
1
CHEMICAL ENGINEERING IN THE PHARMACEUTICAL INDUSTRY: AN INTRODUCTION
David J. am Ende
Nalas Engineering Services, Inc., Centerbrook, CT, USA
Mary T. am Ende*
Pfizer, Inc., Groton, CT, USA
Across the pharmaceutical industry chemical engineers are employed throughout research and development (R&D) to full‐scale manufacturing and packaging in technical and managerial capacities. The chapters in these two volumes provide an emphasis on the application of chemical engineering science to process design, development, and scale‐up for active pharmaceutical ingredients (APIs), drug products (DPs), and biologicals including sections on regulatory considerations such as design space, control strategies, process analytical technology (PAT), and quality by design (QbD). The focus of this introduction is to provide a high‐level overview of bringing a drug to market and highlight industry trends, current challenges, and how chemical engineering skills are an exquisite match to address those challenges.
In general pharmaceuticals are drug delivery systems in which drug‐containing products are designed and manufactured to deliver precise therapeutic responses [1]. The drug is considered the “active,” i.e. active pharmaceutical ingredient (API) or “drug substance,” and the formulated final dosage form is simply referred to as the drug product (DP).
This book focuses on API in volume 1 and DP in volume 2. The API and DP are designed and developed in R&D and then transferred to the commercial manufacturing sites by teams of organic chemists, analytical chemists, pharmaceutical scientists, and chemical engineers. Prior to the transition to the commercial site, co‐development teams are formed with members from R&D and manufacturing working together to define the computational and experimental studies to conduct based on risk and scientific considerations. The outcome of this multidisciplinary team effort forms the regulatory filing strategy for the API and drug products.
Once the commercial API and DP have been established, the co‐development teams support three major regulatory submissions for a global product. A New Drug Application (NDA) is submitted to the US Food and Drug Administration (FDA), whereas in the Europe Union a Marketing Authorization Application (MAA) is submitted to the European Medicines Agency (EMA), and in Japan a Japan New Drug Application (JNDA) is submitted to the Pharmaceuticals and Medical Devices Agency (PMDA). Subsequently, the rest of world regulatory filings are led by the commercial division with no significant involvement by R&D since more commercial experience is available at the site by that time.
In the United States, federal and state laws exist to control the manufacture and distribution of pharmaceuticals. Specifically, the FDA exists by the mandate of the US Congress with the Food, Drug, and Cosmetics Act as the principal law to enforce and constitutes the basis of the drug approval process [1]. Specifically in the United States, “The FDA is responsible for protecting the public health by assuring the safety, efficacy, and security of human and veterinary drugs, biological products, medical devices, our nation's food supply, cosmetics, and products that emit radiation. The FDA is also responsible for advancing the public health where possible by speeding innovations that make medicines and foods more effective, safer, and more affordable. They also serve the public by ensuring accurate, science‐based information on medicines and foods to maintain and improve their health.”1 On 28 March 2018 the FDA announced organizational changes available on their website. Janet Woodcock remains the director of the small molecule division, referred to as Center for Drug Evaluation and Research (CDER).2 Peter W. Marks is the director of the large molecule division, referred to as Center for Biologics Evaluation and Research (CBER).3 Further information can also be easily obtained from the FDA website, including the overall drug review process, current good manufacturing practices (cGMP), International Council on Harmonization (ICH), and mechanisms to comment on draft guidances, recalls, safety alerts, and warning letters that have been issued to companies.4
EMA is a decentralized body of the European Union with headquarters in London whose main responsibility is the protection and promotion of public and animal health, through the evaluation and supervision of medicines for human and veterinary use.5
The Japan Pharmaceutical Affairs Law (JPAL) is a law intended to control and regulate the manufacturing, importation, sale of drugs, and medical devices.6 It exists to assure the quality, efficacy and safety of drugs, cosmetics, and medical devices while improving public health and hygiene. The JPAL also provides guidance to pharmaceutical companies on how to translate their QbD control strategy, which was found to align well with the three levels of criticality initially used in early QbD filings for noncritical, key, and critical process parameters. Japan's Ministry of Health, Labour and Welfare (MHLW) has issued clear guidance in English for three key ministerial ordinances to assure compliance requirements for manufacturers.
Japan, Europe, and United States collaborate as the International Council on Harmonization – Quality (ICH) to establish greater expectations for science and risk‐based approaches to transform the pharmaceutical industry over the past decade. Critical to that transformation were the QbD guidances, Q8, Q9, and Q10 [2–4]. The final versions of the guidances are readily available on the CDER website, including the more recent QbD guidance for drug substance composed in Q11.7
1.1 GLOBAL IMPACT OF THE INDUSTRY
The value of the pharmaceutical industry to the American economy is substantial. In 2016, the industry employed over 854 000 people with each job indirectly supporting an additional 4 jobs. Thus as an aggregate, the industry supported 4.4 million jobs and generated nearly $1.2 trillion in annual economic output when direct, indirect, and induced effects were considered for 2016.8
As an industry sector, the pharmaceutical industry is considered profitable, in spite of the high attrition rate for new chemical entities (NCEs).9 For example, Forbes estimated the profit margin for the health‐care technology industry in 2015 to be approximately 21%, clearly placing near the top for profitable industries.10 The companies that are most profitable in this sector were major pharmaceutical and generics companies. As far as total revenues in pharmaceutical sales, the top 20 pharmaceutical companies are listed in Table 1.1.
TABLE 1.1 Top 20 Pharmaceutical Companies Based on 2017 Revenue as Listed in Wikipedia
Source: From https://en.wikipedia.org/wiki/List_of_largest_pharmaceutical_companies_by_revenue#cite_note‐28. Licensed under CC BY 3.0.
| # | HQ | Company | 2017 (USD Billions) | 2016 (USD Billions) | 2015 (USD Billions) | 2014 (USD Billions) | 2013 (USD Billions) | 2012 (USD Billions) | 2011 (USD Billions) |
| 1 |  |
Johnson & Johnson NYSE: JNJ | 76.50 | 71.89 | 70.10 | 74.30 | 71.31 | 67.20 | 65.00 |
| 2 |  |
Roche OTCQX: RHHBY | 57.37 | 50.11 | 47.70 | 49.86 | 48.53 | 47.80 | 45.21 |
| 3 | |
Pfizer NYSE: PFE | 52.54 | 52.82 | 48.85 | 49.61 | 51.58 | 58.99 | 65.26 |
| 4 | |
Novartis NYSE: NVS | 49.11 | 48.52 | 49.41 | 58.00 | 57.36 | 56.67 | 58.57 |
| 5 |  |
Sanofi NYSE: SNY | 42.91 | 36.57 | 36.73 | 43.07 | 42.08 | 46.41 | 44.34 |
| 6 |  |
GlaxoSmithKline LSE: GSK | 42.05 | 34.79 | 29.84 | 37.96 | 41.61 | 39.93 | 41.39 |
| 7 | |
Merck & Co. NYSE: MRK | 40.10 | 39.80 | 39.50 | 42.24 | 44.03 | 47.27 | 48.05 |
| 8 | |
AbbVie NYSE: ABBV | 28.22 | 25.56 | 22.82 | 19.96 | 18.79 | – | – |
| 9 |  |
Bayer FWB: BAYN | 27.76 | 25.27 | 24.09 | 25.47 | 24.17 | 24.30 | 23.11 |
| 10 | |
Abbott Laboratories NASDAQ: ABT | 27.39 | 20.85 | 20.4 | 20.25 | 21.85 | 39.87 | 38.85 |
| 11 | |
Gilead Sciences NASDAQ: GILD | 25.70 | 30.39 | 32.15 | 24.47 | 10.80 | 9.70 | 8.39 |
| 12 | |
Eli Lilly & Co NYSE: LLY | 22.90 | 21.22 | 20.00 | 19.62 | 23.11 | 22.60 | 24.29 |
| 13 | |
Amgen NASDAQ: AMGN | 22.80 | 22.99 | 21.66 | 20.06 | 18.68 | 17.30 | 15.58 |
| 14 | |
AstraZeneca LSE: AZN | 22.47 | 23.00 | 24.71 | 26.10 | 25.71 | 27.97 | 33.59 |
| 15 |  |
Teva Pharmaceutical Industries NASDAQ: TEVA | 22.40 | 21.90 | 20.00 | 20.27 | 20.31 | 18.31 | 16.12 |
| 16 | |
Boehringer Ingelheim Private | 21.67 | 17.54 | 16.41 | 17.70 | 18.68 | 18.89 | 18.34 |
| 17 | |
Bristol‐Myers Squibb NASDAQ: BMY | 20.80 | 19.43 | 16.56 | 15.88 | 16.39 | 17.62 | 21.24 |
| 18 |  |
Novo Nordisk NYSE: NVO | 18.77 | 16.61 | 16.06 | 15.83 | 14.88 | 13.48 | 11.56 |
| 19 | |
Merck Group ETR: MRK | 15.32 | 15.80 | 13.95 | 14.99 | 14.77 | 13.02 | 12.83 |
| 20 |  |
Shire NASDAQ: SHPG | 14.40 | 11.40 | 6.42 | 6.00 | 4.76 | 4.68 | 3.50 |
Based on revenue, the pharmaceutical and biopharmaceutical companies are based in the following countries: 9 (United States), 2 (Switzerland), 2 (United Kingdom), 1 (France), 3 (Germany), 1 (Israel), 1(Denmark), and 1 (Republic of Ireland). Only 1 company in the top 20 revenue producing is privately held.
Global prescription drug sales are on the order of $800 billion in 2017. These drug sales are forecasted to grow at 6.3% compound annual growth rate (CAGR) between 2016 and 2022 to nearly $1.2 trillion (as shown in Figure 1.1),11 while generic drugs account for approximately 10% of those sales figures.
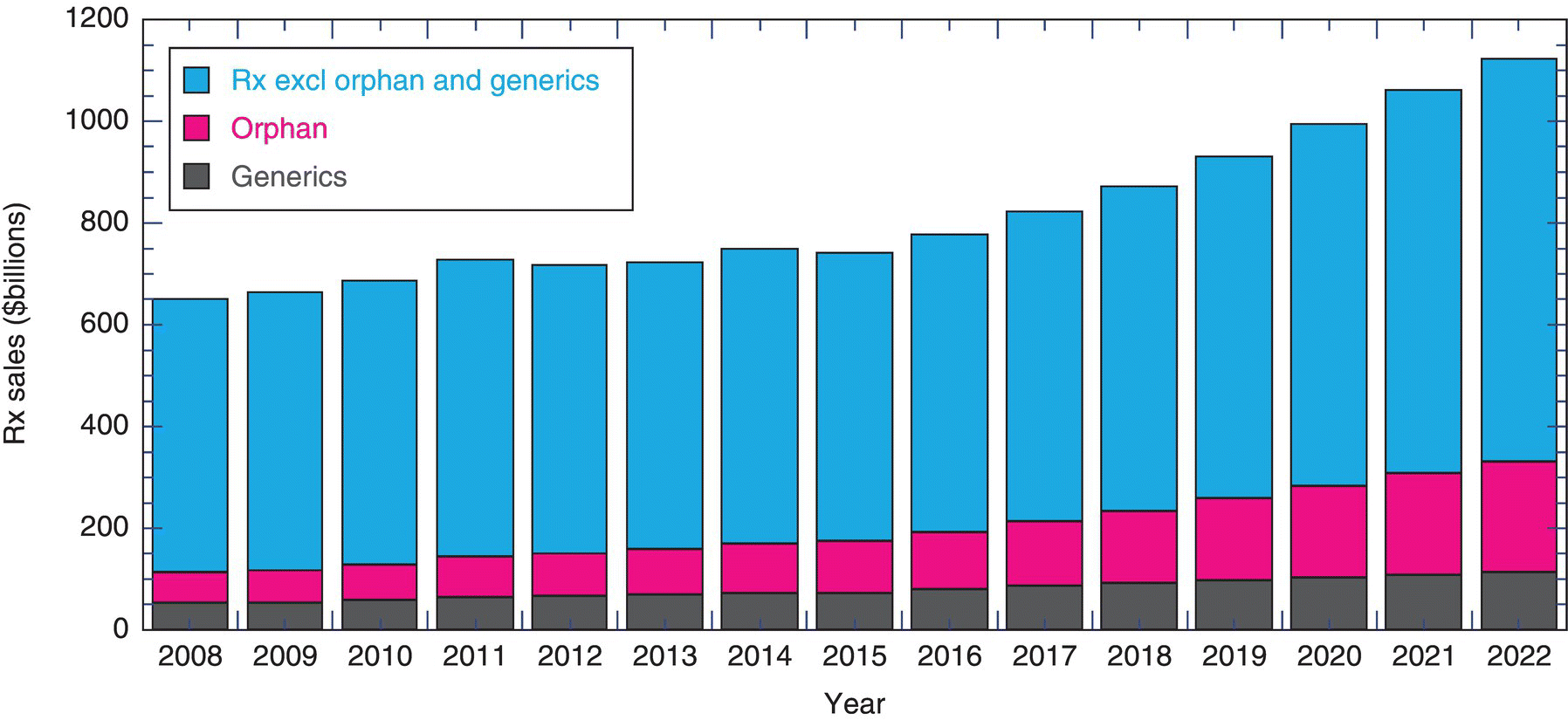
FIGURE 1.1 Global pharmaceutical prescription sales as a function of the type of drug. Global prescription drug sales were on the order of $800 billion in 2017. These drug sales are forecasted to grow at 6.3% compound annual growth rate (CAGR) between 2016 through 2022 to nearly $1.2 trillion while generic drugs account for approximately 10% of those sales figures.
Source: From http://info.evaluategroup.com/rs/607‐YGS‐364/images/wp16.pdf.
There is considerable value in being the first company to deliver a new medicine that treats a new indication (e.g. breakthrough therapy designation from regulators) or uses a new mechanism of action to benefit patients. Therefore, new developments in pharmaceutical R&D that speed quality drug candidates to the market are important investments for the future.
1.2 INVESTMENTS IN PHARMACEUTICAL R&D
R&D is the engine that drives innovation of new drugs and therapies. Significant investment is required to discover and advance potential NCEs and new molecular entities (NMEs). For example, the pharmaceutical industry invested approximately $150 billion into R&D in 2015. Worldwide pharmaceutical R&D spending is expected to grow by 2.8% (CAGR) to $182 billion in 2022 (Figure 1.2).12 The cost of advancing drug candidates and entire pharmaceutical portfolios in R&D is significant. In 2001 the average cost for an approved medicine was estimated to be $802 million, and by the end of 2014, the average cost escalated to $2.6 billion as reported by Tufts Center for the Study of Drug Development.13 Although these figures clearly depend on the drug type, therapeutic area, and speed of development, the bottom line is that the up‐front investments required to reach the market are massive especially when considering the uncertainty whether the up‐front investment will payback.

FIGURE 1.2 World‐wide pharmaceutical R&D spend in 2015 was approximately $150 billion. Growth rate in R&D spend is projected to grow at a rate of 2.8%.
Source: EvaluatePharma®.
Given there might be 10 or more years of R&D costs without any revenue generated on a NCE or NME, the gross margins of a successful drug need to cover prior R&D investments and candidate attrition and to cover the continuing marketing and production costs. Figure 1.3 shows the classic cash flow profile for a new drug developed and marketed. First there is a period of negative cash flow during the R&D phase. When the drug is approved and launched, only then are revenues generated, which have to be priced high enough to recoup the extensive R&D investment and provide a return on the investment.

FIGURE 1.3 A hypothetical cash flow curve for a pharmaceutical product includes 10–15 years of negative cash flows of typically $1–3 billion. Reasonably high margins are needed, once the drug is on the market, if it is to recuperate and provide a positive return on investment (ROI) over its lifecycle.
The net present value (NPV) calculation is one way to assess return on investment with a discount rate of 10–12% generally chosen in the pharmaceutical industry as the rate to value products or programs for investment decisions [6]. The highest revenues for a new drug are achieved during the period of market exclusivity (where no competitors can sell the same drug). So it is in the company's best interest to ensure the best patent protection strategy is in place to maximize the length of market exclusivity. Patents typically have a validity of 20 years from the earliest application grant date base on applications filed after 1995. In some cases the time of market exclusivity can be extended through new indications, new formulations, and devices, which may themselves be patent protected (see Table 1.2).
TABLE 1.2 Periods of Exclusivity Granted by the FDA
Source: Form https://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm079031.htm#What_is_the_difference_between_patents_a
| Specific FDA Applications | Period of Exclusivity |
| New Chemical Entity Exclusivity (NCE) | 5 years |
| Orphan Drug Exclusivity (ODE) | 7 years |
| Generating Antibiotic Incentives Now (GAIN) | 5 years added to certain exclusivities |
| New Clinical Investigation Exclusivity | 3 years |
| Pediatric Exclusivity (PED) | 6 months added to existing patents/exclusivity |
Once market exclusivity ends, generic competition is poised to immediately introduce a alternative cheaper option that will erode sales for the patent owner. A dramatic example of patent cliff can be seen in the sales of Lipitor (Figure 1.4). Peak sales occurred in 2006 with sales nearing $13 billion in revenue, but at the end of patent exclusivity in 2011, sales dropped off precipitously to less than $4 billion in 2012. The trend continued to drop off through 2017 to less than $2 billion.
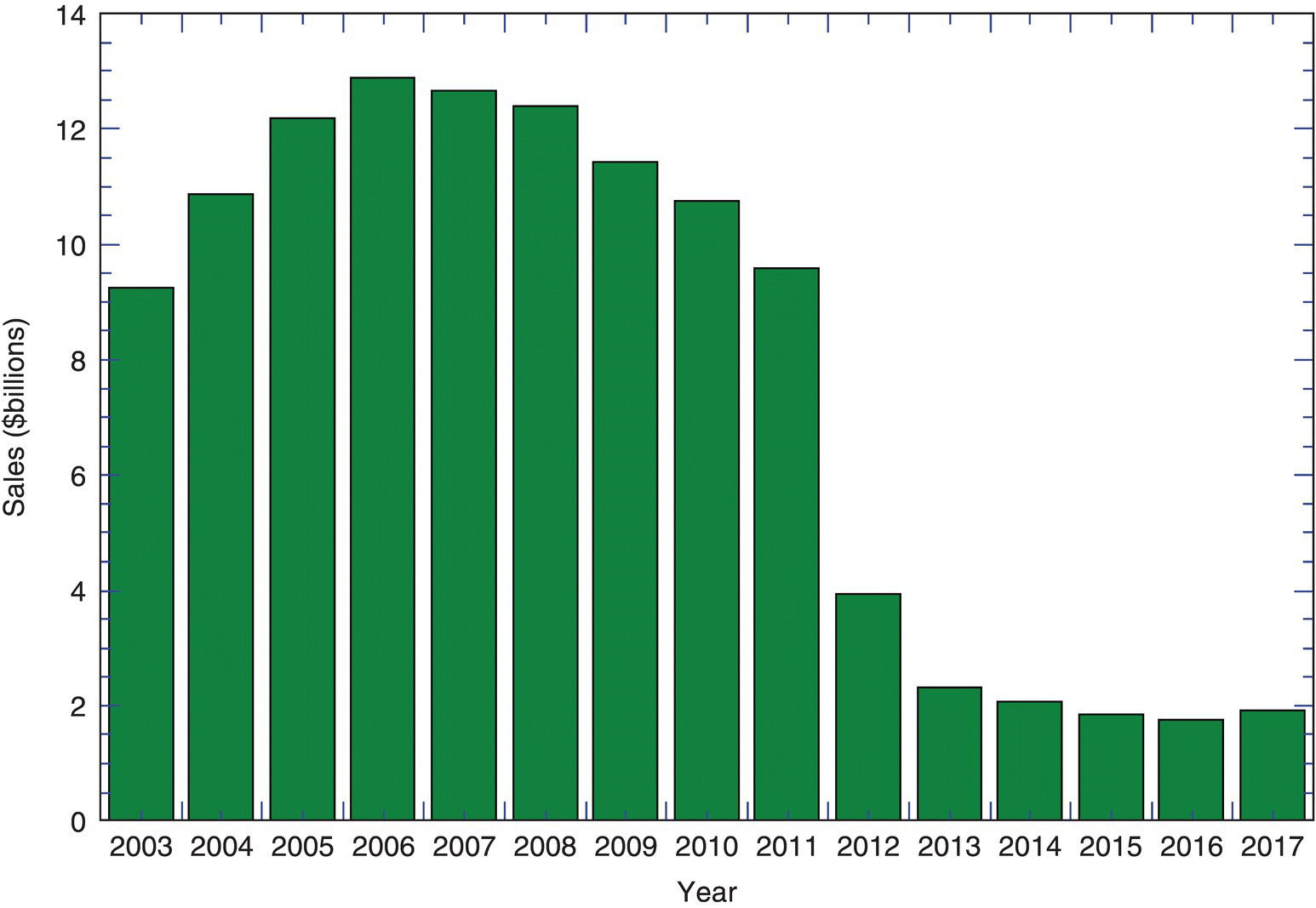
FIGURE 1.4 Sales of Pfizer's Lipitor (atorvastatin) between 2003 and 2017. In 2006 Lipitor generated nearly $13 billion in revenue. Patent exclusivity ended in 2011 and its impact was significant as seen by the significant drop on revenue in subsequent years (known as the “patent cliff”).
Source: Data from www.statista.com.
It now takes 10–15 years for a new medicine to go from the discovery laboratory to the pharmacy. Figure 1.5 shows the typical development activity timeline from discovery to launch. From thousands of compounds evaluated for potential therapeutic effect, very few will clear all the safety, efficacy, and clinical hurdles to make it to approval. Figure 1.5 also shows how a general range of volunteers, and clinical supplies, increases through phases I–III of clinical trials with clinical development typically lasting six years or more.

FIGURE 1.5 Drug research and development can take 10–15 years with one approval from 5 to 10 000 compounds in discovery. BLA, biologics license application; FDA, food and drug administration; IND, investigational new drug; NDA, new drug application.
Source: Adapted from Pharmaceutical Research and Manufacturers of America (PhRMA), publication Pharmaceutical Industry Profile 2009 (www.phrma.org).
Before entering human clinical studies, the drug candidate is tested for safety and efficacy in preclinical studies. When the candidate looks promising for a targeted indication or potential therapeutic effect, the company files an Investigational New Drug Application (IND) for regulatory agency and clinical site approval. At this time, referred to as phase I, the drug candidate will be tested in a few healthy volunteers (n ~ 10's) in single and multiple dose studies to test for safety and understand human pharmacokinetics. If the phase I evaluations are positive, then the candidate can progress to a larger population of healthy volunteers (n ~ 100's) pending approval by the regulatory agency on study design, i.e. doses, route of administration, detection of efficacy, and side effects. If the candidate passes the phases I and II hurdles ensuring safety and efficacy, then the clinical teams will design incrementally larger, broader, and worldwide clinical studies in test patients (phase III, n ~ 1000's).
The two common exceptions to conducting phase II studies in healthy volunteers are for oncology or biological candidates. These candidates proceed directly into the patient population, referred to as phase III, to test treatment of the indicated cancer or to progress the known safe and efficacious candidate derived from human antibodies or viruses, respectively.
After several years of careful study, the drug candidate may be submitted to the regulatory agency (e.g. FDA, EMA, PMDA) for approval. Depending on the type of API, the regulatory submission may need to be filed differently. For example, in the United States, a small molecule is submitted as an NDA, while a biologic is submitted as a Biologics Licensing Application (BLA).
As mentioned, the 2014 cost to advance a NCE or NME to market was estimated at $2.6 billion. The cost of product development that includes the cost to manufacture clinical supplies is estimated to be in the range of 30–35% of the total cost of bringing a NCE/NME to market with the following other cost contributors: discovery 20–25%, safety and toxicology 15–20%, and clinical trials 35–40% [7]. The distribution is graphically displayed in Figure 1.6. Clearly the distribution will depend on the specific drug, its therapeutic area, dose, and specific company.

FIGURE 1.6 Estimated distribution of product development costs within R&D with the total cost to bring a new chemical entity NCE to market in the range of $1–3.5 billion.
Source: Adapted from Suresh and Basu [7].
Chemical engineers, chemists, biologists, pharmaceutical scientists, and others make up the diverse scientific disciplines of product development that include API and formulation development including API and DP manufacture of clinical supplies.
1.3 BEST SELLERS
The top 20 drugs in sales are shown in Table 1.3 with Humira, topping the list with 2017 global sales of $18.43 billion. Interestingly 11 of these top drugs are biologics, 1 is a vaccine, and the remaining 8 are small molecule drugs. The top 20 selling drugs in that year total nearly $135 billion. This has changed significantly since the publication of the original version of this book in 2010 when the majority of top‐selling drugs at that time were small molecules.
TABLE 1.3 Top 20 Global Pharmaceutical Products (2017 Sales)
Source: From https://igeahub.com/2018/04/07/20‐best‐selling‐drugs‐2018
| Rank | Brand Name | API | Marketer | Indication | 2017 Sales ($ Billion) |
| 1 biologic |
Humira | Adalimumab | AbbVie | Autoimmune diseases and rheumatoid arthritis | 18.43 |
| 2 biologic |
Eylea | Aflibercept | Regeneron Pharmaceuticals and Bayer | Macular degeneration | 8.23 |
| 3 | Revlimid | Lenalidomide | Celgene | Multiple myeloma | 8.19 |
| 4 biologic |
Rituxan | Rituximab (MabThera) | Roche and Biogen | Treatment of cancer | 8.11 |
| 5 biologic |
Enbrel | Etanercept | Amgen and Pfizer | Autoimmune diseases including rheumatoid arthritis, psoriasis, and other inflammatory conditions | 7.98 |
| 6 biologic |
Herceptin | Trastuzumab | Roche and Biogen | Treatment of cancer, mainly breast and gastric | 7.55 |
| 7 | Eliquis | Apixaban | BMS and Pfizer | Anticoagulant, mainly used to treat atrial fibrillation and deep vein thrombosis | 7.40 |
| 8 biologic |
Avastin | Bevacizumab | Roche and Biogen | Advanced colorectal, breast, lung, kidney, cervical, and ovarian cancer and relapsed glioblastoma | 7.21 |
| 9 biologic |
Remicade | Infliximab | Johnson & Johnson and Merck | Autoimmune diseases | 7.16 |
| 10 | Xarelto | Rivaroxaban | Bayer and Johnson & Johnson | Anticoagulant | 6.54 |
| 11 | Januvia/Janumet | Sitagliptin | Merck | Treatment of type 2 diabetes | 5.90 |
| 12 biologic |
Lantus | Insulin glargine | Sanofi | Long‐acting human insulin analog for the treatment of diabetes | 5.65 |
| 13 vaccine | Prevnar 13/Prevener | Pneumococcal 13‐valent conjugate vaccine | Pfizer | Pneumococcal vaccine | 5.60 |
| 14 biologic |
Opdivo | Nivolumab | BMS | Melanoma | 4.95 |
| 15 biologic |
Neulasta/Peglasta/Neupogen | (Pegfilgrastim and Filgrastim) | Amgen and Kyowa Hakko Kirin | Neutropenia; decreases the incidence of infection during cancer treatment | 4.56 |
| 16 | Lyrica | Pregabalin | Pfizer | Anti‐epileptic and neuropathic pain | 4.51 |
| 17 | Harvoni | Ledipasvir (sofosbuvir) | Gilead Sciences | HCV/HIV‐1 infection | 4.37 |
| 18 | Advair | Fluticasone and Salmeterol | GlaxoSmithKline | Asthma | 4.36 |
| 19 | Tecfidera | Dimethyl fumarate | Biogen | Multiple sclerosis | 4.21 |
| 20 biologic |
Stelara | Ustekinumab | Johnson & Johnson | Plaque psoriasis | 4.01 |
Shaded row indicates API is a new chemical entity; non‐shaded row indicates API is a biologic.
The majority of the 20 top sellers have remained in similar positions over the past 2 years; however a few have made significant moves in this short time. For instance, Harvoni was the second place with $9.08 billion in sales in 2016 and dropped to seventeenth place in 2017 with $4.37 billion sales. Another interesting move was Eylea from thirteenth to second place from 2016 to 2017 increasing sales from $5.05 to 8.23 billion. It is also noteworthy that 9 of the top 20 products are partnerships, which further illustrates the significant cost to develop DPs are often sharing the risk.
In Table 1.4 the top‐selling drugs of all time were analyzed by Forbes, utilizing the lifetime sales of branded drugs between 1996 and 2012 and company reported sales data between 2013 and 2016. It is noteworthy that the number one in sales, Lipitor, at $148.7 billion is not even on the top 20 drug sales list for 2017 in Table 1.3. While there is a large gap between the top two selling drugs, amounting to $53 billion for Lipitor above Humira, if Humira annual sales continue at $18 billion, it will outperform Lipitor as the all‐time best‐selling drug in just under 3 years. However, the patent expiry for Humira was in 2016, and therefore sales may drop rapidly in the coming years if generics or biosimilars are able to penetrate the market.
TABLE 1.4 Fifteen Top‐Selling Drugs (2013–2016) for Cumulative Sales Through 2016
Source: Data from https://www.fool.com/amp/investing/2017/03/13/the‐19‐best‐selling‐prescription‐drugs‐of‐all‐time.aspx
| Drug/Drug Product Typea | API | Marketer | Approval | $ Billion | |
| 1 | Lipitor Atorvastatin/film‐coated tablet |  |
Pfizer | 1996 | 148.7 |
| 2 | HumiraAdalimumab/solution for injection | Antitumor necrosis factor (TNF) monoclonal antibody | AbbVie | 2003 | 95.6 |
| 3 | Advair (United States)Seretide(EU) fluticasone + salmeterol/dry powder inhaler | 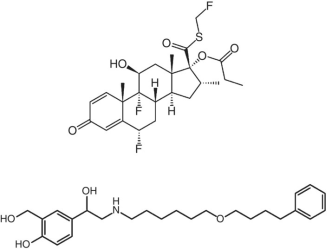 |
GlaxoSmithKline | 2001 | 92.5 |
| 4 | Remicade/lyophilized powder for constitution, solution injection | Anti‐TNF chimeric monoclonal antibody | Janssen | 1998 | 85.5 |
| 5 | Plavix clopidogrel/film‐coated tablet | 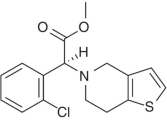 |
Bristol‐Myers Squibb | 1997 | 82.3 |
| 6 | Enbrel Etanercept/subcutaneous injection | Fusion protein produced by recombinant DNA | Amgen/Pfizer | 1998 | 77.2 |
| 7 | Rituxan Rituximab/solution injection | Chimeric monoclonal antibody | RocheGenentech | 1997 | 75.9 |
| 8 | Herceptin Trastuzumab/intravenous (IV) infusion | Monoclonal antibody | RocheGenentech | 1999 | 65.2 |
| 9 | Avastin Bevacizumab/IV infusion | Monoclonal antibody | RocheGenentech | 2004 | 62.3 |
| 10 | Nexium Esomeprazole/delayed release capsule; IV injection | 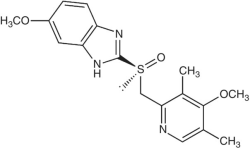 |
AstraZeneca | 2001 | 60.2 |
| 11 | Zyprexa Olanzapine/oral disintegrating tablets; injections | 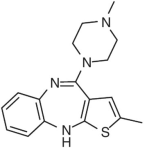 |
Eli Lilly | 1996 | 60.2 |
| 12 | Diovan Valsartan/film‐coated tablet |  |
Novartis | 1997 | 60.1 |
| 13 | LantusInsulin glargine/Subcutaneous Injection | Long‐acting basal analog | Sanofi‐Aventis | 2001 | 58.3 |
| 14 | Crestor Rosuvastatin/film‐coated tablet |  |
AstraZeneca | 2003 | 55.2 |
| 15 | Singulair Montelukast/chewable tablet |  |
Merck | 1998 | 47.4 |
awww.drugs.com source of dosage form type for originator drug.
1.4 PHARMACEUTICAL RESEARCH AND DEVELOPMENT EXPENDITURES
1.4.1 Pharmaceutical Development
In general, pharmaceutical product development is different than most other research intensive industries. Specifically in the pharmaceutical industry, there is the consistent need to ensure that clinical supplies are manufactured and delivered in a timely manner regardless of the current state of development or efficiency of the process. In other words, delivering clinical supplies when they are needed requires using technology that is good enough at the time even if it is not a fully optimized process. However, this is a regulated industry for clinical supplies as well as for commercial.
Further, process development, optimization, and scale‐up historically tends to be an iterative approach [8] – clinical supply demands are met by scale‐ups to kilo lab or pilot plant through phase I, phase II, and phase III, and it is through this period that R&D teams (including analysts/chemist/engineers, referred to as the ACE model) refine, optimize, and understand the API and DP processes to enable them to be eventually transferred to manufacturing. Manufacturing of clinical supplies in kilo lab, pilot plant, and solid dosage plants occurs under the constraints of cGMP conditions, which is discussed further in the chapter on kilo lab and pilot plant. The pilot plant and kilo lab are also sometimes used to “test” the scalability of a process. In this way, pilot plants serve a dual purpose, which make them unique as compared with non‐pharmaceutical pilot plants. In terms of cost, however, large‐scale experimentation in kilo lab or pilot plant can be significant – so there has been a shift toward greater predictability at lab scale to offset the need for pilot plant‐scale “technology demonstration” experiments. Engineers through their training are well suited to scale‐up and scale‐down processes and can effectively model the chemical and physical behaviors in the lab to ensure success on scale. Many chapters in these two volumes discuss how scale‐up/scale‐down of various unit operations is performed. Chemical engineers are well trained in process modeling and optimization that support the reduction of experimentation and rehearsal batches prior to commercialization. This helps to reduce the number of larger‐scale “experiments,” thereby lowering costs during R&D. In this way, with the recent trend toward increasing efficiency and continuous improvement, the pilot plant and kilo labs are preferentially utilized to manufacture supplies for toxicological and clinical supplies rather than being used to “test” or verify that the chemistry or process will work on scale.
A primary focus of process development is to drive down the cost contribution of the API to the final formulated pharmaceutical product cost while at the same time optimizing to ensure quality and process robustness. The impact of API costs on overall manufacturing costs is approximated in Figure 1.7. The cost contribution of API is expected to increase with increasing complexity of molecular structures of APIs, e.g. biologics. It is interesting to note that API molecular complexity can often impact API cost more than formulation or packaging costs. As Federsel points out that, “Given the importance of ‘time to market’ which remains one of the highest priorities of pharmaceutical companies, the need to meet increasingly stretched targets for speed to best route has come to the forefront in process R&D” [9]. In the not too distant past it was considered satisfactory to have a good‐enough synthetic route that was fit for purpose (i.e. could support the quantities of material needed) but not one considered best or lowest cost ($/kg of API). The prevailing view was that the market would bear higher product pricing as compensation for higher cost of goods (COGs). Further cost reduction through new routes could be and were pursued post‐launch with savings realized later in the life cycle. According to Federsel, and evidenced frequently in contemporary R&D organizations, this approach is no longer viable, at least not as a default position. Instead the best synthetic route to API (i.e. route with ultimate lowest cost materials) coupled with best process design and engineering (process with lowest processing costs) must be worked out as early as possible in API process development [9]. The optimal API process developed by the time of launch is necessary to extract additional revenues and respond to reduced COG margins. Achieving this requires continuous improvement in scientific and technical tools as well as multidisciplinary skill sets in the R&D labs, including chemical engineering science. The implementation of process design principles, drawing on the right skill sets, both from chemistry and engineering perspectives during clinical phase II, is considered such an important step toward leaner more cost‐effective processes readied for launch that several portions of this book will expand on this concept.
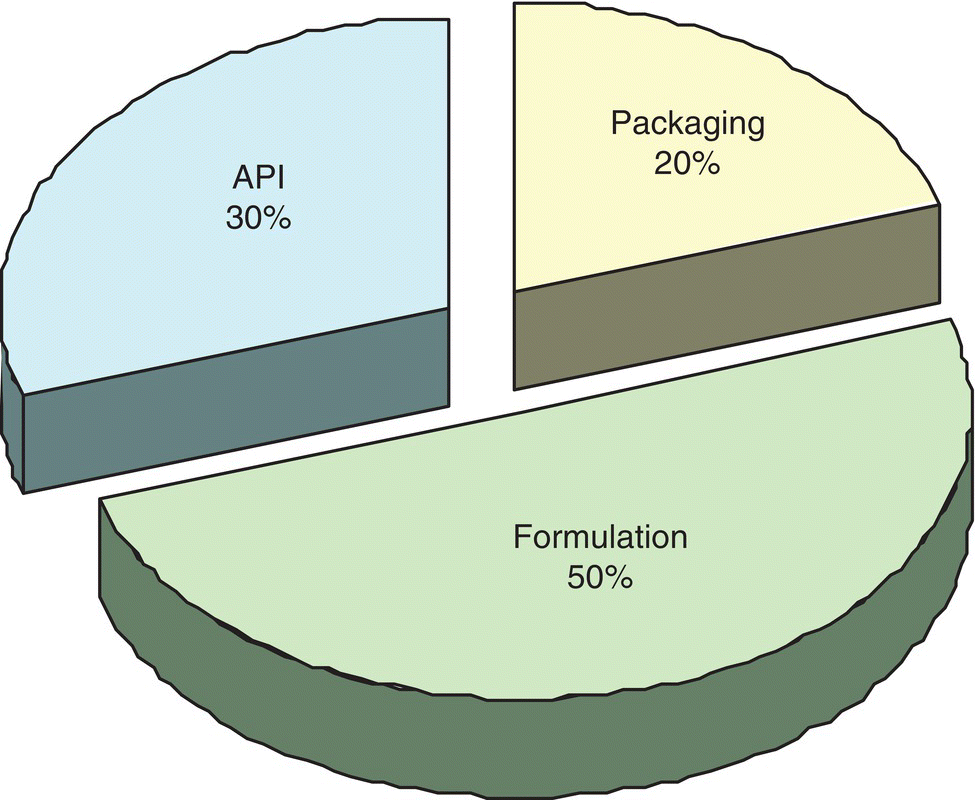
FIGURE 1.7 Average cost of goods (COG's) components in final dosage form across a large product portfolio – may vary widely for individual drugs (e.g. for API from 5 to 40%).
Source: Reprinted with permission from Federsel [9]. Copyright (2006) Elsevier.
1.5 RECENT TRENDS FOR PHARMACEUTICAL DRUG AND MANUFACTURING
During the past decade, the pharmaceutical field has evolved to a science and risk‐based industry. It is now commonplace for the regulatory dossier to contain scientifically rigorous information and descriptions of the risk management approach used for decision making. Now, the industry is undergoing significant changes in the API (from small molecule to biologics), manufacturing (from batch toward continuous), medicinal approach (generalized to personalized), and complexity of manufacturing (from simple dosage forms toward additive manufacturing or 3D printing).
1.5.1 Drug Substances Trend Toward Biologics
Biologic medicines are revolutionizing the treatment of cancer, autoimmune disorders, and rare illnesses and are therefore critical to the future of the pharmaceutical industry. Cancer immunotherapy includes monoclonal antibodies, checkpoint inhibitors, antibody‐drug conjugates (ADCs), and kinase inhibitors, to name a few.
From the 2017 top‐selling drugs shown in Table 1.3, there is a strong trend toward drugs derived from biological origins dominating the market than small molecules. In fact, the majority of best sellers are biologics, often monoclonal antibodies, which treat new diseases such as Crohn's and ulcerative colitis previously unmet medical needs by small molecule APIs.14 It is also evident that the biologics retain their value even after patent exclusivity expires, e.g. Humira sales continue to grow post‐patent expiry in 2016. The current generic industry is skilled in small molecule development but appears to be challenged to rapidly erode sales for biologics. In fact, in the coming years, it appears the first biologic medicine may take over as the all‐time best seller from Lipitor.
Biological drug candidates include many different types of molecules including monoclonal antibodies, vaccines, therapeutic proteins, blood and blood components, and tissues.15 In contrast to chemically synthesized drugs, which have a well‐defined structure and can be thoroughly verified, biologics are derived from living material (human, animal, microorganism, or plant) and are vastly larger and more complex in structure. Biosimilars are versions of biologic products that reference the originator product in applications submitted for marketing approval to a regulatory body and are not exactly generic equivalents. However, biosimilar DPs are far more complex to gain regulatory approval in developed markets than for chemical generics and may involve costly clinical trials. Those that succeed will also have to compete with the originator companies who are unlikely to exit the market considering their expertise and investments. The biosimilars market is expected to increase significantly with the first FDA approval for Sandoz ZARXIO subcutaneous IV injection product in 2015 that helped establish a clear pathway for gaining regulatory approval [10]. Recently, Hospira, a Pfizer company, received FDA approval of their epoetin alfa biosimilar, Retacrit, in May 2018 [11].
Biologic and biosimilar medicines are treating illnesses with unmet needs while retaining value even after post‐exclusivity period. These are clear advantages for the originator, biopharmaceutical company developing biologic medicines, and are expected to continue to increase in the coming years. While the major disciplines making advancements in this area are biologists and chemists, there is a role for chemical and biochemical engineers in the design and development of the processing and purification steps. Chemical engineers are skilled at developing predictive models, and scale‐up/scale‐down principles, which make them a key contributor to this growing field. In fact, for biologics, scale‐down predictive models of process steps were established and helped pave the way for biological products to use them for validation [12].
Chemical engineers that include biochemical engineering are well trained to impact the biotech industry, which utilizes cellular and biomolecular processes for new medicines [13]. Chemical engineers can also support the design of protein recovery, purification, and scaling up from lab to commercial production of the therapeutic proteins.
1.5.2 Lean Manufacturing
Pharmaceutical production of APIs and DPs can be generally characterized as primarily batch‐operated multipurpose manufacturing plants. At these facilities commercial supplies of API intermediates, APIs, and DPs are manufactured before being packaged, labeled, and distributed to customers. Pharmaceutical production plants were typically designed to be flexible to allow a number of different products to be run in separate equipment trains, depending on the demand. Further, these facilities have various degrees of automation, relatively high levels of documentation, and change control to manage reconfigurations, with relatively long downtimes for cleanup and turnover of the plant between product changes [14]. These considerations are in part to meet regulatory requirements for commercial manufacturing. Manufacturing costs or COGs often account for approximately one‐third of the total costs with expenses exceeding that of R&D [15]. For this reason COG's have received considerable focus as an area of opportunity for potential savings [7, 16].
It has been claimed that through adopting QbD principles and principles of lean manufacturing, pharmaceutical companies, as an aggregate, could save in the range of $20–50 billion/year by eliminating inefficiencies in current manufacturing [16]. This translates to 10–25% reduction in current COGs. An early QbD product approval of Chantix afforded an opportune chance to prove the benefits of these lean manufacturing and QbD principles. Chantix (varenicline tablet) was approved as an immediate release tablet commercially manufactured in the Pfizer Illertissen, Germany site. The OEB classification of this product required containment, available at small scale in this facility. Product demand increased dramatically in 1 year by 430%. The site employed lean manufacturing to eliminate process inefficiencies and wastes to increase production from 1 batch/day to 3 batches/day, delivering the desired 900 batches/year in the small‐scale facility [17]. The lean manufacturing was indeed proven when the manufacturing maintained an inventory of only approximately one week lead ahead of demand.
The principles of lean manufacturing are often cited as an approach to reduce COGs in pharmaceutical development and manufacturing. Lean manufacturing describes a management philosophy concerned with improving profitability through the systematic elimination of activities that contribute to waste – thus the central theme to lean manufacturing is the elimination of waste where waste is considered the opposite of value. Based on the work of Taiichi Ohno, creator of the Toyota Production System, wastes are considered based on the following [18]:
- Overproduction
- Waiting
- Transportation
- Unnecessary processing
- Unnecessary inventory
- Unnecessary motion
- Defects
All of these wastes have the effect of increasing the proportion of non‐value‐added activities. Lean thinking is obviously applicable to many industries including pharmaceutical manufacturing as well as pharmaceutical development. Continuous processing, for pharmaceutical APIs and DPs, is one application of lean thinking applied to pharmaceutical manufacturing. The challenge is that batch processing inherently leads to overproduction and specifically the buildup of excess inventory of intermediates and DPs to supply the market. This leads to longer cycle times and is addressed through the concepts of continuous manufacturing (CM).
According to Ohno, “The greatest waste of all is excess inventory” where in simplest terms, excess inventory incurs cost associated with managing, transporting, and storing inventories adding to the waste. Large inventories also tie up large amounts of capital. Excess inventory represents an opportunity cost where capital is held up in the form of work in process (WIP), API finished goods, and formulated finished goods versus what could be invested elsewhere or back into R&D. Implementation of lean manufacturing principles can be used to develop workflows and infrastructures to reduce inventories. One way to reduce inventories is through continuous processing. Several chapters discuss the technical benefits of CM. A reliable steady delivery of product API and DP through small product‐specific continuous plants could potentially reduce the level of inventory required in a dramatic way if the workflows were designed to ensure consistent delivery of product to packaging and distribution. The facilities of continuous production trains tend to be significantly smaller.
The costs of inventory holdings are significant and include both the carrying cost and the cash value of the inventory. The carrying costs of inventory include two main contributions – (i) weighted average cost of capital (WACC) and (ii) overhead [19].
Estimates for the combined carrying cost of WACC and overhead range from 14 to 25% that translates to approximately 20% return for every dollar of inventory eliminated [20]. Technology platforms and new workflows designed to minimize the need for stockpiling API and DP inventories across the industry therefore would seem to offer very rapid payback.
1.5.3 Continuous Manufacturing
For a large pharmaceutical company carrying $5 billion in inventories, the holding cost based on the combined WACC and overhead of 20% is approximately $1 billion/year. Considered another way, technologies that ensure a reliable and steady distribution of product with the result of eliminating the need to build and store massive inventories can return the company cost savings equivalent to a blockbuster drug (generating $billion/year). Indeed one of the three factors having the largest impact on the profitability of a manufacturing organization is inventory with the other two being throughput and operating expense according to Goldratt and Cox [21]. Continuous processing if designed for reliable operations essentially year‐round or in other cases simply “on demand” could potentially eliminate the need to accumulate significant inventories above and beyond two to four weeks of critical safety stocks of finished goods.
CM across API and DP integrated under one roof as a platform technology is one long‐term approach to transforming the way the industry manages their commercial supply chain.
As one reference cites, “Even for very small processes, continuous processes will prove to be less expensive in terms of equipment and operating costs. Dedicated continuous processes often put batch processes out of business” [22]. The real point here is that continuous is one approach to lean manufacturing and to reducing inventories and costs but certainly not the only approach. Other lean systems can be devised that utilize the existing batch facilities as well. Since the publication of the first edition of this book in 2010, there has been a significant wave of interest in considering continuous processing for pharmaceutical API and DP.
In July 2015 FDA granted Vertex Pharmaceuticals approval of the first DP, Orkambi, a cystic fibrosis (CF) drug, to be produced using a CM process. Vertex's second drug, Symdeko, for treating the underlying cause of CF occurred in February 2018. Janssen aims to manufacture 70% of their “highest volume” products using CM within eight years.16 In addition, they intend to increase yield by reducing waste by 33% and reduce manufacturing and testing cycle times by 80% through the use of CM. Their claim is that CM can reduce operating costs by as much as 50%, gain higher throughputs, and significantly reduce waste [23]. Janssen's HIV drug Prezista is also manufactured via a continuous process after obtaining approval to convert from batch to continuous.17 Pfizer and Eli Lilly have made investments in CM and recently submitted an NDA or gained approval for a product, respectively. Merck states that CM will help achieve their goals of well‐controlled processes with flexible sizes to handle small‐to‐large volume products localized closer to the customer.18
Merck Manufacturing Division targets a total lead time of 90 days formulation to the patient, reducing the current timing by one‐quarter. Multiple companies are teaming together to leverage CM, e.g. Novartis‐MIT Center for Continuous Manufacturing. A critical component of CM is that PAT is embedded into the overall plan for monitoring and control of the process. As stated by Kevin Nepveux, “one of the best ways to go about implementing CM processes is to develop the analytics in‐line with the application.”19 In summary, CM requires significant focus by chemical engineers as there is more attention on cost savings and cost efficiencies.20
1.5.4 Personalized Medicine
“One size doesn't fit all is a tenet of personalized medicine, also called precision medicine,” states Lisa Esposito in a recent report [24]. In her article, she highlights the long‐standing personalized medicine approach taken to treat cancer based on the individual patient's disease state and conditions. There is a growing expectation that the pharmaceutical industry should deliver DPs targeted to the individual, tailoring the amount of drug based on their mass, metabolism, genetic factors, and disease state. In this section, we discuss two approaches for manufacturing personalized/precision medicine through a pharm‐on‐demand concept for military personnel and for complex dosage forms using additive manufacturing (referred to as 3D printing).
The Defense Advanced Research Projects Agency (DARPA) Battlefield Medicine program is keenly interested in miniaturized, flexible platforms for end‐to‐end manufacturing of pharmaceuticals to support the troops on location. As discussed in other chapters within this book, advances in continuous flow synthesis, chemistry, biological engineering, and downstream processing, coupled with online analytics, automation, and enhanced process control measures, are proving that such capabilities are ready for implementation. The desire is to have a mobile, on‐demand pharmacy located at the battlefield that could ensure readiness to treat threats of chemical, biological, radiological, and nuclear weapons [25].
1.5.5 Additive Manufacturing
Additive manufacturing, also referred to as three‐dimensional (3D) printing, is an automated process of building layer by layer a complex dosage form personalized and manufactured on demand. FDA approved the first 3D printed DP in August 2015 for Aprecia Pharmaceuticals SPRITAM product as a disintegrating tablet [26].
The 3D printing in this case binds the powders while maintaining a porous structure (without the typical compression of a tablet press), providing a fast dissolving tablet. For example, 1000 mg of levetiracetam dissolves within seconds [27]. Extensions of 3D printing include printing extremely low dose APIs or highly potent APIs, but encapsulated with excipients, thus reducing potential exposure. Norman et al. [27] provide a thorough review of the different modalities of 3D printing for pharmaceutical manufacturing, which includes an analysis of the potential benefits of such products.
1.6 CHEMICAL ENGINEERS SKILLED TO IMPACT FUTURE OF PHARMACEUTICAL INDUSTRY
The fundamental principles taught in the chemical engineering curriculum ensure the chemical engineer is well poised to apply them to solve the coming challenging issues in the pharmaceutical industry. Chemical engineers are uniquely positioned to help address these needs in part derived from their ability to predict using mathematical models and their understanding of equipment and manufacturability. As Wu et al. highlighted, chemical engineers can help transform pharmaceutics from an industry focusing on inventing and testing to a process and product design industry [28]. Significant pressure exists on what used to be a historically high‐margin nature of the pharmaceutical industry to deliver safe, environmentally friendly, and economic processes in increasingly shorter timelines. This means fewer scale‐ups at kilo and pilot plant scale, with expectation that a synthesis or formulation can be designed in the lab to perform as expected (and right the first time) at the desired manufacturing scale.
Chemical engineers are also uniquely positioned to influence regulators by incorporating advancements such as continuous processing coupled with PAT into a highly regulated industry. From R&D through manufacturing within the pharmaceutical industry, chemical engineering can be leveraged to bring competitive advantage to their respective organizations through process and predictive modeling that lead to process understanding, improving speed of development, and developing new technology platforms and leaner manufacturing methods. The chapters in these two volumes are intended to provide examples of chemical engineering principles specifically applied toward relevant problems faced in the pharmaceutical sciences and manufacturing areas. Further the broader goal of this work is to promote the role of chemical engineering within our industry, to promote the breadth of skill sets therein, and to showcase the critical synergy between this discipline and the many scientific disciplines that combine to bring pharmaceutical drugs and therapies to patients in need around the world.
REFERENCES
- 1. Janusz Rzeszotarski, W. (2000). Encyclopedia of Chemical Technology.
- 2. FDA (2009). International conference on harmonisation: guidance on Q8(R1) pharmaceutical development. Federal Register 74 (109): 27325–27326.
- 3. FDA (2006). International conference on harmonisation: guidance on Q9 quality risk management. Federal Register 71 (106): 32105–32106.
- 4. FDA (2009). International conference on harmonisation: guidance on Q10 pharmaceutical quality system. Federal Register 74 (66): 15990–15991.
- 5. DiMasi, J.A., Grabowski, H.G., and Hansen, R.W. (2016). Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics 47: 20–33.
- 6. Gregson, N., Sparrowhawk, K., Mauskopf, J., and Paul, J. (2005). Pricing medicines: theory and practice, challenges and opportunities. Nature Reviews Drug Discovery 4: 121–130.
- 7. Suresh, P. and Basu, P.K. (2008). Improving pharmaceutical product development and manufacturing: impact on cost of drug development and cost of goods sold of pharmaceuticals. Journal of Pharmaceutical Innovation 3: 175–187.
- 8. Dienemann, E. and Osifchin, R. (2000). The role of chemical engineering in process development and optimization. Current Opinion in Drug Discovery and Development 3 (6): 690–698.
- 9. Federsel, H.‐J. (2006). In search of sustainability: process R&D in light of current pharmaceutical R&D challenges. Drug Discovery Today 11 (21/22): 966–974.
- 10. FDA (2015). FDA approves first biosimilar product Zarxio. US Food and Drug Administration (6 March 2015).
- 11. FDA (2018). FDA approves first epoetin alfa biosimilar for the treatment of anemia. US Food and Drug Administration (15 May 2018).
- 12. Li, F., Hashimura, Y., Pendleton, R. et al. (2006). A systematic approach for scale‐down model development and characterization of commercial cell culture processes. Biotechnology Progress 22: 696–703.
- 13. Rovner, S.L. (2011). Process development shines in tough times. Chemical and Engineering News 89 (24): 41–44.
- 14. Behr, A., Brehme, V.A., Ewers, L.J. et al. (2004). New developments in chemical engineering for the production of drug substances. Engineering in Life Sciences 4 (1): 15–24.
- 15. Burns, L.R. (ed.) (2005). The Business of Healthcare Innovation. Cambridge: Cambridge University Press.
- 16. F‐D‐C Reports Inc. (2009). The Gold Sheet, Pharmaceutical & Biotechnology Quality Control, Attention turns to the business case of quality by design. F‐D‐C reports, January 2009.
- 17. am Ende, M.T., Bernhard, G., Lubczyk, V., Dressler, U. et al. (2008). Risk Management and Lifecycle Efficiency within Process Development. Leadership Forum 2008, Washington, DC, 29 May 2008.
- 18. Ohno, T. (1988). Toyota Production System Beyond Large Scale Production. New York: Productivity Press.
- 19. Cogdil, R.P., Knight, T.P., Anderson, C.A., and Drennan, J.K. (2007). The financial returns on investments in process analytical technology and lean manufacturing: benchmarks and case study. Journal of Pharmaceutical Innovation 2: 38–50.
- 20. Lewis, N.A. (2006). A tracking tool for lean solid‐dose manufacturing. Pharmaceutical Technology 30 (10): 94–109.
- 21. Goldratt, E.M. and Cox, J. (2004). The Goal: A Process for Ongoing Improvement, 3e. London: Routledge.
- 22. Biegler, L.T., Grossmann, I.E., and Westerberg, A.W. (1997). Systematic Methods of Chemical Process Design. Upper Saddle River: Prentice Hall.
- 23. Kuehn, S. E. (2015). Janssen embraces continuous manufacturing for Prezista. PharmaceuticalManufacturing.com (8 October 2015), https://www.pharmamanufacturing.com/articles/2015/janssen‐embraces‐continuous‐manufacturing‐for‐prezista/?show=all
- 24. Esposito, L. (2018). What does personalized medicine really mean? US News & World Report (26 January 2018).
- 25. Lewin, J.J., Choi, E.J., and Ling, G. (2016). Pharmacy on demand: new technologies to enable miniaturized and mobile drug manufacturing. American Journal of Health‐System Pharmacy 73 (2): 45–54.
- 26. United States Food and Drug Administration (2015). Highlights of prescribing information: Spritam. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207958s000lbl.pdf
- 27. Norman, J., Madurawe, R.D., Moore, C.M.V. et al. (2017). A new chapter in pharmaceutical manufacturing: 3D‐printed drug products. Advanced Drug Delivery Reviews 108: 39–50.
- 28. Wu, H., Khan, M.A., and Hussain, A.S. (2007). Process control perspective for process analytical technology: integration of chemical engineering practice into semiconductor and pharmaceutical industries. Chemical Engineering Communications 194: 760–779.