.7. Several observations are noted as follows:
- As the particle size (CED, Dv50) increased, ffc improved.
- At a fixed particle size, increasing particle shape factors (ARv50, Cv50, and Sv50) greatly enhanced ffc. As can be seen from the changes in the contour lines, the improvement in ffc became more substantial above a threshold shape factor. For instance, when the ARv50 increased beyond 0.60, the ffc improved significantly.
- Reducing total surface energy had minor influence on ffc, unless the surface energies were substantially low.
Through general inspection of the raw data in Figure 30.7, it is therefore clear that higher ffc are correlated to increase in particle size, shape factors, and reduction in surface energies.
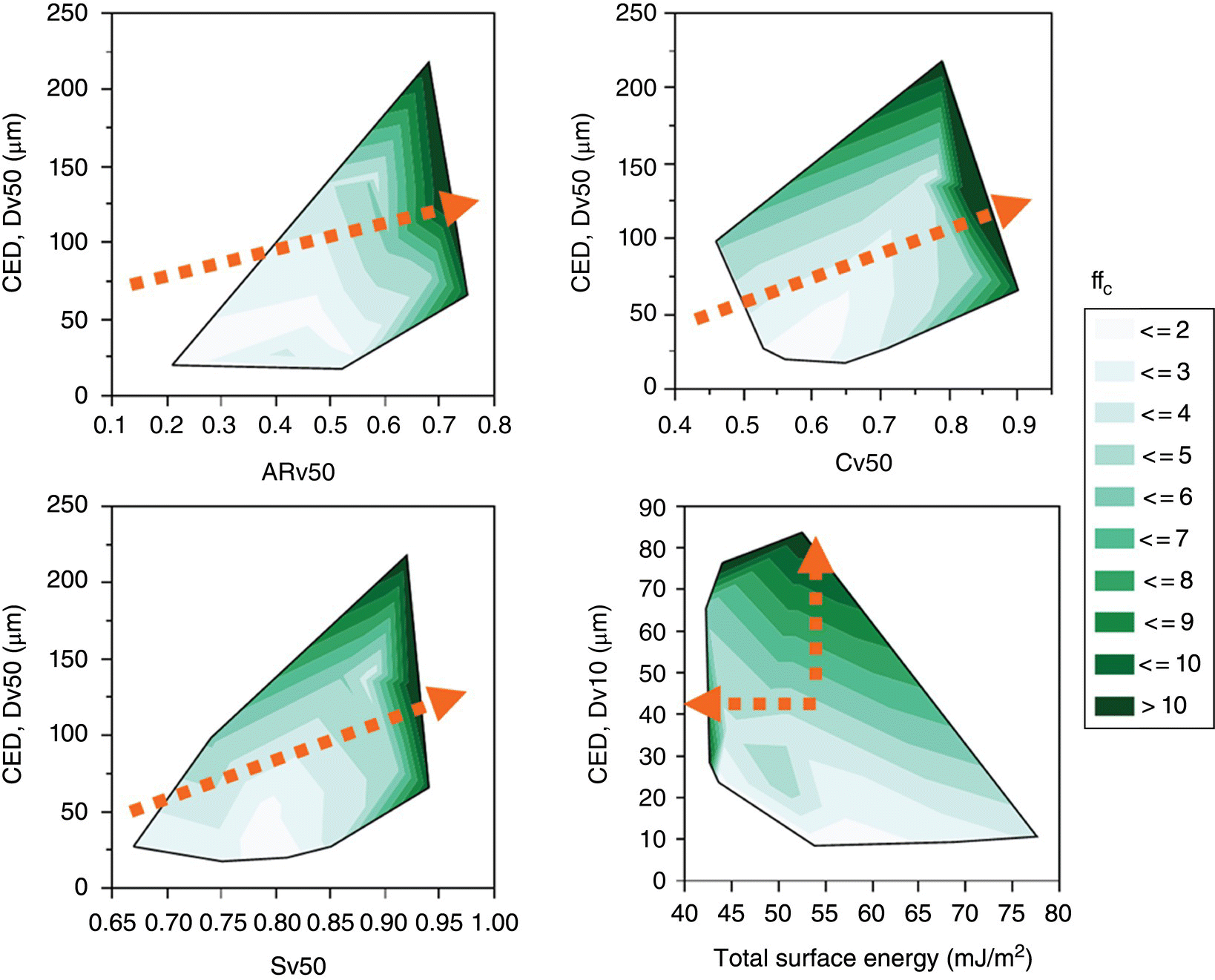
FIGURE 30.7 Contour plots showing relationships between median particle size, median shape factors, total surface energies, and ffc. Arrow indicates the direction of significant ffc improvement.
30.3.4 Partial Least Squares Modeling
In order to further establish the influence of particle size, shape, and surface energies on powder flowability, a PLS model was constructed to link the input variables X (material properties) to the model response Y (ffc). PLS regression can effectively reduce high‐dimensional data matrices into low‐dimensional subspace and provide good estimates for model response. In order to capture the particle size and shape distributions as input variables, the 10th, 50th, and 90th percentiles of the CED, AR, C, and S were selected together with the two surface energy components, γd and γab. The input variables and responses were first centered and scaled to have mean of 0 and standard deviation of 1 to prevent data bias due to range of the magnitude of their numerical values. The model compounds were divided into two subsets of data: a model training set with a total of 11 compounds and a validation data set with a total of 4 compounds. The PLS model was constructed using data from the training set and then validated with the validation data set. The optimum number of latent factors was chosen based on cross‐validation method by minimizing root mean predicted residual sum of squares (PRESS) while maximizing percent variation explained for X and Y. The final PLS showed both excellent X and Y variations explained (94.7 and 95.0%, respectively) and low root mean PRESS (1.31).
Figure 30.8 shows the measured ffc versus predicted ffc from the PLS model covering both the training and validation data set. It can be seen that the final PLS model had an excellent predictability on ffc across the range of compounds studied. The model coefficient and variable importance in projection (VIP) score for each input variable X are tabulated in Table 30.3. The VIP score is a measure of a variable's importance for projecting the data to the X–Y scores. If a variable has a small coefficient and a VIP with a threshold value of less than 0.8, then it can be considered as a candidate for removal from the model. It can be seen from Table 30.3 that most variables have VIP scores larger than the threshold value except for γd, Cv90, and Sv90. Their relatively low VIP scores were likely due to the fact that the variation in these variables for the model compounds was substantially lower than other variables. For instance, organic crystalline solids have very similar dispersive surface energy component because of their similar London, Debye, and Keesom interactions but widely different acid–base surface energy due to vast differences in the surface propensity for hydrogen bonding, coulombic, and other acid–base interactions. According to the model coefficients, the most significant contributor to ffc was Dv10, followed by ARv10, γab, and Dv90 (excluding those variables with lower than threshold VIP scores). It is known from previous studies that increasing particle size would increase ffc [60–63], but our model also showed that reducing volume percent of small particles or fines (increase in Dv10) and a reduction of the span of the PSD (as defined by difference of Dv90 and Dv10 divided by Dv50) would also promote an increase in powder flowability. In terms of shape factors, the most significant contributor to ffc came from particles that were most acicular or needlelike (low ARv10). Reducing the volume of these particles significantly contributed to the increase in powder flowability. In fact, our model showed that an increase in shape factors (ARv, Sv, or Cv) would generally increase the ffc. This means that particles are preferably more spherical, non‐needlelike, and macroscopically smooth to minimize frictional effects and mechanical interlocking that are detrimental to powder flow. In the context of surface properties, the model revealed that a reduction in γTOTAL would generally lead to increase in ffc, when a single γTOTAL was used as an input variable rather than the surface energy components. In addition, when considering the surface chemistry, an increase in surface hydrophilicity (increase in γab and decrease in γd) would enhance powder flowability.

FIGURE 30.8 Measured versus predicted ffc by the PLS model (training data set are black circles; validation data set are empty circles).
TABLE 30.3 Model Coefficients and VIPs for Centered and Scaled Data
| Model Parameters | Coefficients | VIP |
| Intercept | 0.00 | |
| CED, Dv10 | 0.56 | 1.163 |
| CED, Dv50 | 0.05 | 0.968 |
| CED, Dv90 | −0.25 | 0.863 |
| Dispersive surface energy | −0.13 | 0.706 |
| Acid–base surface energy | 0.26 | 0.897 |
| ARv10 | 0.37 | 1.268 |
| ARv50 | 0.11 | 1.165 |
| ARv90 | −0.22 | 1.112 |
| Cv10 | 0.09 | 1.065 |
| Cv50 | −0.16 | 0.956 |
| Cv90 | 0.29 | 0.747 |
| Sv10 | 0.15 | 1.160 |
| Sv50 | 0.06 | 1.009 |
| Sv90 | 0.09 | 0.703 |
30.3.5 Key Insights to API Process Development to Optimize Flow
From the PLS model, it is obvious that both bulk and surface properties of particles are intrinsically related to powder flowability. In our study, several key insights to API process development in the context of powder flow optimization can be drawn (Table 30.4):
- The elimination of fines should be the primary objective as shown by the PLS model. A reduction of fines would significantly improve powder flowability even when it is difficult to promote median particle size increase. This could be achieved, for instance, by adding heat–cool cycles at the appropriate stage in the crystallization process to promote Oswald ripening for systems which display temperature-dependent solubility. Careful selection of the milling equipment and design of operation based on the breakage behavior of the API particles can also be used to reduce fines generation and negative consequences for flow.
- While increasing particle size can improve flowability, consideration should also be given to the shape of the PSD with the objective of achieving more uniform PSD. The use of wet milling in conjunction with heat–cool cycles described above is generally applicable solutions to tighten the span of size distribution. Continuous crystallization offers a distinct avenue toward tightening size distribution and achieving consistency.
- Spherical, non‐acicular, and macroscopically smooth particles exhibit better flowability, but reducing the amount of the longest needles would also enhance flow. Understanding the impact of solvent and supersaturation on crystal shape can help design crystallization processes that intrinsically provide optimum crystal shape for the selected polymorphic form. A combination of in silico and in vitro screening experiments along with direct mechanistic understanding of relative growth rates and mechanism, for instance, via in situ AFM, can be used to build high level of confidence in the scalability and consistency of the shape control. Process‐focused optimization techniques mentioned above, including in situ wet milling combined with heat–cool cycles during crystallization, can help further optimize crystal morphology.
- Both surface energies and surface chemistry were shown to play a role in powder flowability. API becomes more cohesive and exhibits higher surface energies primarily due to exposed surface functional groups (as dictated by its shape), surface defect sites, and disorders. From the PLS model, the surface chemistry effect on powder flow was favored by more hydrophilic surfaces such that hydrophobic cohesive interactions were reduced. While attempts can be made to focus the morphology optimization efforts described above toward increasing the relative surface area of hydrophilic surfaces, such attempt may not always be practical when all the other constraints for process design are considered. In such cases, it is of paramount importance that the multiscale understanding of the API physical properties feeds into the formulation selection process, so that appropriate composition and/or processing choices are made to reduce the risk to overall drug substance and DP processing and performance.
TABLE 30.4 Key Physical Insights from the PLS Model of API Physical Properties and ffc
| Model Prediction | Direction | Physical Meaning | Effect |
| Dv10 | Increase | Reduction in fines | Significant |
| Dv10, Dv90 | Increase and decrease, respectively | Reduction in PSD span (narrower PSD) | Intermediate |
| Shape factors (ARv, Cv, and Sv) | Generally increase | Favored cubical, spherical, and macroscopically smooth particles | Intermediate |
| Total surface energies | Decrease | Reduction in surface cohesive forces | Intermediate |
| Surface chemistry | Increase in acid–base and decrease in dispersive | Increased hydrophilicity | Intermediate |
30.3.6 Conclusions
In this study, the concept of MST was applied to extract property–performance relationships to provide critical insights for the development of API isolation processes in the context of powder flow optimization. This case study demonstrated that fundamental physical characteristics of API for better powder flowability are limited not necessarily only to larger PSD and more ideal shape factors but also amount of fines, shape of the PSD, and the surface properties such as surface intermolecular interactions and chemistry.
30.4 IMPACT OF API INTRINSIC MECHANICAL PROPERTIES ON PROCESS‐INDUCED DISORDER
30.4.1 Introduction
During the manufacturing of DP containing crystalline APIs, drug crystals are often subjected to unit operations such as milling. It is well documented that this pharmaceutical process can generate various types of defects in API crystals [64, 65]. These crystal defects represent regions of higher disorder and higher energy relative to the average overall energy of the crystalline material [66]. These high‐energy regions can ultimately affect a number of important pharmaceutical properties of APIs including dissolution rate [67–69], chemical stability [70–72], mechanical properties [73], and moisture sorption [74]. Historically, a range of “bulk” analytical techniques have been used to understand the extent of disorders in pharmaceutical solids including X‐ray powder diffraction [75], density [76], heat of solution [77], infrared spectroscopy [78], dissolution rate [79], Raman spectroscopy [80], solid‐state NMR [81], dynamic mechanical analysis [82], differential scanning calorimetry [83], and water vapor sorption. The disorders generated during milling are, however, more local in nature, and are not always easily detectable with the aforementioned methods. It is, therefore, important for pharmaceutical industry to investigate and develop analytical methods that are capable of accurately detecting and assessing the extents of subtle changes in API local structures that can be linked with product stability and performance.
In this case study, a collection of advanced complimentary characterization and analysis tools including synchrotron‐based total scattering pair distribution function (TS‐PDF), nano‐indentation, in situ SPM, and single‐crystal X‐ray diffraction were applied to holistically characterize and correlate the mechanical properties and crystal structural orders of two API crystals, PARP inhibitor compound 1, 2‐([R]‐2‐methylpyrrolidin‐2‐yl)‐1H‐benzimidazole‐4‐carboxamide, and S1P1 antagonist compound 2, ((1R,3S)‐1‐amino‐3‐(4‐(7‐methoxyheptyl) phenyl)cyclopentyl)methanol(S)‐mandelate, following milling under various process conditions [84]. The chemical structures of the two compounds are depicted in Scheme 30.1.

SCHEME 30.1 Chemical structures of compound 1 and 2.
30.4.2 Mechanical Properties
Nano‐indentation (Triboindenter TI900, Hysitron, Minneapolis, MN) equipped with a three-sided pyramidal cube corner probe was used to differentiate the intrinsic mechanical properties of 1 and 2. Figure 30.9 shows the characteristic force–displacement profiles of the two compounds subjected to maximum load force (Pmax) of up to 9 000 μN. Compound 1 required much larger force to reach the same indent depth compared with compound 2, suggesting that compound 1 is an intrinsically harder material. In situ SPM images (Triboindenter TI900, Hysitron, Minneapolis, MN) obtained immediately after nano‐indentation for compounds 1 and 2 are displayed in Figure 30.10. For compound 1, cracking lines and material “sink‐in” around the indents, especially at high load force, are clearly visible. The cracking of the crystal and the collapse of material inward are indicative of the brittle nature of the material [54]. Compared with compound 1, indents of compound 2 were clear triangular indents. Around the indents, the presence of material “pileup” suggests that this material is much softer, and deformation of this material can occur via a plastic flow mechanism [85, 86]. The shallower slopes of the unloading curves for compound 2 also suggest that this material is less stiff compared with compound 1.
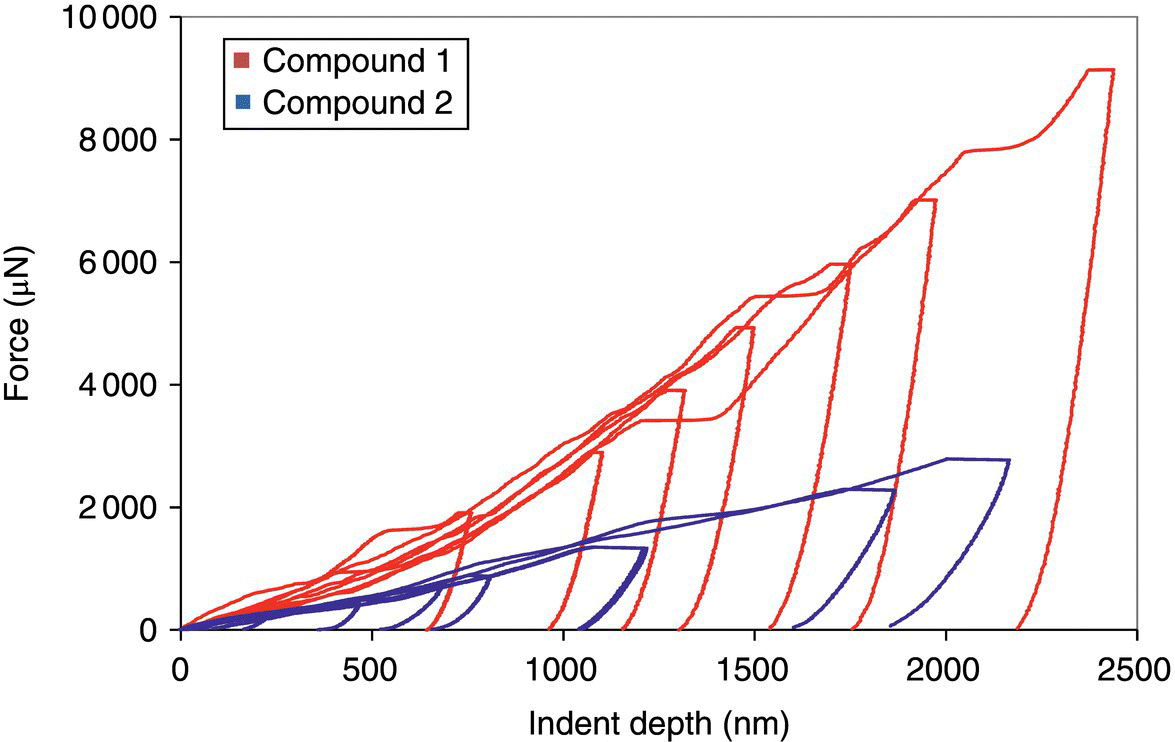
FIGURE 30.9 Representative force–displacement curves of different Pmax for compounds 1 and 2.

FIGURE 30.10 Scanning probe microscopy gradient (left) and topography (right) profiles of (a) compound 1 and (b) compound 2 immediately post‐nano‐indentation. Crystal cracking and material “sink‐in” (as indicated by arrows) are evident in compound 1, whereas material “pileup” (as indicated by arrows) is evident in compound 2.
Figure 30.11 shows a comparison of particle reduced elastic modulus and hardness as a function of indentation depth for compounds 1 and 2. The hardness and reduced elastic modulus of the crystals were determined using the method described by Oliver and Pharr [87]. Young's modulus, E, was determined, with Poisson's ratio, v, of 0.30 via
Although the H and Er are not constant within the indentation depth range explored, compound 1 displays much larger H and Er compared with compound 2. At constant indent depth, Young's modulus (E) and hardness (H) of compound 1 are almost four times greater than those of compound 2 (Table 30.5). The calculated mechanical properties are consistent with the observed behavior of the materials from SPM: compound 1 is a hard, brittle material, whereas compound 2 is a soft, plastic material.
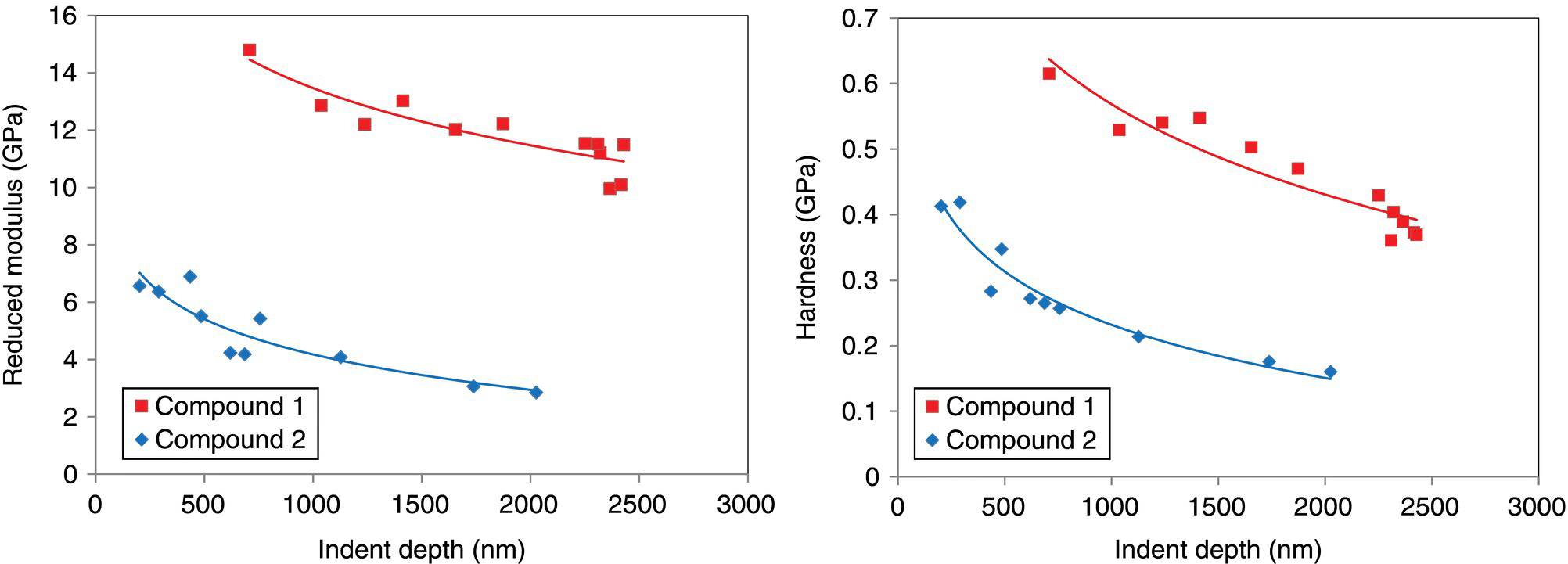
FIGURE 30.11 Reduced modulus and hardness as a function of indent depth for compounds 1 and 2.
TABLE 30.5 Summary of Mechanical Properties and Behavior for Compounds 1 and 2
| Compound | E (GPa) | H (GPa) | Observed Events |
| 1 | 10.4 | 0.43 | Cracking and material “sink‐in” around indents show that material exhibits fracture upon indentation (brittleness) |
| 2 | 2.7 | 0.15 | “Pileup” events indicate that material undergoes plastic flow upon deformation (softness) |
Young's modulus and hardness values were calculated at an indent depth of 2000 nm.
30.4.3 Single‐Crystal Structures
In the crystal structure of compound 1 (Figure 30.12), both intramolecular and intermolecular hydrogen bonds are present. The intramolecular and intermolecular hydrogen bonds form a cross‐linked three‐dimensional network with comparable intermolecular interactions in the three orthogonal directions that is a structure feature known for hard and brittle molecular crystals [7]. Nano‐indentation of compound 1 was carried out on its dominant (103) face. As shown in Table 30.6, the attachment energy and interplanar spacing of the various crystallographic faces are quite similar in different directions for compound 1 (Eatt ranging from −16.6 to −22.7 kcal/mol and d‐spacing from 5.5 to 9.0 Å). This is consistent to the bipyramidal crystal morphology and the isotropic structural feature of the compound. Because of its isotropic lattice packing, the intrinsic mechanical properties (Young's modulus, E, and hardness, H) measured on (103) face are considered a good indicator of the bulk mechanical properties of compound 1 crystals [88, 89].

FIGURE 30.12 Single‐crystal structure of compound 1 viewed along b‐axis. The triangle represents the indentation direction.
TABLE 30.6 Interplanar Spacing (dhkl) and Calculated Attachment Energies of the Compounds 1 and 2 Crystals
| Compound | Lattice Plane (hkl) | dhkl (Å) | Eatt (kcal/mol) |
| 1 | (101) | 8.0 | −16.6 |
| (102) | 7.4 | −17.9 | |
| (004) | 9.0 | −19.6 | |
| (110) | 5.8 | −19.9 | |
| (103) | 6.8 | −20.5 | |
| (111) | 5.7 | −21.4 | |
| (112) | 5.5 | −22.3 | |
| (104) | 6.0 | −22.7 | |
| 2 | (001) | 23.9 | −10.7 |
| (010) | 9.0 | −18.6 | |
| (011) | 8.3 | −18.6 | |
| (110) | 5.6 | −21.8 | |
| (111) | 5.6 | −22.3 | |
| (012) | 7.1 | −22.7 | |
| (101) | 5.9 | −23.4 | |
| (100) | 6.0 | −26.0 |
Compound 2 exhibits a thin‐plate morphology with (001) as its dominant face. The crystal structure of compound 2 is depicted in Figure 30.13. Unlike compound 1, no cross‐linked three‐dimensional hydrogen‐bond network exists in compound 2. Instead, there exist only 2D hydrogen‐bond networks parallel to the a–b plane. Molecular bilayers are formed based on the 2D hydrogen‐bond networks. The packing of compound 2 lattice is anisotropic in such a way that strong and weak interaction patterns are present in nearly orthogonal directions, a structural characteristic typically present in soft and plastic organic crystals [7]. As shown in Table 30.6, the attachment energies of the different faces of compound 2 and their corresponding interplanar distances vary quite significantly in different directions. The (001) face has lowest absolute value of attachment energy (−10.7 kcal/mol), and its corresponding interplanar distance is 23.9 Å. Nano‐indentation of compound 2 was carried out on its dominant (001) face that coincides with the primary slip plane of the crystal. Because of the significant anisotropy of compound 2 crystals (thin plate), it was only possible to indent the crystals in a direction normal to (001) face. Nevertheless, the low Young's modulus, E, and hardness, H, obtained on the (001) face are still considered indicative of the overall soft and plastic nature of the compound.
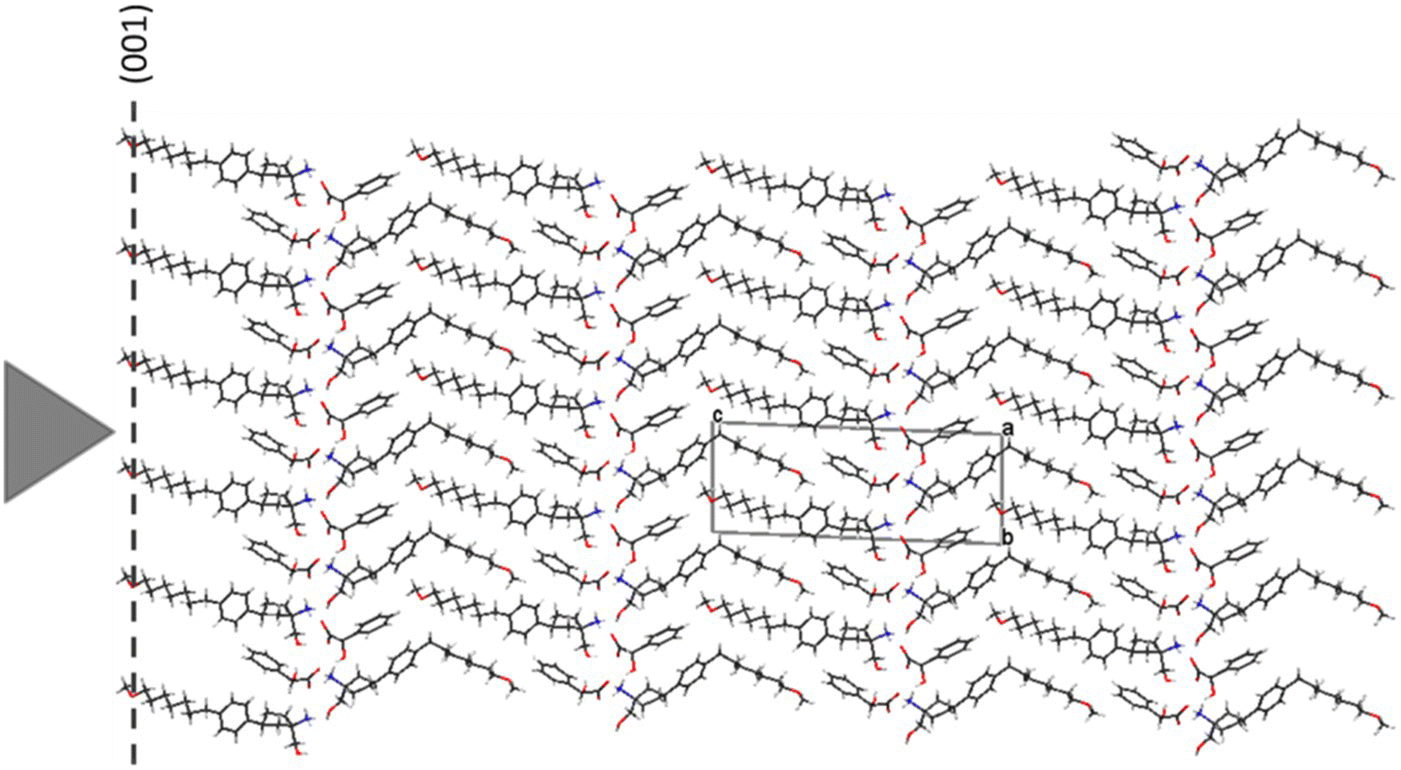
FIGURE 30.13 Single‐crystal structure of compound 2 viewed along a‐axis. The triangle represents the indentation direction.
30.4.4 Pair Distribution Function Analysis
Traditionally, powder X‐ray diffraction analysis has been focused on the assessment of position and intensity of Bragg reflection peaks to provide information about the long‐range order (or the average structure) of a given crystalline phase. The method of TS analysis, on the other hand, uses both the Bragg reflections and diffuse scattering on an equal basis. Assessment of the diffuse scattering component of the powder data provides quantifiable information regarding deviations from the average lattice properties by revealing differences in the local and short‐range structure of the material (Figure 30.14) [90]. This structural information is obtained by mathematically treating the TS data to obtain PDF. The pair distribution function, G(r), provides information on local molecular packing arrangement by giving the number of atoms in a spherical shell of unit thickness at a distance r from any reference atom. The crystalline samples were filled into 1 mm diameter Kapton capillary tubes. High‐energy X‐ray (58 keV, λ = 0.2128 Å) scattering measurements were performed at beamline 11‐ID‐B of the Advanced Photon Source (APS) located at Argonne National Laboratory (ANL). A PerkinElmer amorphous Si 2D image plate detector was placed perpendicular to the high‐energy X‐ray beam, 170 mm behind the capillary samples. Scattering patterns for the samples were collected as a function of the magnitude of the scattered wave vector Q, which is given by Q = 4π sin(θ)/λ. To obtain high accuracy and adequate real‐space resolution of PDF, the scattering data were measured at a scattered angle, 2θ, up to 55°. The corresponding Qmax was greater than 27 Å−1 (compared with a value of ~8 Å−1, equivalent to 160° 2θ, typically achieved with a lab Cu‐based power X‐ray diffraction). To achieve good statistic counts, each sample was collected for 600 seconds.
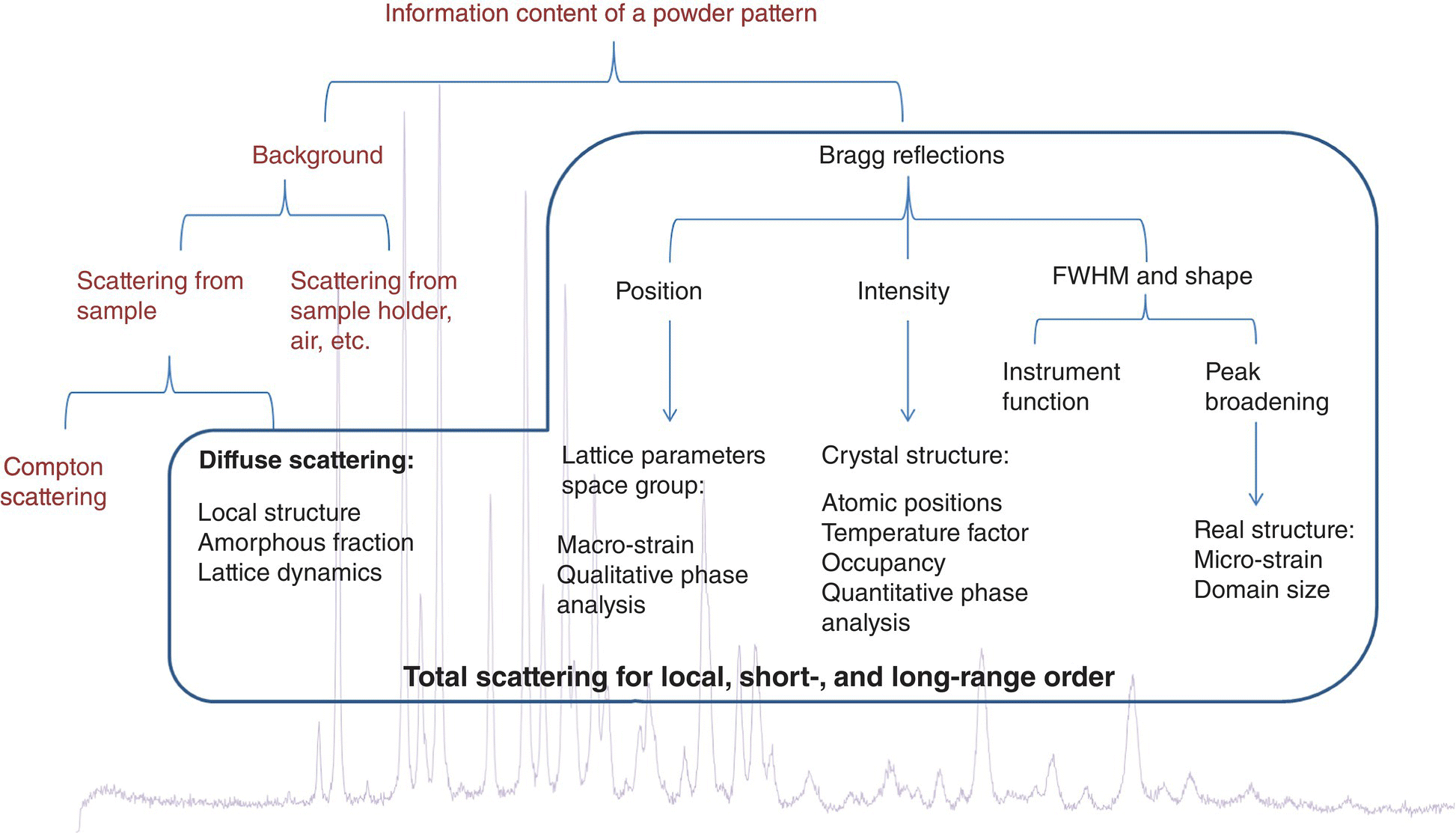
FIGURE 30.14 Information content of a powder X‐ray diffraction pattern.
Compounds 1 and 2 crystalline materials were milled under four different milling conditions with increasing intensity and time: low‐speed 9 000 rpm pin mill (9 000 rpm pm), high‐speed 36 000 rpm pin mill (36 000 rpm pm), short‐time (1 minute) cryomill (1 min cm), and long‐time (7 minutes) cryomill (7 min cm). For pin milling, the residence time of sample powder in the mill was less than one minute. The TS‐PDFs from compound 1 samples obtained under different milling conditions are shown in Figure 30.15. The PDF of unmilled compound 1 was added for comparison. The PDFs of compound 1 milling samples, with the exception of the seven minute cryomilling sample, were visually identical to that of the unmilled, suggesting the hard crystals of compound 1 exhibit very little or essentially no disorders under typical pin milling (9 000–36 000 rpm) and short‐time (1 minute) cryomilling conditions. Only when subjected to extensive milling, such as long‐time (seven minute) cryomilling, crystals of compound 1 began to show discernible changes in its PDF plot. The PDF plots of compound 1 milling samples indicate that while the particle size of compound 1 was reduced with increasing milling intensity and duration, its internal structure was left largely intact.

FIGURE 30.15 Comparison of total scattering pair distribution functions from samples of compound 1 prepared under different milling conditions: unmilled, 9 000 rpm pin milling, 36 000 rpm pin milling, 1 minute cryomilling, and 7 minutes cryomilling.
A similar analysis was also carried out for compound 2. The TS‐PDFs of compound 2 milling samples are shown in Figure 30.16. Compared with the unmilled, the PDF plots of milling samples showed not only obvious changes in peak amplitudes but also small and noticeable differences in peak positions and shape, especially at radial distances r of about 6, 8, 10, 23, and 26 Å. An advantage of PDF G(r) analysis is that the amplitude of the oscillations in PDF plot gives a direct measure of the structural coherence of the sample. In crystalline samples with some degree of disorder, the amplitude of G(r) falls off faster than dictated by the resolution of scattered wave factor Q (Q = 4π sin(θ)/λ). As the positions of the PDF peaks give the separations of pairs of atoms in the structure directly and the width and shape of the PDF peaks contain information about the real atomic probability distribution [89], the PDF peak position and shape changes observed in compound 2 milling samples indicate substantial changes in local structural orders. Moreover, as the longest intramolecular atom–atom distance in compound 2 crystals is about 17 Å and the shortest non‐H‐bonding intermolecular distance is about 4 Å, the PDF difference seen above between the unmilled and milled samples suggests that there are likely structural changes in both the molecular configuration and the intermolecular stacking.
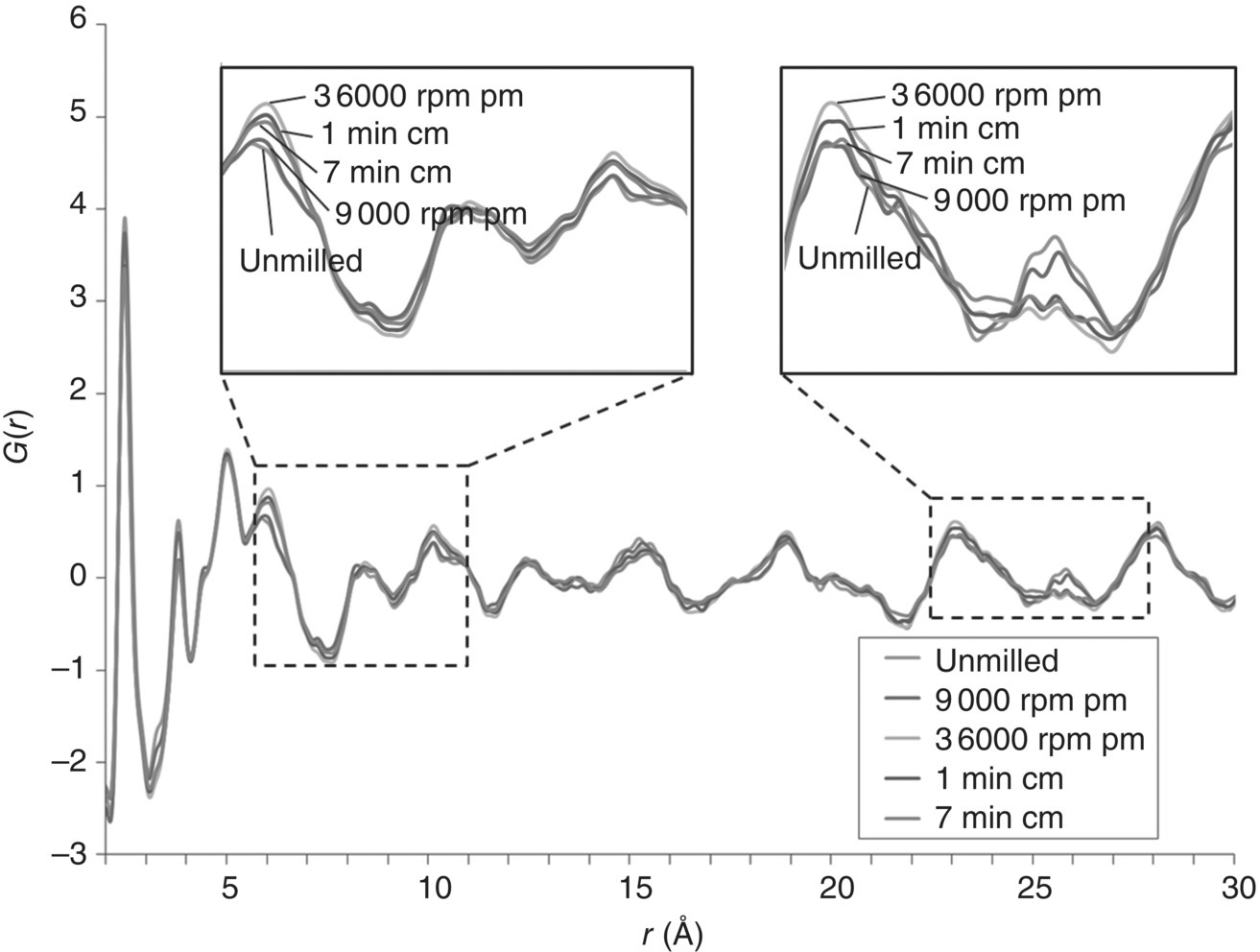
FIGURE 30.16 Comparison of total scattering pair distribution functions from samples of compound 2 prepared under different milling conditions: unmilled, 9 000 rpm pin milling, 36 000 rpm pin milling, 1 minute cryomilling, and 7 minutes cryomilling.
The different milling behaviors of the two compounds, as detected through the PDF analysis, are consistent with their crystal structure features and the different mechanical properties measured by nano‐indentation. For the hard and brittle material of compound 1, the molecules are interlocked through the cross‐linked three‐dimensional hydrogen‐bond network, resulting in a very rigid isotropic crystal lattice. Compound 1 is, therefore, more resistant to the destructive milling force. On the other hand, for the soft and plastic material of compound 2, the 2D hydrogen‐bond networks hold the molecules together in bilayers, and the long methoxyheptyl tails among the stacking bilayers lack strong interactions with each other, particularly along the c‐axis. Therefore the compound has a higher propensity to change its molecular configuration and stacking structure upon milling.
30.4.5 Conclusions
In this case study, nano‐indentation was successfully used to determine the intrinsic mechanical properties, hardness, and reduced modulus of two API compounds 1 and 2. Synchrotron TS and pair distribution function analysis was successfully applied to assess the effects of different milling conditions on the structural disorders of the two drug crystals. The study showed that the hard compound 1 crystals exhibited very little to essentially no structural disorders under typical pharmaceutical milling (pin mill speed of 9 000–36 000 rpm) conditions. To create discernible disorders in hard crystals of compound 1, long‐time cryomilling had to be used. The soft crystals of compound 2, on the other hand, displayed obvious disorders under milling conditions as mild as 9 000 rpm pin milling. The different milling behaviors of the two compounds were found consistent with their contrasting mechanical properties and crystal structure features. As the extent of disorders in API crystals is often tied to their physical and chemical stabilities, the fact that compound 1 does not exhibit noticeable structural disorders under typical milling and compaction conditions allows one to conclude that the risk of hard crystals like compound 1 showing process‐related stability issues is low. On the other hand, for soft crystals like compound 2 that present noticeable structural disorders after milling, their stability should be monitored closely.
ACKNOWLEDGMENTS
Raimundo Ho, Yujin Shin, Laura Poloni, Yinshan Chen, Shuang Chen, and Ahmad Sheikh are employees of AbbVie. The design, study conduct, and financial support for this research was provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
REFERENCES
- 1. Sun, C.C. (2009). Materials science tetrahedron—a useful tool for pharmaceutical Research and Development. J. Pharm. Sci. 98 (5): 1671–1687.
- 2. Yang, P. and Tarascon, J.‐M. (2012). Towards systems materials engineering. Nat. Mater. 11: 560.
- 3. Brittain, H.G. (2009). Polymorphism in Pharmaceutical Solids, 2e. New York: Informa Healthcare USA, Inc.
- 4. Beyer, T., Day, G.M., and Price, S.L. (2001). The prediction, morphology, and mechanical properties of the polymorphs of paracetamol. J. Am. Chem. Soc. 123 (21): 5086–5094.
- 5. Khomane, K.S. and Bansal, A.K. (2013). Weak hydrogen bonding interactions influence slip system activity and compaction behavior of pharmaceutical powders. J. Pharm. Sci. 102 (12): 4242–4245.
- 6. Yadav, J.A., Khomane, K.S., Modi, S.R. et al. (2017). Correlating single crystal structure, nanomechanical, and bulk compaction behavior of Febuxostat polymorphs. Mol. Pharm. 14 (3): 866–874.
- 7. Reddy, C.M., Padmanabhan, K.A., and Desiraju, G.R. (2006). Structure−property correlations in bending and brittle organic crystals. Cryst. Growth Des. 6 (12): 2720–2731.
- 8. Bag, P.P., Chen, M., Sun, C.C., and Reddy, C.M. (2012). Direct correlation among crystal structure, mechanical behaviour and tabletability in a trimorphic molecular compound. CrystEngComm 14 (11): 3865–3867.
- 9. York, P. (1983). Solid‐state properties of powders in the formulation and processing of solid dosage forms. Int. J. Pharm. 14 (1): 1–28.
- 10. Rasenack, N. and Muller, B.W. (2002). Properties of ibuprofen crystallised under various conditions: a comparative study. Drug Dev. Ind. Pharm. 28 (9): 1077–1089.
- 11. Hooper, D., Clarke, F.C., Docherty, R. et al. (2017). Effects of crystal habit on the sticking propensity of ibuprofen—a case study. Int. J. Pharm. 531 (1): 266–275.
- 12. Chow, A.H.L., Hsia, C.K., Gordon, J.D. et al. (1995). Assessment of wettability and its relationship to the intrinsic dissolution rate of doped phenytoin crystals. Int. J. Pharm. 126 (1): 21–28.
- 13. Heng, J.Y.Y., Bismarck, A., Lee, A.F. et al. (2006). Anisotropic surface energetics and wettability of macroscopic form I paracetamol crystals. Langmuir 22 (6): 2760–2769.
- 14. Lippold, B.C. and Ohm, A. (1986). Correlation between wettability and dissolution rate of pharmaceutical powders. Int. J. Pharm. 28 (1): 67–74.
- 15. Clint, J.H. (2001). Adhesion and components of solid surface energies. Curr. Opin. Colloid Interface Sci. 6 (1): 28–33.
- 16. Lovette, M.A., Browning, A.R., Griffin, D.W. et al. (2008). Crystal shape engineering. Ind. Eng. Chem. Res. 47 (24): 9812–9833.
- 17. Ho, R., Dilworth, S.E., Williams, D.R., and Heng, J.Y.Y. (2011). Role of surface chemistry and energetics in high shear wet granulation. Ind. Eng. Chem. Res. 50 (16): 9642–9649.
- 18. Newell, H.E., Buckton, G., Butler, D.A. et al. (2001). The use of inverse phase gas chromatography to measure the surface energy of crystalline, amorphous, and recently milled lactose. Pharm. Res. 18 (5): 662–666.
- 19. Rumondor, A.C.F., Marsac, P.J., Stanford, L.A., and Taylor, L.S. (2009). Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol. Pharm. 6 (5): 1492–1505.
- 20. Fichtner, F., Mahlin, D., Welch, K. et al. (2008). Effect of surface energy on powder compactibility. Pharm. Res. 25 (12): 2750–2759.
- 21. Chow, A.H.L., Tong, H.H.Y., Chattopadhyay, P., and Shekunov, B.Y. (2007). Particle engineering for pulmonary drug delivery. Pharm. Res. 24 (3): 411–437.
- 22. Ho, R. and Heng, J.Y.Y. (2013). A review of inverse gas chromatography and its development as a tool to characterize anisotropic surface properties of pharmaceutical solids. KONA Powder Part. J. 30: 164–180.
- 23. Das, S.C., Larson, I., Morton, D.A.V., and Stewart, P.J. (2011). Determination of the polar and total surface energy distributions of particulates by inverse gas chromatography. Langmuir 27 (2): 521–523.
- 24. Ho, R., Heng, J.Y.Y., Dilworth, S.E. et al. (2009). Determination of surface heterogeneity of D‐mannitol by sessile drop contact angle and inverse gas chromatography. Int. J. Pharm. 387 (1–2): 79–86.
- 25. Smith, R.R., Williams, D.R., Burnett, D.J., and Heng, J.Y.Y. (2014). A new method to determine dispersive surface energy site distributions by inverse gas chromatography. Langmuir 30 (27): 8029–8035.
- 26. Khoo, J.Y., Williams, D.R., and Heng, J.Y.Y. (2010). Dehydration kinetics of pharmaceutical hydrate: effects of environmental conditions and crystal forms. Dry. Technol. 28 (10): 1164–1169.
- 27. Vitez, I.M., Newman, A.W., Davidovich, M., and Kiesnowski, C. (1998). The evolution of hot‐stage microscopy to aid solid‐state characterizations of pharmaceutical solids. Thermochim. Acta 324 (1): 187–196.
- 28. Eddleston, M.D., Bithell, E.G., and Jones, W. (2010). Transmission electron microscopy of pharmaceutical materials. J. Pharm. Sci. 99 (9): 4072–4083.
- 29. Klang, V., Valenta, C., and Matsko, N.B. (2013). Electron microscopy of pharmaceutical systems. Micron 44 (Supplement C): 45–74.
- 30. Liao, X. and Wiedmann, T.S. (2004). Characterization of pharmaceutical solids by scanning probe microscopy. J. Pharm. Sci. 93 (9): 2250–2258.
- 31. Shur, J. and Price, R. (2012). Advanced microscopy techniques to assess solid‐state properties of inhalation medicines. Adv. Drug Deliv. Rev. 64 (4): 369–382.
- 32. Pygall, S.R., Whetstone, J., Timmins, P., and Melia, C.D. (2007). Pharmaceutical applications of confocal laser scanning microscopy: the physical characterisation of pharmaceutical systems. Adv. Drug Deliv. Rev. 59 (14): 1434–1452.
- 33. Martin, D.C., Chen, J., Yang, J. et al. (2005). High resolution electron microscopy of ordered polymers and organic molecular crystals: recent developments and future possibilities. J. Polym. Sci. B Polym. Phys. 43 (14): 1749–1778.
- 34. Poloni, L.N., Zhong, X., Ward, M.D., and Mandal, T. (2017). Best practices for real‐time in situ atomic force and chemical force microscopy of crystals. Chem. Mater. 29 (1): 331–345.
- 35. Poloni, L.N., Zhu, Z., Garcia‐Vázquez, N. et al. (2017). Role of molecular recognition in l‐cystine crystal growth inhibition. Cryst. Growth Des. 17 (5): 2767–2781.
- 36. Poloni, L.N., Ford, A.P., and Ward, M.D. (2016). Site discrimination and anisotropic growth inhibition by molecular imposters on highly dissymmetric crystal surfaces. Cryst. Growth Des. 16 (9): 5525–5541.
- 37. Maugis, D. (2000). Contact, Adhesion and Rupture of Elastic Solids. Berlin, Heidelberg: Springer‐Verlag.
- 38. Mandal, T. and Ward, M.D. (2013). Determination of specific binding interactions at l‐cystine crystal surfaces with chemical force microscopy. J. Am. Chem. Soc. 135 (15): 5525–5528.
- 39. Shekunov, B.Y., Chattopadhyay, P., Tong, H.H.Y., and Chow, A.H.L. (2007). Particle size analysis in pharmaceutics: principles, methods and applications. Pharm. Res. 24 (2): 203–227.
- 40. Nalluri, V.R., Schirg, P., Gao, X. et al. (2010). Different modes of dynamic image analysis in monitoring of pharmaceutical dry milling process. Int. J. Pharm. 391 (1): 107–114.
- 41. Yu, W. and Hancock, B.C. (2008). Evaluation of dynamic image analysis for characterizing pharmaceutical excipient particles. Int. J. Pharm. 361 (1–2): 150–157.
- 42. Bika, D.G., Gentzler, M., and Michaels, J.N. (2001). Mechanical properties of agglomerates. Powder Technol. 117 (1): 98–112.
- 43. Reynolds, G.K., Fu, J.S., Cheong, Y.S. et al. (2005). Breakage in granulation: a review. Chem. Eng. Sci. 60 (14): 3969–3992.
- 44. Bika, D.G., Gentzler, M., and Michaels, J.N. (2001). Mechanical properties of agglomerates. Powder Technol. 117 (1–2): 98–112.
- 45. Hancock, B.C., Clas, S.D., and Christensen, K. (2000). Micro‐scale measurement of the mechanical properties of compressed pharmaceutical powders. 1: The elasticity and fracture behavior of microcrystalline cellulose. Int. J. Pharm. 209 (1–2): 27–35.
- 46. Kawakita, K. and Lüdde, K.‐H. (1971). Some considerations on powder compression equations. Powder Technol. 4 (2): 61–68.
- 47. Adams, M.J., Mullier, M.A., and Seville, J.P.K. (1994). Agglomerate strength measurement using a uniaxial confined compression test. Powder Technol. 78 (1): 5–13.
- 48. Amidon, G.E., Secreast, P.J., and Mudie, D. (2009). Particle, powder, and compact characterization. In: Developing Solid Oral Dosage Forms (ed. Y. Qiu, Y. Chen, G.G.Z. Zhang, et al.), 163–186. San Diego: Academic Press.
- 49. Vogel, L. and Peukert, W. (2002). Characterisation of grinding‐relevant particle properties by inverting a population balance model. Part. Part. Syst. Charact. 19: 149–157.
- 50. Vogel, L. and Peukert, W. (2003). Breakage behaviour of different materials—construction of a mastercurve for the breakage probability. Powder Technol. 129 (1): 101–110.
- 51. Vogel, L. and Peukert, W. (2005). From single particle impact behaviour to modelling of impact mills. Chem. Eng. Sci. 60 (18): 5164–5176.
- 52. de Vegt, O., Vromans, H., Faassen, F., and van der Voort Maarschalk, K. (2005). Milling of organic solids in a jet mill. Part 1: Determination of the selection function and related mechanical material properties. Part. Part. Syst. Charact. 22 (2): 133–140.
- 53. Meier, M., John, E., Wieckhusen, D. et al. (2009). Influence of mechanical properties on impact fracture: prediction of the milling behaviour of pharmaceutical powders by nanoindentation. Powder Technol. 188 (3): 301–313.
- 54. Taylor, L.J., Papadopoulos, D.G., Dunn, P.J. et al. (2004). Predictive milling of pharmaceutical materials using nanoindentation of single crystals. Org. Process. Res. Dev. 8 (4): 674–679.
- 55. Bhatt, P.M., Ravindra, N.V., Banerjee, R., and Desiraju, G.R. (2005). Saccharin as a salt former. Enhanced solubilities of saccharinates of active pharmaceutical ingredients. Chem. Commun. (8): 1073–1075. https://doi.org/10.1039/B416137H.
- 56. Varughese, S., Kiran, M.S.R.N., Ramamurty, U., and Desiraju, G.R. (2013). Nanoindentation in crystal engineering: quantifying mechanical properties of molecular crystals. Angew. Chem. Int. Ed. 52 (10): 2701–2712.
- 57. Feng, Y., Grant, D.J.W., and Sun, C.C. (2007). Influence of crystal structure on the tableting properties of n‐alkyl 4‐hydroxybenzoate esters (parabens). J. Pharm. Sci. 96 (12): 3324–3333.
- 58. Jager, P.D., Bramante, T., and Luner, P.E. (2015). Assessment of pharmaceutical powder flowability using shear cell‐based methods and application of Jenike's methodology. J. Pharm. Sci. 104 (11): 3804–3813.
- 59. Behringer, R., Louge, M., McElwaine, J. et al. (2017). Report of the IFPRI Powder Flow Working Group SAR30–08.
- 60. Capece, M., Ho, R., Strong, J., and Gao, P. (2015). Prediction of powder flow performance using a multi‐component granular bond number. Powder Technol. 286 (Supplement C): 561–571.
- 61. Fu, X., Huck, D., Makein, L. et al. (2012). Effect of particle shape and size on flow properties of lactose powders. Particuology 10 (2): 203–208.
- 62. Sandler, N. and Wilson, D. (2010). Prediction of granule packing and flow behavior based on particle size and shape analysis. J. Pharm. Sci. 99 (2): 958–968.
- 63. Yu, W., Muteki, K., Zhang, L., and Kim, G. (2011). Prediction of bulk powder flow performance using comprehensive particle size and particle shape distributions. J. Pharm. Sci. 100 (1): 284–293.
- 64. Dialer, V. and Kuessner, K. (1973). High surface energy absorption in vibration milling and influence in conditions of fine grinding. Kolloid‐Z.U.Z. Poly. 251: 710–715.
- 65. Saleki‐Gerhardt, A., Ahlneck, C., and Zografi, G. (1994). Assessment of disorder in crystalline solids. Int. J. Pharm. 101 (3): 237–247.
- 66. Zhang, J., Ebbens, S., Chen, X. et al. (2006). Determination of the surface free energy of crystalline and amorphous lactose by atomic force microscopy adhesion measurement. Pharm. Res. 23 (2): 401–407.
- 67. Burt, H.M. and Mitchell, A.G. (1981). Crystal defects and dissolution. Int. J. Pharm. 9 (2): 137–152.
- 68. Tawashi, R. (1968). Dissolution rates of crystalline drugs. J Mondial de Pharmacie 11: 371–379.
- 69. Ward, G.H. and Schultz, R.K. (1995). Process‐induced crystallinity changes in albuterol sulfate and its effect on powder physical stability. Pharm. Res. 12 (5): 773–779.
- 70. Byrn, S.R., Pfeiffer, R.R., Stephenson, G. et al. (1994). Solid‐state pharmaceutical chemistry. Chem. Mater. 6 (8): 1148–1158.
- 71. Byrn, S.R., Xu, W., and Newman, A.W. (2001). Chemical reactivity in solid‐state pharmaceuticals: formulation implications. Adv. Drug Deliv. Rev. 48 (1): 115–136.
- 72. Shalaev, E., Shalaeva, M., and Zografi, G. (2002). The effect of disorder on the chemical reactivity of an organic solid, tetraglycine methyl ester: change of the reaction mechanism. J. Pharm. Sci. 91 (2): 584–593.
- 73. Wildfong, P.L.D., Hancock, B.C., Moore, M.D., and Morris, K.R. (2006). Towards an understanding of the structurally based potential for mechanically activated disordering of small molecule organic crystals. J. Pharm. Sci. 95 (12): 2645–2656.
- 74. Ahlneck, C. and Zografi, G. (1990). The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int. J. Pharm. 62 (2): 87–95.
- 75. Klung, H. and Alexander, L. (1974). X‐Ray Diffraction Procedures for Polycrystalline and Amorphous Materials. New York: Wiley.
- 76. Duncan‐Hewitt, W.C. and Grant, D.J.W. (1986). True density and thermal expansivity of pharmaceutical solids: comparison of methods and assessment of crystallinity. Int. J. Pharm. 28 (1): 75–84.
- 77. Pikal, M.J., Lukes, A.L., Lang, J.E., and Gaines, K. (1978). Quantitative crystallinity determinations for β‐lactam antibiotics by solution calorimetry: correlations with stability. J. Pharm. Sci. 67 (6): 767–773.
- 78. Black, D.B. and Lovering, E.G. (1977). Estimation of the degree of crystallinity in digoxin by X‐ray and infrared methods. J. Pharm. Pharmacol. 29 (1): 684–687.
- 79. Hendriksen, B.A. (1990). Characterization of calcium fenoprofen 1. Powder dissolution rate and degree of crystallinity. Int. J. Pharm. 60 (3): 243–252.
- 80. Strachan, C.J., Rades, T., Gordon, K.C., and Rantanen, J. (2007). Raman spectroscopy for quantitative analysis of pharmaceutical solids. J. Pharm. Pharmacol. 59 (2): 179–192.
- 81. Offerdahl, T.J., Salsbury, J.S., Dong, Z. et al. (2005). Quantitation of crystalline and amorphous forms of anhydrous neotame using 13C CPMAS NMR spectroscopy. J. Pharm. Sci. 94 (12): 2591–2605.
- 82. Royall, P.G., Huang, C.‐Y., Tang, S.‐W.J. et al. (2005). The development of DMA for the detection of amorphous content in pharmaceutical powdered materials. Int. J. Pharm. 301 (1): 181–191.
- 83. Zimper, U., Aaltonen, J., McGoverin, M.C. et al. (2010). Quantification of process induced disorder in milled samples using different analytical techniques. Pharmaceutics 2 (1): 30–49.
- 84. Chen, S., Sheikh, A.Y., and Ho, R. (2014). Evaluation of effects of pharmaceutical processing on structural disorders of active pharmaceutical ingredient crystals using nanoindentation and high‐resolution total scattering pair distribution function analysis. J. Pharm. Sci. 103 (12): 3879–3890.
- 85. Cao, X., Morganti, M., Hancock, B.C., and Masterson, V.M. (2010). Correlating particle hardness with powder compaction performance. J. Pharm. Sci. 99 (10): 4307–4316.
- 86. Ramos, K.J. and Bahr, D.F. (2011). Mechanical behavior assessment of sucrose using nanoindentation. J. Mater. Res. 22 (7): 2037–2045.
- 87. Oliver, W.C. and Pharr, G.M. (1992). An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 7 (6): 1564–1583.
- 88. Egart, M., Ilić, I., Janković, B. et al. (2014). Compaction properties of crystalline pharmaceutical ingredients according to the Walker model and nanomechanical attributes. Int. J. Pharm. 472 (1): 347–355.
- 89. Egart, M., Janković, B., Lah, N. et al. (2015). Nanomechanical properties of selected single pharmaceutical crystals as a predictor of their bulk behaviour. Pharm. Res. 32 (2): 469–481.
- 90. Billinge, S. (2008). Local structure from total scattering and atomic pair distribution function (PDF) analysis. In: Powder Diffraction: Theory and Practice (ed. R. Dinnebier and S. Billinge), 464–493. Cambridge: The Royal Society of Chemistry.